
A Thermal-Analysis-Technique-Based Mechanistic Approach toward the Release of Omeprazole from Solid Dosage Forms


 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Granules’ Production
2.2.2. Formulation Production (Capsule Filling and Tablet Production)
2.2.3. Tablet Characterization (Tablets’ Weight, Thickness, and Crushing Strength Uniformity)
2.2.4. Image Analysis
2.2.5. Dissolution Testing
2.2.6. Dissolution Profile Comparison
2.2.7. Data Analysis
2.2.8. Model-Independent Analysis of Dissolution Profiles
2.2.9. Thermal Analysis Techniques
3. Results
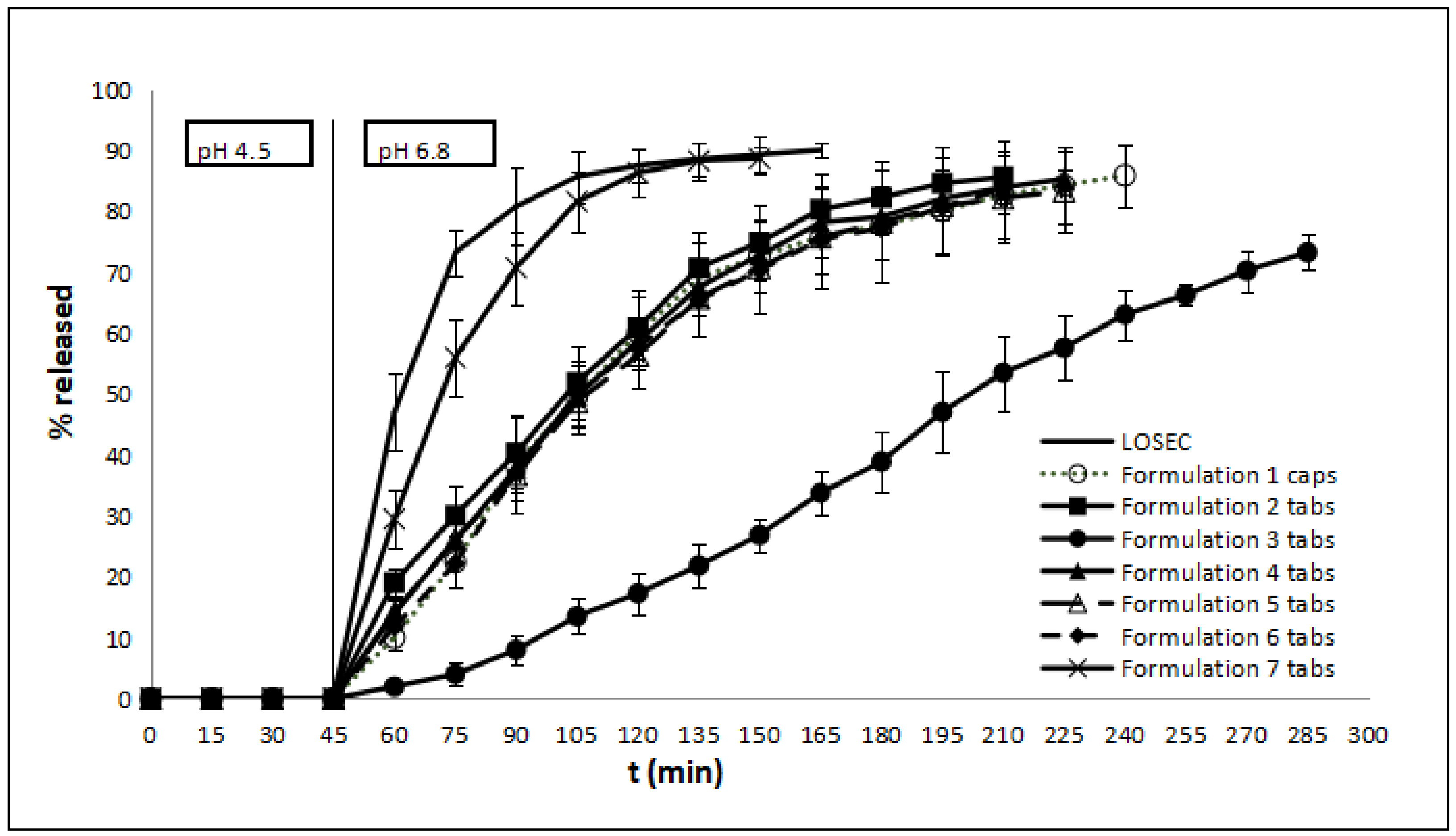
3.1. Release Kinetics of OME from Different Formulations
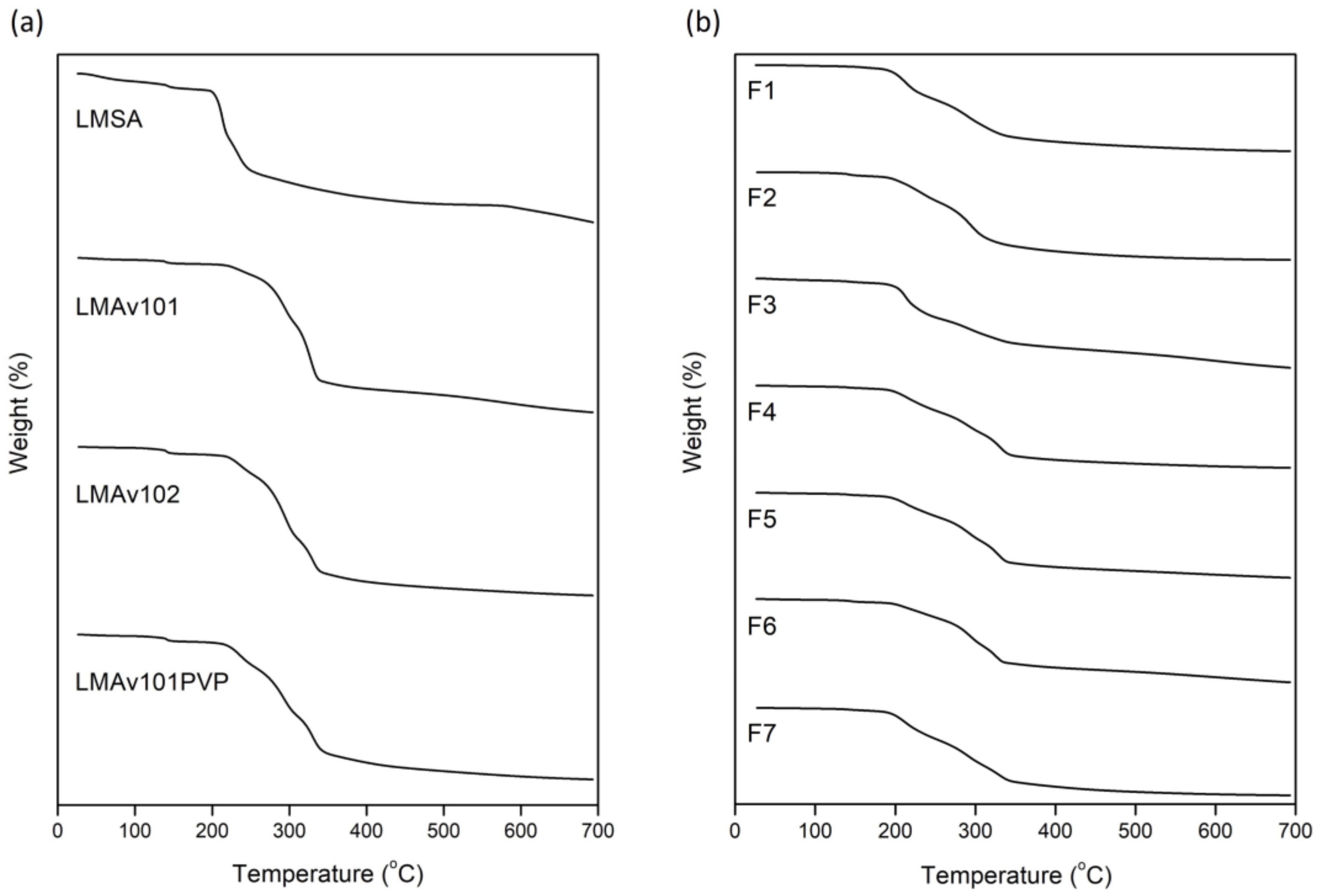
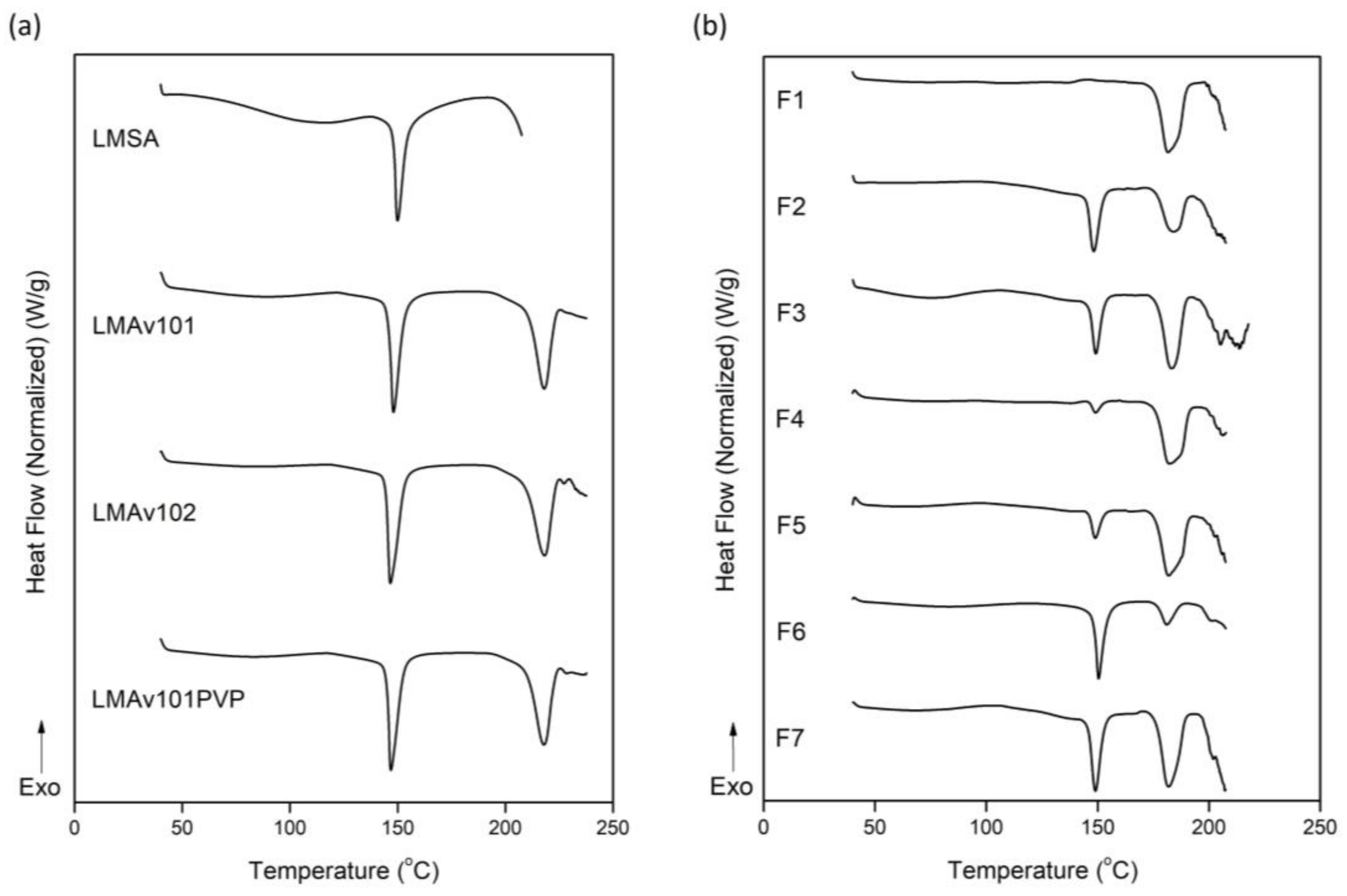
3.2. Thermotropic Behavior of OME Formulations
4. Discussion
Mechanistic Explanation of OME Release Kinetics: The Role of the Excipients
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brandstrom, A.; Lindberg, P.; Junggren, U. Structure-activity relationships of substituted benzimidazoles. Scand. J. Gastroenterol. Suppl. 1985, 108, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Massoomi, F.; Savage, J.; Destache, C.J. Omeprazole: A Comprehensive Review. Pharmacotherapy 1993, 13, 46–59. [Google Scholar] [CrossRef]
- Filho, V.J.T.; Andreazza, I.F.; Sato, M.E.O.; Murakami, F.S. Development of a multiparticulate system containing enteric release mini-tablets of omeprazole. Braz. J. Pharm. Sci. 2014, 50, 505–511. [Google Scholar] [CrossRef]
- El-Sayed, A.; Boraie, N.A.; Ismail, F.A.; El-Khordagui, L.K.; Khalil, S.A. Assessment of the pharmaceutical quality of omeprazole capsule brands marketed in Egypt. East. Mediterr. Health J. 2007, 13, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Sachs, G.; Shin, J.M.; Howden, C.W. Review article: The clinical pharmacology of proton pump inhibitors. Aliment. Pharmacol. Ther. 2006, 23, 2–8. [Google Scholar] [CrossRef]
- Shin, J.M.; Sachs, G. Pharmacology of Proton Pump Inhibitors. Curr. Gastroenterol. Rep. 2008, 10, 528–534. [Google Scholar] [CrossRef]
- Strand, D.S.; Kim, D.; Peura, D.A. 25 Years of Proton Pump Inhibitors: A Comprehensive Review. Gut Liver 2017, 11, 27–37. [Google Scholar] [CrossRef]
- El-Rouby, N.; Lima, J.J.; Johnson, J.A. Proton pump inhibitors: From CYP2C19 pharmacogenetics to precision medicine. Expert Opin. Drug Metab. Toxicol. 2018, 14, 447–460. [Google Scholar] [CrossRef]
- Farley, D. Making it easier to read prescriptions. FDA Consum. 1995, 29, 25–27. [Google Scholar]
- Lundell, L. The physiological background behind and course of development of the first proton pump inhibitor. Scand. J. Gastroenterol. 2015, 50, 680–684. [Google Scholar] [CrossRef]
- Perrie, Y.; Rades, T. FASTtrack: Pharmaceutics—Drug Delivery and Targeting, 2nd ed.; Pharmaceutical Press: London, UK, 2012; pp. 1–24. [Google Scholar]
- Shargel, L.; Wu-Pong, S.; Yu, A.B.C. Applied Biopharmaceutics & Pharmacokinetics, 6th ed.; McGraw-Hill Education: New York, NY, USA, 2012; 5p. [Google Scholar]
- Saha, S. Immediate Release Drug Delivery Systems: A Current Update. J. Appl. Pharm. Res. 2018, 6, 1–9. [Google Scholar] [CrossRef]
- Nyol, S.; Gupta, M.M. Immediate Drug Release Dosage Form: A Review. J. Drug Deliv. Ther. 2013, 3, 155–161. [Google Scholar] [CrossRef]
- Trenfield, S.J.; Basit, A.W. Nanotechnology for Oral Drug Delivery: From Concept to Applications, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 177–197. [Google Scholar]
- Montero Mirabet, M.; Skalsky, B. Advanced approaches for delayed-release formulations. ONdrugDelivery Mag. 2017, 77, 4–9. [Google Scholar]
- Chakravarthy, K.K.; Younus, M.; Shaik, S.; Pisipati, S.V.V. Formulation and Evaluation of Enteric Coated Pellets of Omeprazole. Int. J. Drug Dev. Res. 2012, 4, 257–264. [Google Scholar]
- Ronchi, F.; Sereno, A.; Paide, M.; Sacre, P.; Guillaume, G.; Stephenne, V.; Goole, J.; Amighi, K. Development and evaluation of an omeprazole-based delayed-release liquid oral dosage form. Int. J. Pharm. 2019, 567, 118416. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.T.; Porter, S.C. Coatings for Controlled-Release Drug Delivery Systems. Drug Dev. Ind. Pharm. 1998, 24, 1139–1154. [Google Scholar] [CrossRef]
- Thakral, S.; Thakral, N.K.; Majumdar, D.K. Eudragit®: A technology evaluation. Expert Opin. Drug Deliv. 2013, 10, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, J.; Da Silva, G.S.; Velho, M.C.; Beck, R.C.R. Eudragit®: A Versatile Family of Polymers for Hot Melt Extrusion and 3D Printing Processes in Pharmaceutics. Pharmaceutics 2021, 13, 1424. [Google Scholar] [CrossRef]
- Dallas, P. Handbook of Laboratory Exercises for the Undergraduate Pharmacy Students in National and Kapodistrian University of Athens; National and Kapodistrian University of Athens: Athens, Greece, 2023. [Google Scholar]
- Brockmeier, D.; Voegele, D.; Von Hattingberg, H. In vitro-in vivo correlation, a time scaling problem? Basic techniques for testing equivalence. Arzneimittelforschung 1983, 33, 598–601. [Google Scholar]
- Moore, J.W.; Flanner, H.H. Mathematical comparison of dissolution profiles. Pharm. Technol. 1996, 20, 64–74. [Google Scholar] [CrossRef]
- USP 29-NF24; Pharmacopoeia US. USP: Rockville, MD, USA, 2005.
- Ruiz, M.A.; Reyes, I.; Parera, A.; Gallardo, V. Determination of the stability of omeprazole by means of differential scanning calorimetry. J. Therm. Anal. Calorim. 1998, 51, 29–35. [Google Scholar] [CrossRef]
- Markovic, N.; Agotonovic-Kustrin, S.; Glass, B.; Prestidge, C.A. Physical and thermal characterisation of chiral omeprazole sodium salts. J. Pharm. Biomed. Anal. 2006, 42, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Murakami, F.S.; Lang, K.L.; Mendes, C.; Cruz, A.P.; Carvalho-Filho, M.A.; Silva, M.A. Physico-chemical solid-state characterization of omeprazole sodium: Thermal, spectroscopic and crystallinity studies. J. Pharm. Biomed. Anal. 2009, 49, 72–80. [Google Scholar] [CrossRef]
- Yohana Chaerunisaa, A.; Sriwidodo, S.; Abdassah, M. Microcrystalline Cellulose as Pharmaceutical Excipient. In Pharmaceutical Formulation Design—Recent Practices, 1st ed.; Ahmad, U., Akhtar, J., Eds.; IntechOpen: London, UK, 2020; 21p. [Google Scholar]
- Vlachou, Μ.; Pippa, Ν.; Siamidi, A.; Kyrili, A. Thermal analysis studies on the compatibility of furosemide with solid state and liquid crystalline excipients. Hem. Ind. 2020, 74, 15–23. [Google Scholar] [CrossRef]
- Soares, J.P.; Santos, J.E.; Chierice, G.O.; Cavalheiro, E.T.G. Thermal behavior of alginic acid and its sodium salt. Eclét. Quim. 2004, 29, 57–63. [Google Scholar] [CrossRef]
- Chadha, R.; Bhandari, S. Drug-excipient compatibility screening-Role of thermoanalytical and spectroscopic techniques. J. Pharm. Biom. Anal. 2014, 87, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, J.; Chen, T.; Sun, C.C.; Zheng, Y. Tablets of multi-unit pellet system for controlled drug delivery. J. Control. Release 2017, 262, 222–231. [Google Scholar] [CrossRef]
- Murthy Dwibhashyam, V.S.; Ratna, J.V. Key formulation variables in tableting of coated pellets. Indian J. Pharm. Sci. 2008, 70, 555–564. [Google Scholar] [CrossRef]
- Türkoğlu, M.; Varol, H.; Çelikok, M. Tableting and stability evaluation of enteric-coated omeprazole pellets. Eur. J. Pharm. Biopharm. 2004, 57, 279–286. [Google Scholar] [CrossRef]
- Tan, X.; Hu, J. Investigation for the quality factors on the tablets containing medicated pellets. Saudi Pharm. J. 2016, 24, 507–514. [Google Scholar] [CrossRef]
- Bodmeier, R. Tableting of coated pellets. Eur. J. Pharm. Biopharm. 1997, 43, 1–8. [Google Scholar] [CrossRef]
- Efentakis, M.; Buckton, G. The effect of erosion and swelling on the dissolution of theophylline from low and high viscosity sodium alginate matrices. Pharm. Dev. Technol. 2002, 7, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Holte, Ø.; Onsøyen, E.; Myrvold, R.; Karlsen, J. Sustained release of watersoluble drug from directly compressed alginate tablets. Eur. J. Pharm. Sci. 2003, 20, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Ghahramanpoor, M.K.; Najafabadi, S.A.H.; Abdouss, M.; Bagheri, F.; Eslaminejad, M.B. A hydrophobically-modified alginate gel system: Utility in the repair of articular cartilage defects. J. Mater. Sci. Mater. Med. 2011, 22, 2365–2375. [Google Scholar] [CrossRef]
- Draget, K.I.; Skjåk-Bræek, G.; Smidsrød, O. Alginate based new materials. Int. J. Biol. Macromol. 1997, 21, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Liew, C.V.; Chan, L.W.; Ching, A.L.; Heng, P.W.S. Evaluation of sodium alginate as drug release modifier in matrix tablets. Int. J. Pharm. 2006, 309, 25–37. [Google Scholar] [CrossRef]
- Enobakhare, B.; Bader, D.L.; Lee, D.A. Concentration and M/G ratio influence the physiochemical and mechanical properties of alginate constructs for tissue engineering. J. Appl. Biomech. 2006, 4, 87–96. [Google Scholar] [CrossRef]
- Martinsen, A.; Storra, I.; Skajak-Braek, G. Alginate as immobilization material: III. Diffusion properties. Biotechnol. Bioeng. 1992, 39, 186–194. [Google Scholar] [CrossRef]
- Yamagiwa, K.; Kozawa, T.; Ohkawa, A. Effects of alginate composition and gelling conditions on diffusional and mechanical properties of calcium alginate gel beads. J. Chem. Eng. Jpn. 1992, 28, 462–467. [Google Scholar] [CrossRef]
- Rowe, R.C.; Sheskey, P.J.; Quinn, M.E. Handbook of Pharmaceutical Excipients, 6th ed.; RPS Publishing: London, UK, 2009; 917p. [Google Scholar]
- Vlachou, M.; Tsiakoulia, A.; Eikosipentaki, A. Controlled release of the pineal hormone melatonin from hydroxypropylmethylcellulose/sodium alginate matrices in aqueous media containing dioctyl sulfosuccinate. Curr. Drug. Discov. Technol. 2007, 4, 31–38. [Google Scholar] [CrossRef]
- Narang, A.S.; Desai, D.; Badawy, S. Impact of excipient interactions on solid dosage form stability. Pharm. Res. 2012, 29, 2660–2683. [Google Scholar] [CrossRef]
- Franco, P.; De Marco, I. The Use of Poly(N-vinyl pyrrolidone) in the Delivery of Drugs: A Review. Polymers 2020, 12, 1114. [Google Scholar] [CrossRef]
- Mandal, S.; Basu, S.K.; Sa, B. Sustained release of a water-soluble drug from alginate matrix tablets prepared by wet granulation method. AAPS PharmSciTech 2009, 10, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Patra, C.H.N.; Pandit, H.K.; Singh, S.P.; Devi, M.V. Applicability and comparative evaluation of wet granulation and direct compression technology to Rauwolfia serpentina root powder: A technical note. AAPS PharmSciTech 2008, 9, 100–104. [Google Scholar] [CrossRef]
- Reza, M.M.M.; Bhuiyan, M.A.; Quadir, M.A. Comparative Evaluation of Wet Granulation and Direct Compression methods for preparation of Compressed tablets using Avicel pH 101. Bangladesh Pharm. J. 2002, 12, 19–22. [Google Scholar]
- Park, J.H.; Kwon, D.Y.; Heo, J.Y.; Park, S.H.; Park, J.Y.; Lee, B.; Kim, J.H.; Kim, M.S. Effect of Drug Carrier melting points on drug release of dexamethasone-loaded microspheres. Tissue Eng. Regen. Med. 2017, 14, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kang, H.J.; Kwon, D.Y.; Lee, B.K.; Lee, B.; Jang, J.W.; Chun, H.J.; Kim, J.H.; Kim, M.S. Biodegradable poly(lactide-co-glycolide-co-ε-caprolactone) block copolymers—Evaluation as drug carriers for a localized and sustained delivery system. J. Mater. Chem. B 2015, 3, 8143–8153. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, B.K.; Park, S.H.; Kim, M.G.; Lee, J.W.; Lee, H.Y.; Lee, H.B.; Kim, J.H.; Kim, M.S. Preparation of Biodegradable and Elastic Poly(ε-caprolactone-co-lactide) Copolymers and Evaluation as a Localized and Sustained Drug Delivery Carrier. Int. J. Mol. Sci. 2017, 18, 671. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients (mg) | 1 Caps | 2 Tabs | 3 Tabs | 4 Tabs | 5 Tabs | 6 Tabs | 7 Tabs |
|---|---|---|---|---|---|---|---|
| Omeprazole pellets (8.5% w/w) 1 | 235 | 235 | 235 | 235 | 235 | 235 | 235 |
| Lactose Monohydrate | - | 235 | 117.5 | 117.5 | 117.5 | - | 143 |
| Sodium alginate (medium viscosity) | - | - | 117.5 | - | - | - | - |
| MCC (Avicel® PH-102) | - | - | - | 117.5 | - | - | - |
| MCC (Avicel® PH-101) | - | - | - | - | 117.5 | - | 85 |
| Granules (36.3% Avicel® PH-101 60.7% Lactose 3% PVP K-30) | - | - | - | - | - | 235 | - |
| PVP Κ-30 | - | - | - | - | - | - | 7 |
| Magnesium Stearate | - | 4.70 | 4.70 | 4.70 | 4.70 | 4.70 | 4.70 |
| Total | 235 | 474.70 | 474.70 | 474.70 | 474.70 | 474.70 | 474.70 |
| No. of Tablet Formulation | Diameter (mm) | Weight (mg) (± SD) | Mean Tablet Height (mm) (± SD) | Crushing Strength (Ν) |
|---|---|---|---|---|
| F2 | 10 | 475.2 ± 1.3 | 4.3 ± 0.00 | 50 |
| F3 | 10 | 474.3 ± 1.1 | 4.3 ± 0.00 | 55 |
| F4 | 10 | 475.5 ± 0.8 | 4.3 ± 0.00 | 53 |
| F5 | 10 | 475.2 ± 1.2 | 4.3 ± 0.00 | 51 |
| F6 | 10 | 474.7 ± 1.9 | 4.3 ± 0.00 | 81 |
| F7 | 10 | 475.1 ± 1.3 | 4.3 ± 0.00 | 62 |
| Formulation | t20% | t50% | t90% | MDT |
|---|---|---|---|---|
| Losec® | 52 | 62 | 165 | 64.96 |
| F1 | 72 | 105 | * | 93.73 |
| F2 | 62 | 102 | * | 89.89 |
| F3 | 129 | 202 | * | 112.59 |
| F4 | 72 | 105 | * | 92.41 |
| F5 | 68 | 108 | * | 92.33 |
| F6 | 72 | 105 | * | 93.11 |
| F7 | 55 | 72 | 155 | 72.48 |
| Formulation | Zero Order | First Order | Higuchi | Korsmeyer-Peppas | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R2 | Y0 | K0 | R2 | Y1 | K1 | R2 | KH | R2 | KKP | n | |
| Losec® | 0.51 | 46.71 | 0.32 | 0.46 | 55.17 | −0.003 | 0.57 | 7.85 | 0.86 | 1.25 | 0.92 |
| F1 | 0.88 | 5.58 | 0.37 | 0.78 | 26.21 | −0.005 | 0.76 | 5.21 | 0.87 | 1.40 | 0.77 |
| F2 | 0.93 | 0.74 | 0.45 | 0.84 | 24.37 | −0.006 | 0.75 | 5.57 | 0.93 | 0.64 | 0.93 |
| F3 | 0.99 | −22.25 | 0.35 | 0.92 | 7.58 | −0.008 | 0.57 | 3.15 | 0.98 | 0.01 | 1.76 |
| F4 | 0.90 | 0.66 | 0.43 | 0.80 | 24.42 | −0.006 | 0.74 | 5.39 | 0.91 | 0.63 | 0.93 |
| F5 | 0.91 | 0.52 | 0.42 | 0.81 | 23.78 | −0.006 | 0.74 | 5.27 | 0.91 | 0.61 | 0.93 |
| F6 | 0.92 | −6.17 | 0.47 | 0.82 | 20.43 | −0.007 | 0.69 | 5.21 | 0.91 | 0.35 | 1.04 |
| F7 | 0.83 | 10.12 | 0.59 | 0.76 | 32.32 | −0.007 | 0.73 | 7.21 | 0.85 | 1.63 | 0.82 |
| Formulation | f1 | f2 |
|---|---|---|
| Losec® vs. 7 | 18.93 | 56.06 |
| 1 vs. 2 | 6.22 | 69.75 |
| 1 vs. 4 | 2.85 | 84.48 |
| 1 vs. 5 | 3.28 | 82.22 |
| 1 vs. 6 | 2.32 | 87.85 |
| 2 vs. 4 | 4.77 | 78.29 |
| 2 vs. 5 | 7.15 | 71.53 |
| 2 vs. 6 | 7.80 | 68.55 |
| 4 vs. 5 | 2.68 | 89.59 |
| 4 vs. 6 | 3.00 | 84.99 |
| 5 vs. 6 | 2.00 | 88.79 |
| Sample | T1 (°C) | ΔH1 (J/g) | T2 (°C) | ΔH2 (J/g) |
|---|---|---|---|---|
| LMSA | 150.0 | 49.4 | - | - |
| LMAv101 | 148.0 | 57.1 | 218.0 | 57.7 |
| LMAv102 | 146.6 | 77.3 | 218.2 | 83.9 |
| LMAv101PVP | 146.8 | 71.2 | 217.9 | 78.9 |
| F1 | - | - | 181.6 | 65.2 |
| F2 | 148.3 | 27.8 | 183.7 | 34.9 |
| F3 | 149.2 | 13.5 | 183.2 | 33.5 |
| F4 | 149.1 | 3.8 | 182.3 | 52.5 |
| F5 | 148.9 | 8.5 | 181.8 | 45.1 |
| F6 | 150.4 | 49.7 | 181.0 | 19.4 |
| F7 | 149.0 | 21.3 | 181.8 | 39.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agapakis, G.; Siamidi, A.; Kikionis, S.; Vlachou, M.; Pippa, N. A Thermal-Analysis-Technique-Based Mechanistic Approach toward the Release of Omeprazole from Solid Dosage Forms. Sci. Pharm. 2024, 92, 8. https://doi.org/10.3390/scipharm92010008
Agapakis G, Siamidi A, Kikionis S, Vlachou M, Pippa N. A Thermal-Analysis-Technique-Based Mechanistic Approach toward the Release of Omeprazole from Solid Dosage Forms. Scientia Pharmaceutica. 2024; 92(1):8. https://doi.org/10.3390/scipharm92010008
Chicago/Turabian StyleAgapakis, Georgios, Angeliki Siamidi, Stefanos Kikionis, Marilena Vlachou, and Natassa Pippa. 2024. "A Thermal-Analysis-Technique-Based Mechanistic Approach toward the Release of Omeprazole from Solid Dosage Forms" Scientia Pharmaceutica 92, no. 1: 8. https://doi.org/10.3390/scipharm92010008
APA StyleAgapakis, G., Siamidi, A., Kikionis, S., Vlachou, M., & Pippa, N. (2024). A Thermal-Analysis-Technique-Based Mechanistic Approach toward the Release of Omeprazole from Solid Dosage Forms. Scientia Pharmaceutica, 92(1), 8. https://doi.org/10.3390/scipharm92010008