
Human Target Proteins for Benzo(a)pyrene and Acetaminophen (And Its Metabolites): Insights from Inverse Molecular Docking and Molecular Dynamics Simulations

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
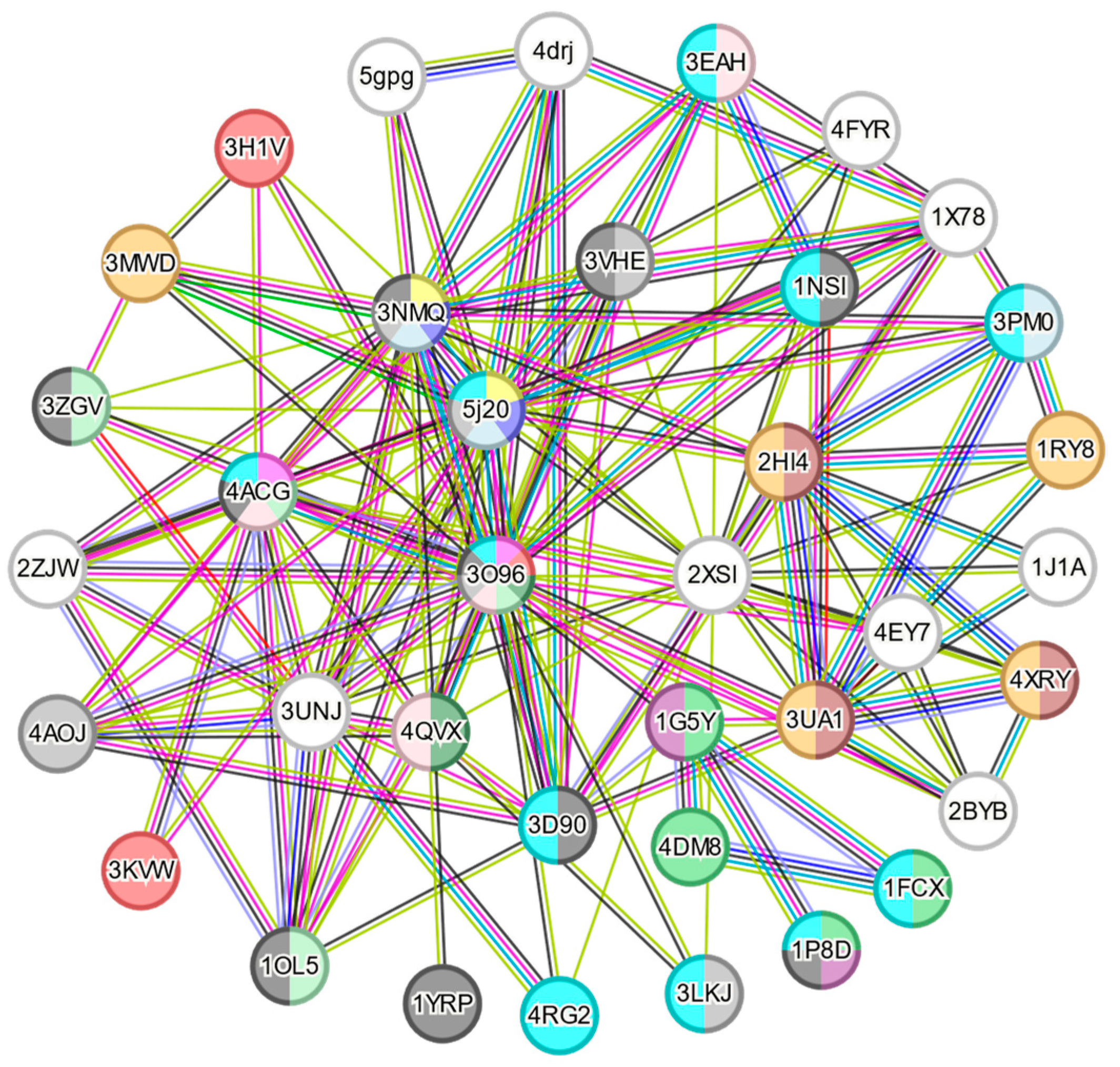
2.1. Virtual Screening for Selection of Human Proteins
2.2. Molecular Docking with AutoDock Vina
2.2.1. Compounds’ Structures
2.2.2. Preparation of Protein Structures
2.2.3. Docking Calculations
2.3. Protein–Ligand Interaction Prediction
2.4. Validation of Docking Protocol
2.5. Molecular Dynamics Simulations (MDS)
3. Results
3.1. Virtual Screening by Automatic Docking Generation
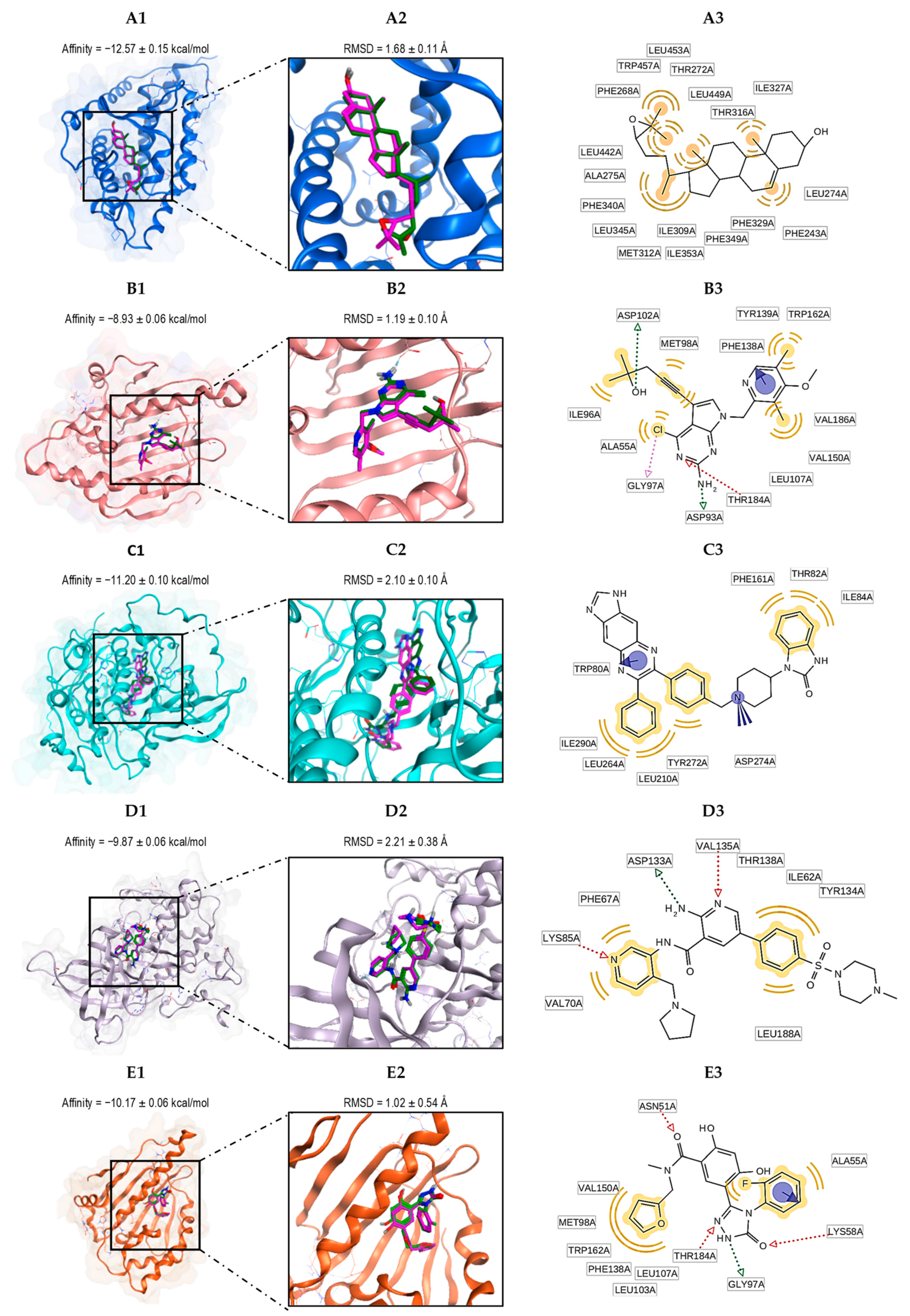
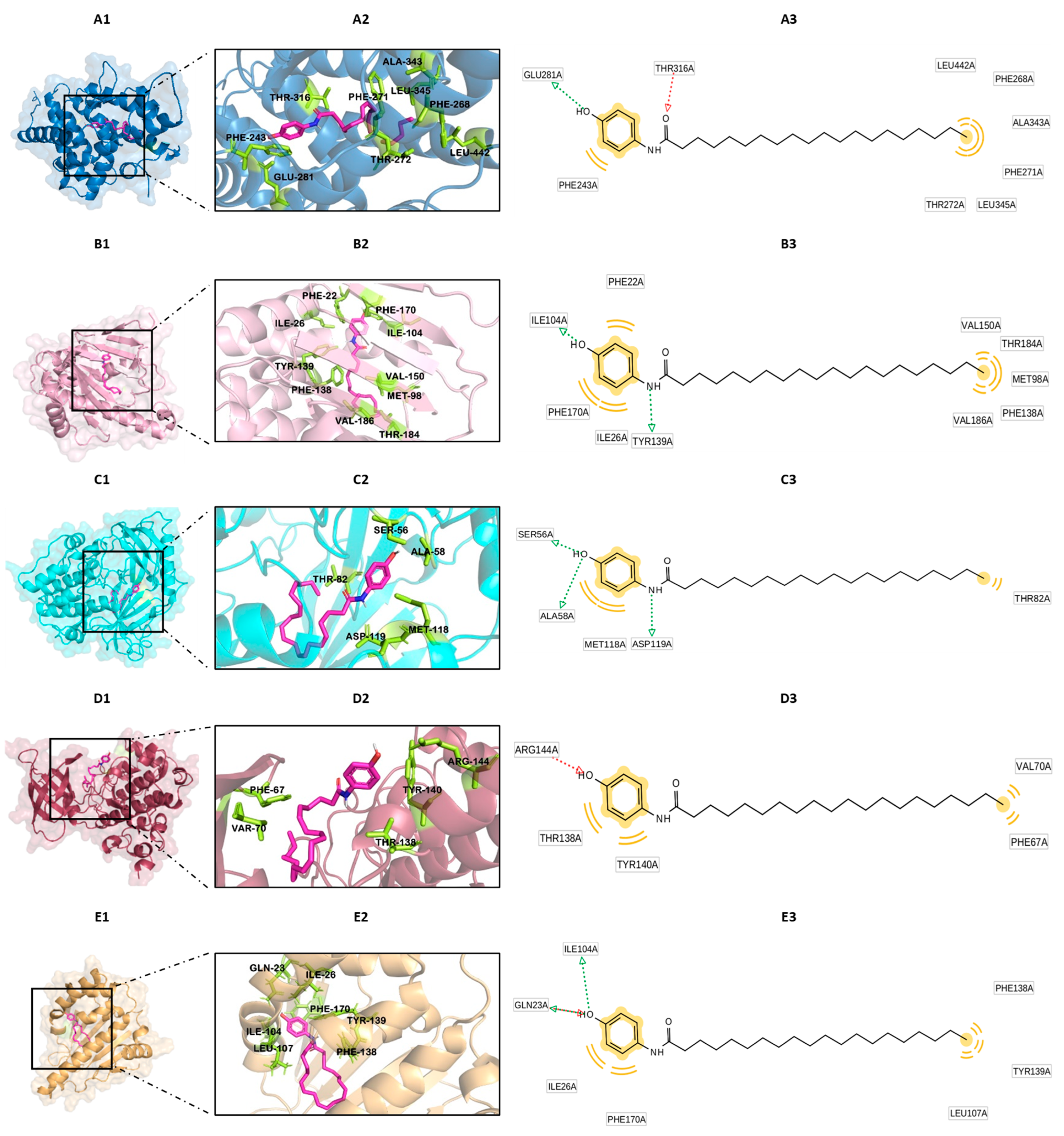
3.2. Docking Analysis
3.3. The Nature of the Interactions of Protein–Ligand Complexes
3.4. Docking Validation
3.5. Molecular Dynamics Simulations
3.5.1. Root Mean Square Deviations (RSMD)
3.5.2. Root Mean Square Fluctuation (RMSF)
3.5.3. Hydrogen Bond Analysis
3.5.4. Molecular Mechanics Energies Combined with Surface Area Continuum Solvation (MMGBSA)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
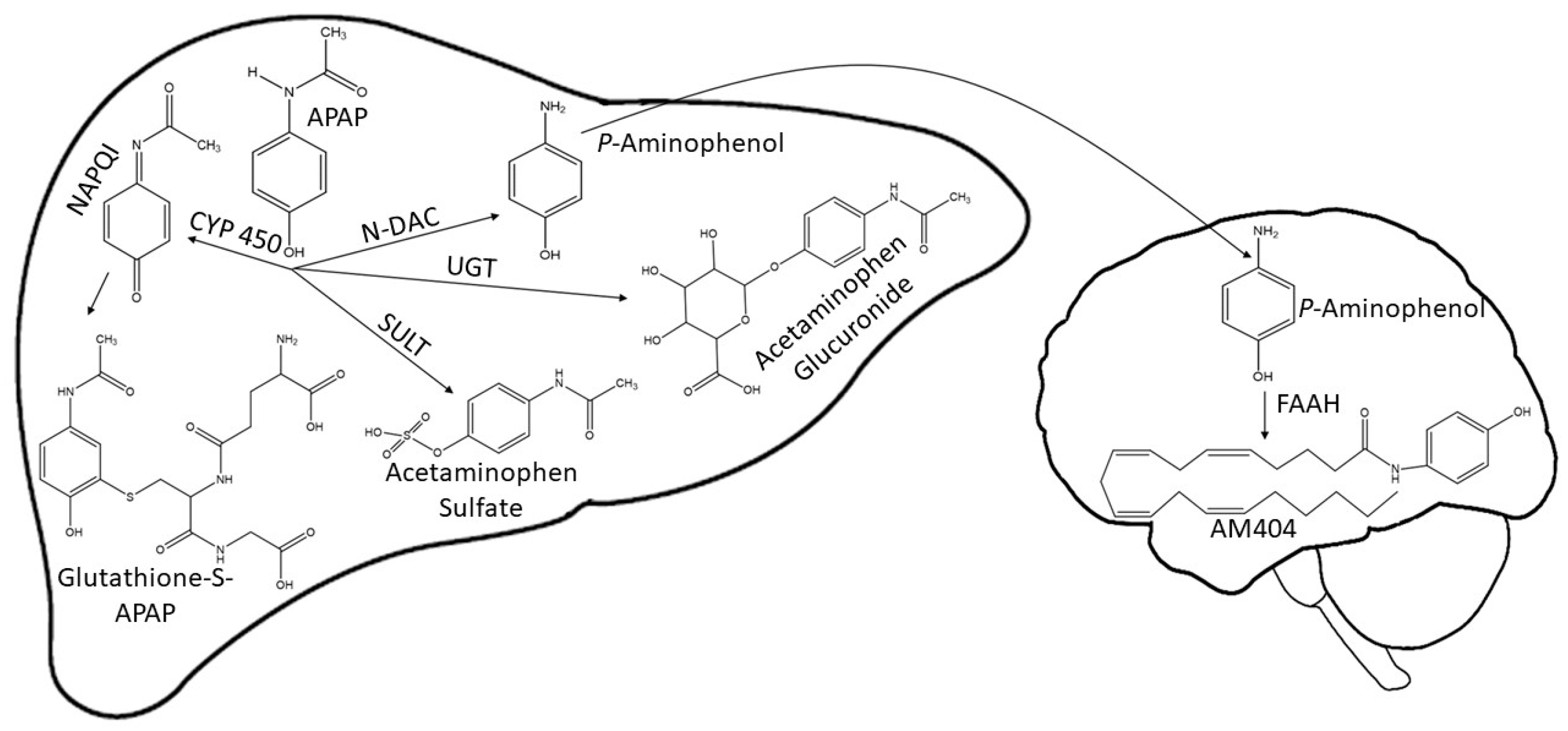
- Mallet, C.; Desmeules, J. An Updated Review on the Metabolite (AM404)-Mediated Central Mechanism of Action of Paracetamol (Acetaminophen): Experimental Evidence and Potential Clinical Impact. J. Pain. Res. 2023, 16, 1081–1094. [Google Scholar] [CrossRef]
- Hurwitz, J.; Sands, S.; Davis, E.; Nielsen, J.; Warholak, T. Patient knowledge and use of acetaminophen in over-the-counter medications. J. Am. Pharm. Assoc. 2014, 54, 19–26. [Google Scholar] [CrossRef]
- Chidiac, A.S.; Buckley, N.A.; Noghrehchi, F.; Cairns, R. Paracetamol (acetaminophen) overdose and hepatotoxicity: Mechanism, treatment, prevention measures, and estimates of burden of disease. Expert. Opin. Drug Metab. Toxicol. 2023, 19, 297–317. [Google Scholar] [CrossRef]
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet Genom. 2015, 25, 416–426. [Google Scholar] [CrossRef]
- Ben-Shachar, R.; Chen, Y.; Luo, S.; Hartman, C.; Reed, M.; Nijhout, H.F. The biochemistry of acetaminophen hepatotoxicity and rescue: A mathematical model. Theor. Biol. Med. Model. 2012, 9, 55. [Google Scholar] [CrossRef]
- Gloor, Y.; Schvartz, D.; Samer, C.F. Old problem, new solutions: Biomarker discovery for acetaminophen liver toxicity. Expert. Opin. Drug Metab. Toxicol. 2019, 15, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Huang, L.; Zhang, Z. The molecular mechanisms of acetaminophen-induced hepatotoxicity and its potential therapeutic targets. Exp. Biol. Med. 2023, 248, 412–424. [Google Scholar] [CrossRef]
- Sampaio, G.R.; Guizellini, G.M.; da Silva, S.A.; de Almeida, A.P.; Pinaffi-Langley, A.C.C.; Rogero, M.M.; de Camargo, A.C.; Torres, E. Polycyclic Aromatic Hydrocarbons in Foods: Biological Effects, Legislation, Occurrence, Analytical Methods, and Strategies to Reduce Their Formation. Int. J. Mol. Sci. 2021, 22, 6010. [Google Scholar] [CrossRef]
- Bukowska, B.; Mokra, K.; Michałowicz, J. Benzo[a]pyrene-Environmental Occurrence, Human Exposure, and Mechanisms of Toxicity. Int. J. Mol. Sci. 2022, 23, 6348. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Chemical Agents and Related Occupations. Lyon (FR): International Agency for Research on Cancer; (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, No. 100F.) Benzo[a]pyrene. Available online: https://www.ncbi.nlm.nih.gov/books/NBK304415/ (accessed on 4 October 2018).
- Carlson, E.A.; Li, Y.; Zelikoff, J.T. Benzo[a]pyrene-induced immunotoxicity in Japanese medaka (Oryzias latipes): Relationship between lymphoid CYP1A activity and humoral immune suppression. Toxicol. Appl. Pharmacol. 2004, 201, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Usal, M.; Veyrenc, S.; Darracq-Ghitalla-Ciock, M.; Regnault, C.; Sroda, S.; Fini, J.B.; Canlet, C.; Tremblay-Franco, M.; Raveton, M.; Reynaud, S. Transgenerational metabolic disorders and reproduction defects induced by benzo[a]pyrene in Xenopus tropicalis. Env. Pollut. 2021, 269, 116109. [Google Scholar] [CrossRef]
- Kim, J.T.; Park, J.E.; Lee, S.J.; Yu, W.J.; Lee, H.J.; Kim, J.M. Benzo[a]pyrene Cytotoxicity Tolerance in Testicular Sertoli Cells Involves Aryl-hydrocarbon Receptor and Cytochrome P450 1A1 Expression Deficiencies. Dev. Reprod. 2021, 25, 15–24. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, Y.; Pan, L.; Xu, R.; Li, D. Benzo[a]pyrene exposure induced reproductive endocrine-disrupting effects via the steroidogenic pathway and estrogen signaling pathway in female scallop Chlamys farreri. Sci. Total Env. 2020, 726, 138585. [Google Scholar] [CrossRef]
- Ayoub, S.S. Paracetamol (acetaminophen): A familiar drug with an unexplained mechanism of action. Temperature 2021, 8, 351–371. [Google Scholar] [CrossRef]
- Bukowska, B.; Duchnowicz, P. Molecular Mechanisms of Action of Selected Substances Involved in the Reduction of Benzo[a]pyrene-Induced Oxidative Stress. Molecules 2022, 27, 1379. [Google Scholar] [CrossRef]
- Deng, C.; Dang, F.; Gao, J.; Zhao, H.; Qi, S.; Gao, M. Acute benzo[a]pyrene treatment causes different antioxidant response and DNA damage in liver, lung, brain, stomach and kidney. Heliyon 2018, 4, e00898. [Google Scholar] [CrossRef]
- Fu, C.; Li, Y.; Xi, H.; Niu, Z.; Chen, N.; Wang, R.; Yan, Y.; Gan, X.; Wang, M.; Zhang, W.; et al. Benzo(a)pyrene and cardiovascular diseases: An overview of pre-clinical studies focused on the underlying molecular mechanism. Front. Nutr. 2022, 9, 978475. [Google Scholar] [CrossRef]
- Stevens, E.A.; Mezrich, J.D.; Bradfield, C.A. The aryl hydrocarbon receptor: A perspective on potential roles in the immune system. Immunology 2009, 127, 299–311. [Google Scholar] [CrossRef]
- Schuran, F.A.; Lommetz, C.; Steudter, A.; Ghallab, A.; Wieschendorf, B.; Schwinge, D.; Zuehlke, S.; Reinders, J.; Heeren, J.; Lohse, A.W.; et al. Aryl Hydrocarbon Receptor Activity in Hepatocytes Sensitizes to Hyperacute Acetaminophen-Induced Hepatotoxicity in Mice. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 371–388. [Google Scholar] [CrossRef] [PubMed]
- Montero-Pérez, Y.; Olivero-Verbel, J. Exposure to Benzo(a)pyrene Enhances Acetaminophen-Induced Liver Injury in Mice at Non-Hepatotoxic Doses. Sci. Pharm. 2024, 92, 30. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.D.; Keith, T.A.; Millam, J.M. GaussView 5.0; Gaussian Inc.: Wallingford, CT, USA, 2008; p. 340. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- O'Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Maldonado-Rojas, W.; Olivero-Verbel, J.; Marrero-Ponce, Y. Computational fishing of new DNA methyltransferase inhibitors from natural products. J. Mol. Graph. Model. 2015, 60, 43–54. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Bashford, D.; Bellott, M.; Dunbrack, R.L., Jr.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Miller, B.R., III.; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Collins, K. The biogenesis and regulation of telomerase holoenzymes. Nat. Rev. Mol. Cell Biol. 2006, 7, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhi, W.; Wang, L. Role of Tau Protein in Neurodegenerative Diseases and Development of Its Targeted Drugs: A Literature Review. Molecules 2024, 29, 2812. [Google Scholar] [CrossRef] [PubMed]
- Lorberbaum, D.S.; Kishore, S.; Rosselot, C.; Sarbaugh, D.; Brooks, E.P.; Aragon, E.; Xuan, S.; Simon, O.; Ghosh, D.; Mendelsohn, C.; et al. Retinoic acid signaling within pancreatic endocrine progenitors regulates mouse and human β cell specification. Development 2020, 147, dev189977. [Google Scholar] [CrossRef]
- Roach, P.J.; Depaoli-Roach, A.A.; Hurley, T.D.; Tagliabracci, V.S. Glycogen and its metabolism: Some new developments and old themes. Biochem. J. 2012, 441, 763–787. [Google Scholar] [CrossRef]
- Ameur, A.; Enroth, S.; Johansson, A.; Zaboli, G.; Igl, W.; Johansson, A.C.; Rivas, M.A.; Daly, M.J.; Schmitz, G.; Hicks, A.A.; et al. Genetic adaptation of fatty-acid metabolism: A human-specific haplotype increasing the biosynthesis of long-chain omega-3 and omega-6 fatty acids. Am. J. Hum. Genet. 2012, 90, 809–820. [Google Scholar] [CrossRef]
- Duan, Y.; Gong, K.; Xu, S.; Zhang, F.; Meng, X.; Han, J. Regulation of cholesterol homeostasis in health and diseases: From mechanisms to targeted therapeutics. Signal Transduct. Target. Ther. 2022, 7, 265. [Google Scholar] [CrossRef]
- Madhavan, H. Simple Laboratory methods to measure cell proliferation using DNA synthesis property. J. Stem Cells Regen. Med. 2007, 3, 12–14. [Google Scholar] [CrossRef]
- Wiman, K.G.; Zhivotovsky, B. Understanding cell cycle and cell death regulation provides novel weapons against human diseases. J. Intern. Med. 2017, 281, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Cyr, A.R.; Domann, F.E. The redox basis of epigenetic modifications: From mechanisms to functional consequences. Antioxid. Redox Signal 2011, 15, 551–589. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Mouzat, K.; Baron, S.; Marceau, G.; Caira, F.; Sapin, V.; Volle, D.H.; Lumbroso, S.; Lobaccaro, J.-M. Emerging roles for LXRs and LRH-1 in female reproduction. Mol. Cell. Endocrinol. 2013, 368, 47–58. [Google Scholar] [CrossRef]
- Prodromou, C. Mechanisms of Hsp90 regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef]
- Huo, X.; Sun, H.; Liu, Q.; Ma, X.; Peng, P.; Yu, M.; Zhang, Y.; Cao, D.; Shen, K. Clinical and Expression Significance of AKT1 by Co-expression Network Analysis in Endometrial Cancer. Front. Oncol. 2019, 9, 1147. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhang, Y.; Jia, Y.; Chen, X.; Niu, T.; Chatterjee, A.; He, P.; Hou, G. Heat shock protein 90: Biological functions, diseases, and therapeutic targets. MedComm 2024, 5, e470. [Google Scholar] [CrossRef]
- Duggal, S.; Jailkhani, N.; Midha, M.K.; Agrawal, N.; Rao, K.V.S.; Kumar, A. Defining the Akt1 interactome and its role in regulating the cell cycle. Sci. Rep. 2018, 8, 1303. [Google Scholar] [CrossRef]
- Cao, Q.; Lu, X.; Feng, Y.-J. Glycogen synthase kinase-3β positively regulates the proliferation of human ovarian cancer cells. Cell Res. 2006, 16, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, R.L.; Gill, N.S.; Pugh, W.; Lee, J.P.; Koeberlein, B.; Furth, E.E.; Polonsky, K.S.; Naji, A.; Birnbaum, M.J. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat. Med. 2001, 7, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Elmadbouh, O.H.M.; Pandol, S.J.; Edderkaoui, M. Glycogen Synthase Kinase 3β: A True Foe in Pancreatic Cancer. Int. J. Mol. Sci. 2022, 23, 14133. [Google Scholar] [CrossRef] [PubMed]
- Codenotti, S.; Zizioli, D.; Mignani, L.; Rezzola, S.; Tabellini, G.; Parolini, S.; Giacomini, A.; Asperti, M.; Poli, M.; Mandracchia, D.; et al. Hyperactive Akt1 Signaling Increases Tumor Progression and DNA Repair in Embryonal Rhabdomyosarcoma RD Line and Confers Susceptibility to Glycolysis and Mevalonate Pathway Inhibitors. Cells 2022, 11, 2859. [Google Scholar] [CrossRef]
- Liu, H.; Remedi, M.S.; Pappan, K.L.; Kwon, G.; Rohatgi, N.; Marshall, C.A.; McDaniel, M.L. Glycogen Synthase Kinase-3 and Mammalian Target of Rapamycin Pathways Contribute to DNA Synthesis, Cell Cycle Progression, and Proliferation in Human Islets. Diabetes 2009, 58, 663–672. [Google Scholar] [CrossRef]
- Noguchi, M.; Hirata, N.; Suizu, F. The links between AKT and two intracellular proteolytic cascades: Ubiquitination and autophagy. Biochim. Biophys. Acta (BBA) Rev. Cancer 2014, 1846, 342–352. [Google Scholar] [CrossRef]
- Kim, R.; Kin, T.; Beck, W.T. Impact of Complex Apoptotic Signaling Pathways on Cancer Cell Sensitivity to Therapy. Cancers 2024, 16, 984. [Google Scholar] [CrossRef]
- Beurel, E.; Jope, R.S. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006, 79, 173–189. [Google Scholar] [CrossRef]
- Edwards, P.A.; Kennedy, M.A.; Mak, P.A. LXRs; oxysterol-activated nuclear receptors that regulate genes controlling lipid homeostasis. Vasc. Pharmacol. 2002, 38, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Komati, R.; Spadoni, D.; Zheng, S.; Sridhar, J.; Riley, K.E.; Wang, G. Ligands of Therapeutic Utility for the Liver X Receptors. Molecules 2017, 22, 88. [Google Scholar] [CrossRef]
- Premalatha, R.; Srikumar, K.; Vijayalaksmi, D.; Kumar, G.N.; Mathur, P.P. 28-Homobrassinolide: A novel oxysterol transactivating LXR gene expression. Mol. Biol. Rep. 2014, 41, 7447–7461. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Cai, H.; Wang, J.; Yang, Q.; Guan, J.; Deng, J.; Chen, Z. Molecular pathogenesis of acetaminophen-induced liver injury and its treatment options. J. Zhejiang Univ. Sci. B 2022, 23, 265–285. [Google Scholar] [CrossRef] [PubMed]
- Salamat, N.; Derakhshesh, N. Oxidative stress in liver cell culture from mullet, Liza klunzingeri, induced by short-term exposure to benzo[a]pyrene and nonylphenol. Fish. Physiol. Biochem. 2020, 46, 1183–1197. [Google Scholar] [CrossRef]
- Guo, B.; Feng, D.; Xu, Z.; Qi, P.; Yan, X. Acute benzo[a]pyrene exposure induced oxidative stress, neurotoxicity and epigenetic change in blood clam Tegillarca granosa. Sci. Rep. 2021, 11, 18744. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Lathwal, A.; Nagpal, G.; Kumar, V.; Raghav, P.K. Chapter 1—Impact of chemoinformatics approaches and tools on current chemical research. In Chemoinformatics and Bioinformatics in the Pharmaceutical Sciences; Sharma, N., Ojha, H., Raghav, P.K., Goyal, R.K., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 1–26. [Google Scholar]
- Wong, J.; Quinn, C.M.; Brown, A.J. Synthesis of the oxysterol, 24(S), 25-epoxycholesterol, parallels cholesterol production and may protect against cellular accumulation of newly-synthesized cholesterol. Lipids Health Dis. 2007, 6, 10. [Google Scholar] [CrossRef]
- Gerin, I.; Dolinsky, V.W.; Shackman, J.G.; Kennedy, R.T.; Chiang, S.H.; Burant, C.F.; Steffensen, K.R.; Gustafsson, J.A.; MacDougald, O.A. LXRbeta is required for adipocyte growth, glucose homeostasis, and beta cell function. J. Biol. Chem. 2005, 280, 23024–23031. [Google Scholar] [CrossRef]
- Duan, J.; Chen, C.; Li, H.; Ju, G.; Gao, A.; Sun, Y.; Zhang, W. Multifaceted Protective Effects of Hesperidin by Aromatic Hydrocarbon Receptor in Endothelial Cell Injury Induced by Benzo[a]Pyrene. Nutrients 2022, 14, 574. [Google Scholar] [CrossRef]
- Saini, S.P.; Zhang, B.; Niu, Y.; Jiang, M.; Gao, J.; Zhai, Y.; Hoon Lee, J.; Uppal, H.; Tian, H.; Tortorici, M.A.; et al. Activation of liver X receptor increases acetaminophen clearance and prevents its toxicity in mice. Hepatology 2011, 54, 2208–2217. [Google Scholar] [CrossRef]
- Li, Z.N.; Luo, Y. HSP90 inhibitors and cancer: Prospects for use in targeted therapies. Oncol. Rep. 2023, 49, 6. [Google Scholar] [CrossRef]
- Obermann, W.M.; Sondermann, H.; Russo, A.A.; Pavletich, N.P.; Hartl, F.U. In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J. Cell Biol. 1998, 143, 901–910. [Google Scholar] [CrossRef]
- Zuehlke, A.D.; Moses, M.A.; Neckers, L. Heat shock protein 90: Its inhibition and function. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160527. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, A.; Kent, C.N.; Balch, M.; Peng, S.; Mishra, S.J.; Deng, J.; Day, V.W.; Liu, W.; Subramanian, C.; Cohen, M.; et al. Structure-guided design of an Hsp90β N-terminal isoform-selective inhibitor. Nat. Commun. 2018, 9, 425. [Google Scholar] [CrossRef] [PubMed]
- Hoxie, R.S.; Street, T.O. Hsp90 chaperones have an energetic hot-spot for binding inhibitors. Protein Sci. 2020, 29, 2101–2111. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Xu, W.; Neckers, L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 2003, 3, 213–217. [Google Scholar] [CrossRef]
- Kamal, A.; Boehm, M.F.; Burrows, F.J. Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol. Med. 2004, 10, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Abdul, N.S.; Ahmad Alrashed, N.; Alsubaie, S.; Albluwi, H.; Badr Alsaleh, H.; Alageel, N.; Ghaleb Salma, R. Role of Extracellular Heat Shock Protein 90 Alpha in the Metastasis of Oral Squamous Cell Carcinoma: A Systematic Review. Cureus 2023, 15, e38514. [Google Scholar] [CrossRef]
- Galam, L.; Hadden, M.K.; Ma, Z.; Ye, Q.Z.; Yun, B.G.; Blagg, B.S.; Matts, R.L. High-throughput assay for the identification of Hsp90 inhibitors based on Hsp90-dependent refolding of firefly luciferase. Bioorg Med. Chem. 2007, 15, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Birbo, B.; Madu, E.E.; Madu, C.O.; Jain, A.; Lu, Y. Role of HSP90 in Cancer. Int. J. Mol. Sci. 2021, 22, 10317. [Google Scholar] [CrossRef]
- Abbasi, M.; Sadeghi-Aliabadi, H.; Amanlou, M. Prediction of new Hsp90 inhibitors based on 3,4-isoxazolediamide scaffold using QSAR study, molecular docking and molecular dynamic simulation. Daru 2017, 25, 17. [Google Scholar] [CrossRef]
- Rezvani, S.; Ebadi, A.; Razzaghi-Asl, N. In silico identification of potential Hsp90 inhibitors via ensemble docking, DFT and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2022, 40, 10665–10676. [Google Scholar] [CrossRef]
- Magwenyane, A.M.; Ugbaja, S.C.; Amoako, D.G.; Somboro, A.M.; Khan, R.B.; Kumalo, H.M. Heat Shock Protein 90 (HSP90) Inhibitors as Anticancer Medicines: A Review on the Computer-Aided Drug Discovery Approaches over the Past Five Years. Comput. Math. Methods Med. 2022, 2022, 2147763. [Google Scholar] [CrossRef] [PubMed]
- Gruszczyk, J.; Grandvuillemin, L.; Lai-Kee-Him, J.; Paloni, M.; Savva, C.G.; Germain, P.; Grimaldi, M.; Boulahtouf, A.; Kwong, H.-S.; Bous, J.; et al. Cryo-EM structure of the agonist-bound Hsp90-XAP2-AHR cytosolic complex. Nat. Commun. 2022, 13, 7010. [Google Scholar] [CrossRef]
- Su, M.; Zhou, S.; Li, J.; Lin, N.; Chi, T.; Zhang, M.; Lv, X.; Hu, Y.; Bai, T.; Chang, F. Benzo(a)pyrene regulates chaperone-mediated autophagy via heat shock protein 90. Toxicol. Lett. 2023, 383, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Guo, C.; Su, M.; Wu, X.; Li, R. Biocharacterization of Heat Shock Protein 90 in Acetaminophen-Treated Livers Without Conspicuous Drug Induced Liver Injury. Cell Physiol. Biochem. 2017, 43, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, T.; Zhang, M.; Chen, Q.; Su, M.; Bai, T.; Hu, Y.; Li, J.; Chang, F.; Huangfu, W. Study on molecular mechanism of benzo (ɑ) pyrene on CMA by HSP90ɑ and HIF-1ɑ. Toxicol. Vitr. 2022, 83, 105372. [Google Scholar] [CrossRef]
- Milovanović, M.R.; Stanković, I.M.; Živković, J.M.; Ninković, D.B.; Hall, M.B.; Zarić, S.D. Water: New aspect of hydrogen bonding in the solid state. IUCrJ 2022, 9, 639–647. [Google Scholar] [CrossRef]
- Vaidyanathan, R.; Murugan Sreedevi, S.; Ravichandran, K.; Vinod, S.M.; Hari Krishnan, Y.; Babu, L.K.; Parthiban, P.S.; Basker, L.; Perumal, T.; Rajaraman, V.; et al. Molecular docking approach on the binding stability of derivatives of phenolic acids (DPAs) with Human Serum Albumin (HSA): Hydrogen-bonding versus hydrophobic interactions or combined influences? JCIS Open 2023, 12, 100096. [Google Scholar] [CrossRef]
- Van Lommel, R.; Bettens, T.; Barlow, T.M.A.; Bertouille, J.; Ballet, S.; De Proft, F. A Quantum Chemical Deep-Dive into the π-π Interactions of 3-Methylindole and Its Halogenated Derivatives-Towards an Improved Ligand Design and Tryptophan Stacking. Pharmaceuticals 2022, 15, 935. [Google Scholar] [CrossRef]
- Li, Q.; Gao, C.; Deng, H.; Song, Q.; Yuan, L. Benzo[a]pyrene induces pyroptotic and autophagic death through inhibiting PI3K/Akt signaling pathway in HL-7702 human normal liver cells. J. Toxicol. Sci. 2019, 44, 121–131. [Google Scholar] [CrossRef]
- Ye, Y.; Jiang, S.; Du, T.; Ding, M.; Hou, M.; Mi, C.; Liang, T.; Zhong, H.; Xie, J.; Xu, W.; et al. Correction: Environmental Pollutant Benzo[a]pyrene Upregulated Long Non-coding RNA HZ07 Inhibits Trophoblast Cell Migration by Inactivating PI3K/AKT/MMP2 Signaling Pathway in Recurrent Pregnancy Loss. Reprod. Sci. 2023, 30, 728. [Google Scholar] [CrossRef]
- Martorana, F.; Motta, G.; Pavone, G.; Motta, L.; Stella, S.; Vitale, S.R.; Manzella, L.; Vigneri, P. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front. Pharmacol. 2021, 12, 662232. [Google Scholar] [CrossRef] [PubMed]
- Kometani, T.; Yoshino, I.; Miura, N.; Okazaki, H.; Ohba, T.; Takenaka, T.; Shoji, F.; Yano, T.; Maehara, Y. Benzo[a]pyrene promotes proliferation of human lung cancer cells by accelerating the epidermal growth factor receptor signaling pathway. Cancer Lett. 2009, 278, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, S.; Cheng, H.; Lv, H.; Cheng, G.; Ci, X. Nrf2-mediated liver protection by esculentoside A against acetaminophen toxicity through the AMPK/Akt/GSK3β pathway. Free Radic. Biol. Med. 2016, 101, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Zhu, J.; Wu, Y.; Xu, L.; Jin, J. Theoretical Studies on the Selectivity Mechanisms of Glycogen Synthase Kinase 3β (GSK3β) with Pyrazine ATP-competitive Inhibitors by 3DQSAR, Molecular Docking, Molecular Dynamics Simulation and Free Energy Calculations. Curr. Comput. Aided Drug Des. 2020, 16, 17–30. [Google Scholar] [CrossRef]
- Berg, S.; Bergh, M.; Hellberg, S.; Högdin, K.; Lo-Alfredsson, Y.; Söderman, P.; von Berg, S.; Weigelt, T.; Ormö, M.; Xue, Y.; et al. Discovery of novel potent and highly selective glycogen synthase kinase-3β (GSK3β) inhibitors for Alzheimer's disease: Design, synthesis, and characterization of pyrazines. J. Med. Chem. 2012, 55, 9107–9119. [Google Scholar] [CrossRef]
- Dendelé, B.; Tekpli, X.; Sergent, O.; Dimanche-Boitrel, M.T.; Holme, J.A.; Huc, L.; Lagadic-Gossmann, D. Identification of the couple GSK3α/c-Myc as a new regulator of hexokinase II in benzo[a]pyrene-induced apoptosis. Toxicol. Vitr. 2012, 26, 94–101. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Hoek, J.B.; Shulga, N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005, 65, 10545–10554. [Google Scholar] [CrossRef]
- Bhushan, B.; Poudel, S.; Manley, M.W., Jr.; Roy, N.; Apte, U. Inhibition of Glycogen Synthase Kinase 3 Accelerated Liver Regeneration after Acetaminophen-Induced Hepatotoxicity in Mice. Am. J. Pathol. 2017, 187, 543–552. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO-Term | Biological Process | Count in Network | Strength | False Discovery Rate |
|---|---|---|---|---|
| GO:1905323 | Telomerase holoenzyme complex assembly | 2 of 4 | 2.43 | 0.0036 |
| GO:1902949 | Positive regulation of tau protein kinase activity | 2 of 7 | 2.18 | 0.0073 |
| GO:0048384 | Retinoic acid receptor signaling pathway | 4 of 18 | 2.07 | 2.39 × 10−5 |
| GO:2000074 | Regulation of type B pancreatic cell development | 2 of 9 | 2.07 | 0.0100 |
| GO:0045725 | Positive regulation of glycogen biosynthetic process | 3 of 15 | 2.03 | 0.00058 |
| GO:0070989 | Oxidative demethylation | 3 of 19 | 1.92 | 0.00097 |
| GO:0090201 | Negative regulation of release of cytochrome c from mitochondria | 2 of 21 | 1.71 | 0.0333 |
| GO:0010875 | Positive regulation of cholesterol efflux | 2 of 26 | 1.61 | 0.0444 |
| GO:0006633 | Fatty acid biosynthetic process | 5 of 119 | 1.35 | 0.00043 |
| GO:0032436 | Positive regulation of proteasomal ubiquitin-dependent protein catabolic process | 4 of 96 | 1.35 | 0.0029 |
| GO:2001237GO:2001237 | Negative regulation of extrinsic apoptotic signaling pathway | 4 of 97 | 1.34 | 0.0030 |
| GO:2000573 | Positive regulation of DNA biosynthetic process | 3 of 73 | 1.34 | 0.0181 |
| GO:0033674 | Positive regulation of kinase activity | 6 of 494 | 0.81 | 0.0150 |
| GO:0010629 | Negative regulation of gene expression | 10 of 899 | 0.77 | 0.00056 |
| GO:0010628 | Positive regulation of gene expression | 11 of 1146 | 0.71 | 0.00065 |
| Average Affinity Values (kcal/mol) | ||||||
|---|---|---|---|---|---|---|
| PDB ID | Description | B[a]P | APAP | AM404 | NAPQI | APAP- GLUCURONIDE |
| 1P8D | Oxysterol receptor LXR-beta | −12.7 ± 0 | −6.3 ± 0.1 | −9.0 ± 0.2 | −6.7 ± 0.1 | −9.0 ± 0.1 |
| 3NMQ | Heat shock protein HSP 90-beta | −11.7 ± 0 | −6.4 ± 0.2 | −8.3 ± 0.1 | −6.6 ± 0.1 | −7.4 ± 0.2 |
| 3O96 | RAC-alpha serine/ threonine protein kinase | −12.5 ± 0 | −6.4 ± 0 | −9.1 ± 0.1 | −6.7 ± 0 | −8.2 ± 0 |
| 4ACG | Glycogen synthase kinase-3 beta | −10.6 ± 0.2 | −5.6 ± 0 | −7.0 ± 0.1 | −5.6 ± 0 | −6.7 ± 0 |
| 5J20 | Heat shock protein HSP 90-alpha | −11.0 ± 0 | −6.5 ± 0 | −8.1 ± 0.6 | −6.7 ± 0.1 | −7.6 ± 0.3 |
| Protein | Ligand | Total Binding Free Energy (kcal/mol) | van der Waals Energy (kcal/mol) | Electrostatic Energy (kcal/mol) | Polar Solvation Energy (kcal/mol) | SASA Energy (kcal/mol) | Delta G Gas | Delta G Solv |
|---|---|---|---|---|---|---|---|---|
| 1P8D | CO1 | −48.55 ± 0.32 | −59.61 ± 0.26 | −7.48 ± 0.51 | 25.4 ± 0.50 | −6.85 ± 0.02 | −67.09 ± 0.60 | 18.55 ± 0.50 |
| AM404 | −37.95 ± 0.66 | −51.93 ± 0.47 | −13.9 ± 0.63 | 35.58 ± 0.59 | −7.69 ± 0.04 | −65.84 ± 0.82 | 27.89 ± 0.58 | |
| BP | −19.79 ± 0.31 | −32.45 ± 0.32 | −2.09 ± 0.25 | 18.77 ± 0.37 | −4.02 ± 0.03 | −34.53 ± 0.42 | 14.74 ± 0.36 | |
| 3NMQ | 7PP | −17.08 ± 0.48 | −49.4 ± 0.38 | −19.73 ± 1.25 | 58.22 ± 1.23 | −6.16 ± 0.03 | −69.14 ± 1.29 | 52.05 ± 1.21 |
| AM404 | −40.52 ± 0.77 | −54.12 ± 0.67 | −9.423 ± 0.46 | 30.72 ± 0.40 | −7.69 ± 0.07 | −63.55 ± 0.86 | 23.03 ± 0.38 | |
| B(a)P | −20.41 ± 0.32 | −34.14 ± 0.39 | −2.81 ± 0.26 | 20.38 ± 0.30 | −3.84 ± 0.03 | −36.95 ± 0.44 | 16.54 ± 0.29 | |
| 3O96 | 1QO | −37.97 ± 0.39 | −73.43 ± 0.33 | −26.8 ± 0.82 | 70.37 ± 0.75 | −8.11 ± 0.02 | −100.23 ± 0.83 | 62.3 ± 0.73 |
| AM404 | −38.28 ± 0.59 | −52.8 ± 0.44 | −27.22 ± 0.74 | 42.72 ± 0.48 | −7.99 ± 0.05 | −80.02 ± 0.66 | 41.73 ± 0.48 | |
| B(a)P | −20.46 ± 0.20 | −35.48 ± 0.24 | −2.29 ± 0.19 | 21.24 ± 0.22 | −3.93 ± 0.02 | −37.77 ± 0.30 | 17.3 ± 0.22 | |
| 4ACG | 6LQ | −41.67 ± 0.88 | −53.29 ± 0.51 | −31.99 ± 0.95 | 50.49 ± 0.57 | −6.88 ± 0.04 | −85.28 ± 0.92 | 43.61 ± 0.57 |
| AM404 | −35.32 ± 0.86 | −44.99 ± 0.54 | −14.11 ± 1.59 | 30.44 ± 0.96 | −6.65 ± 0.06 | −59.1 ± 1.66 | 23.79 ± 0.95 | |
| B(a)P | −20.46 ± 0.32 | −30.65 ± 0.38 | −4.31 ± 0.29 | 18.27 ± 0.22 | −3.77 ± 0.04 | −34.97 ± 0.42 | 14.5 ± 0.20 | |
| 5J20 | 6FJ | −27.11 ± 0.43 | −44.18 ± 0.38 | −37.73 ± 0.82 | 60.48 ± 0.68 | −5.68 ± 0.03 | −81.91 ± 0.80 | 54.8 ± 0.66 |
| AM404 | −40.66 ± 0.62 | −56.72 ± 0.50 | −15.3 ± 0.68 | 39.51 ± 0.51 | −8.15 ± 0.04 | −72.02 ± 0.87 | 31.36 ± 0.50 | |
| B(a)P | −22.89 ± 0.21 | −36.09 ± 0.20 | −3.49 ± 0.24 | 20.6 ± 0.26 | −3.92 ± 0.017 | −39.59 ± 0.27 | 16.69 ± 0.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Published by MDPI on behalf of the Österreichische Pharmazeutische Gesellschaft. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montero-Pérez, Y.; Pájaro-Castro, N.; Coronado-Posada, N.; Ahumedo-Monterrosa, M.; Olivero-Verbel, J. Human Target Proteins for Benzo(a)pyrene and Acetaminophen (And Its Metabolites): Insights from Inverse Molecular Docking and Molecular Dynamics Simulations. Sci. Pharm. 2024, 92, 55. https://doi.org/10.3390/scipharm92040055
Montero-Pérez Y, Pájaro-Castro N, Coronado-Posada N, Ahumedo-Monterrosa M, Olivero-Verbel J. Human Target Proteins for Benzo(a)pyrene and Acetaminophen (And Its Metabolites): Insights from Inverse Molecular Docking and Molecular Dynamics Simulations. Scientia Pharmaceutica. 2024; 92(4):55. https://doi.org/10.3390/scipharm92040055
Chicago/Turabian StyleMontero-Pérez, Yina, Nerlis Pájaro-Castro, Nadia Coronado-Posada, Maicol Ahumedo-Monterrosa, and Jesus Olivero-Verbel. 2024. "Human Target Proteins for Benzo(a)pyrene and Acetaminophen (And Its Metabolites): Insights from Inverse Molecular Docking and Molecular Dynamics Simulations" Scientia Pharmaceutica 92, no. 4: 55. https://doi.org/10.3390/scipharm92040055
APA StyleMontero-Pérez, Y., Pájaro-Castro, N., Coronado-Posada, N., Ahumedo-Monterrosa, M., & Olivero-Verbel, J. (2024). Human Target Proteins for Benzo(a)pyrene and Acetaminophen (And Its Metabolites): Insights from Inverse Molecular Docking and Molecular Dynamics Simulations. Scientia Pharmaceutica, 92(4), 55. https://doi.org/10.3390/scipharm92040055