
Comparative Analysis of Adverse Effects: Protein Kinase Inhibitors Versus Traditional Anticancer Therapies



Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of the Cancer Drugs
2.2. Preparation of AE Database for Approved Anticancer Drugs
2.3. Data Analysis
2.4. Cluster Analysis
3. Results
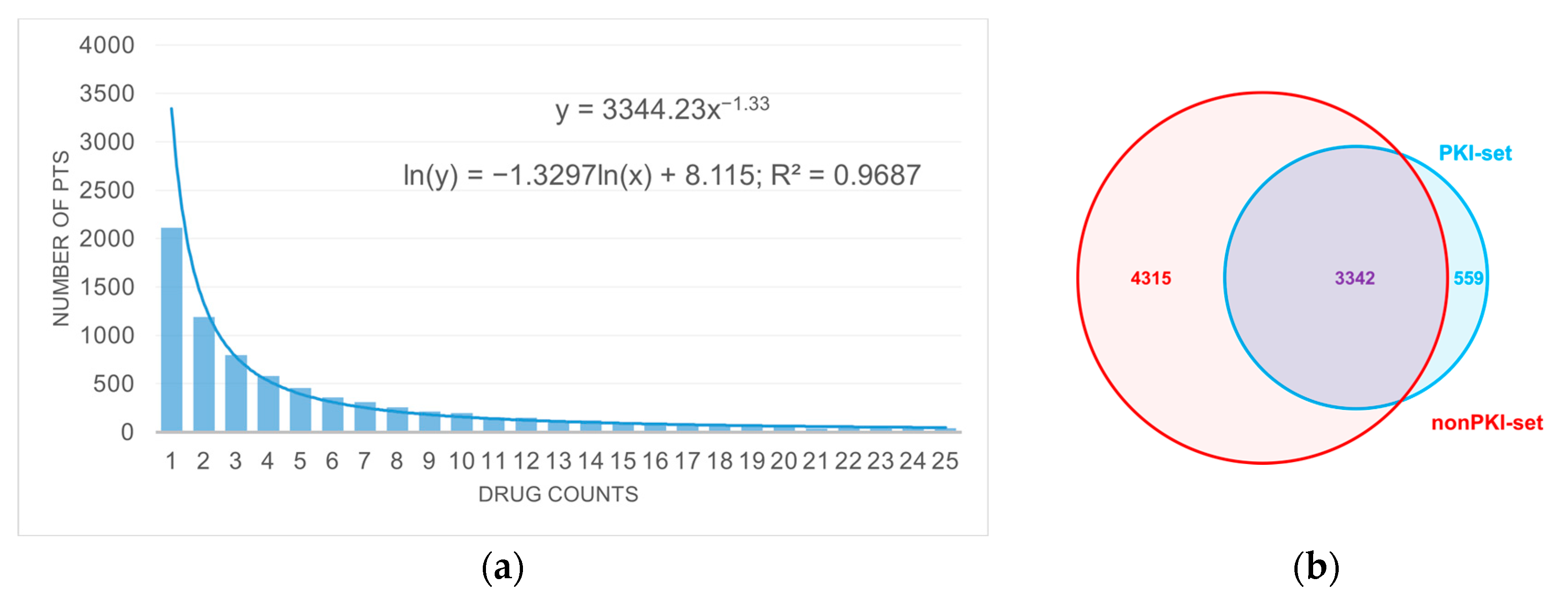
3.1. Anticancer Drug Dataset
3.2. The Analysis of the PKI Dataset
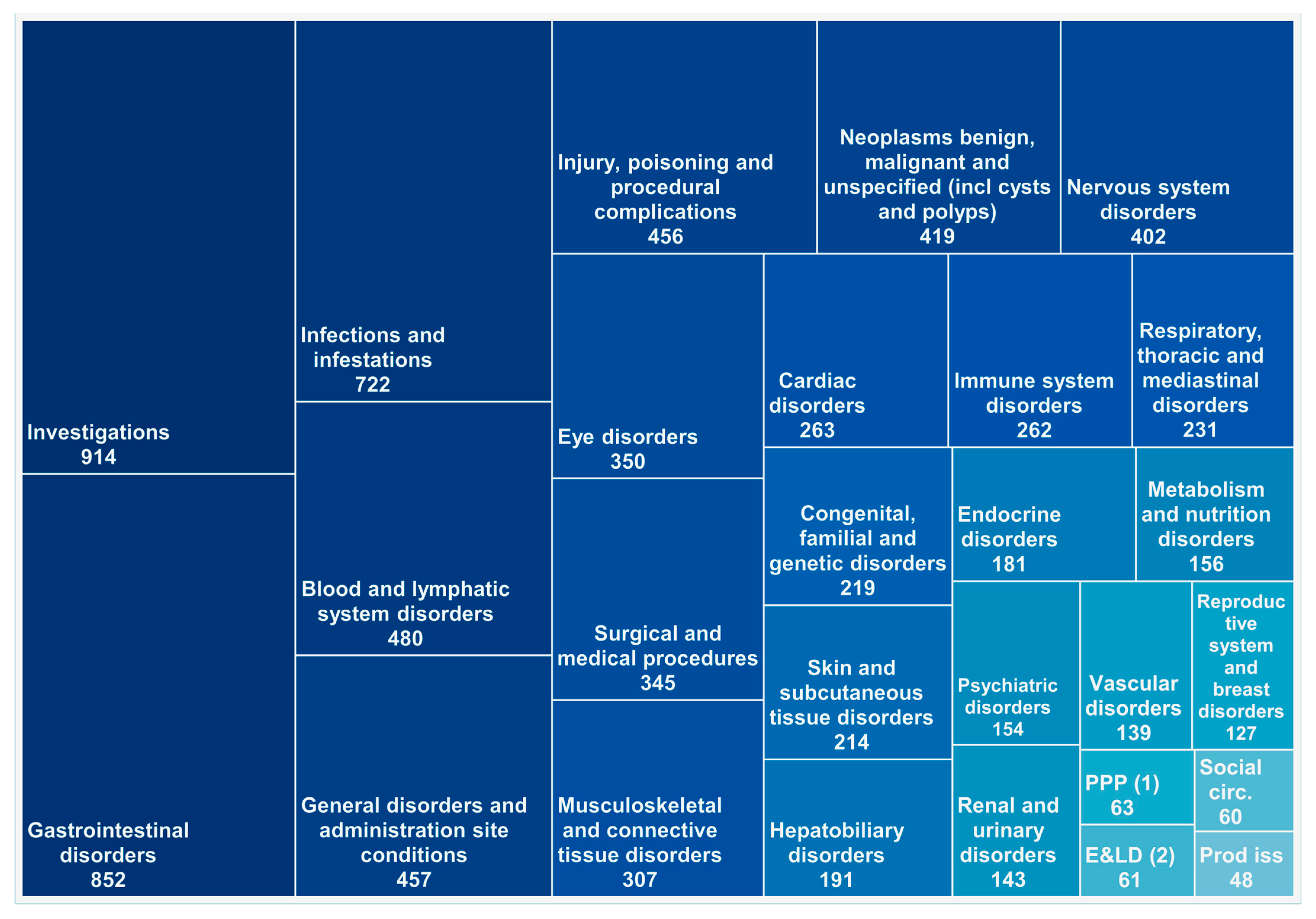
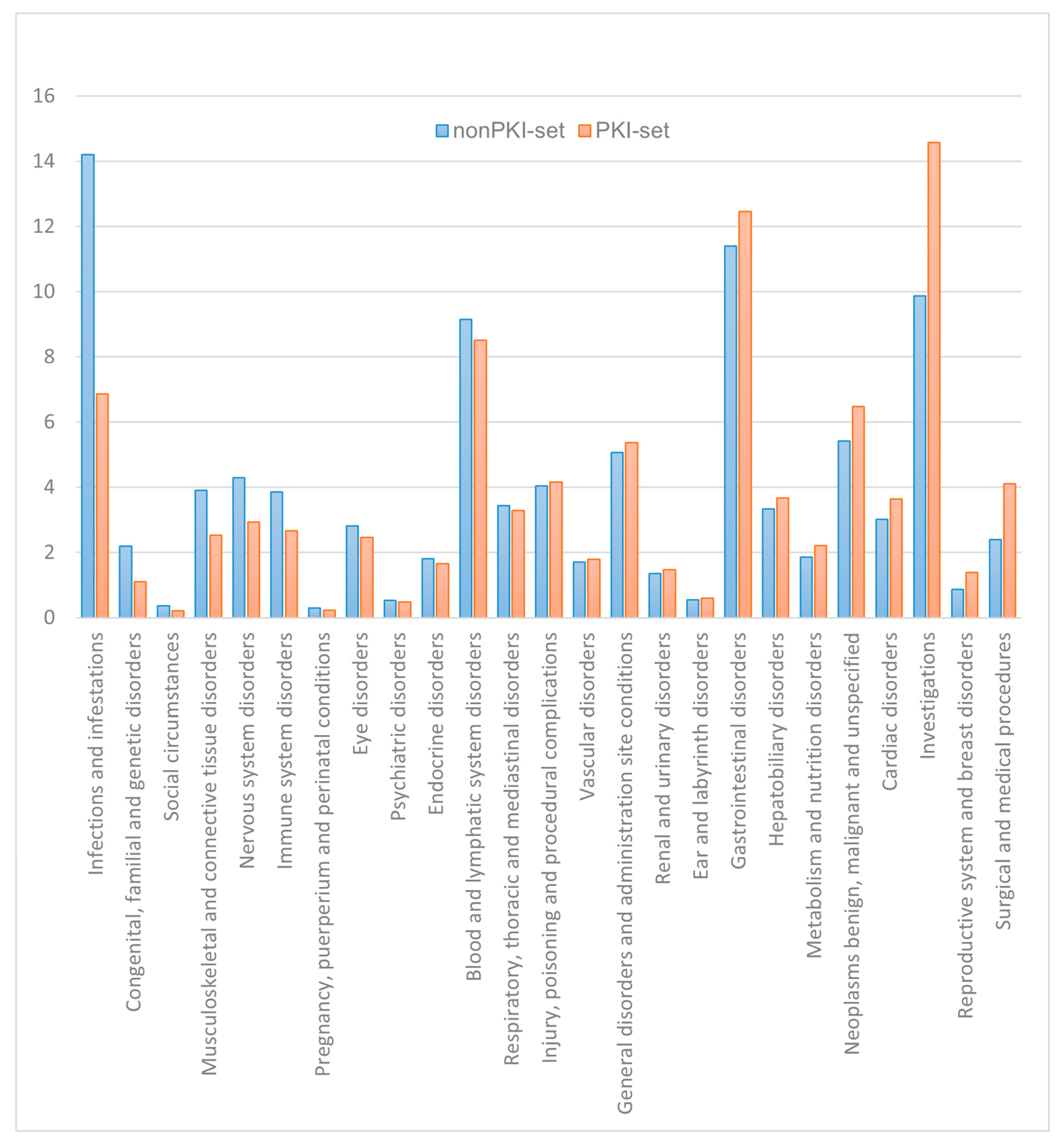
3.3. The Analysis Based on SOC Classification
3.4. Cluster Analysis of the Anticancer Drugs Based on Their AE Spectrum
3.4.1. PKI Set
3.4.2. NonPKI Set
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Franciosa, G.; Locard-Paulet, M.; Jensen, L.J.; Olsen, J.V. Recent Advances in Kinase Signaling Network Profiling by Mass Spectrometry. Curr. Opin. Chem. Biol. 2023, 73, 102260. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Choithramani, A.; Shard, A. A Molecular Perspective for the Use of Type IV Tyrosine Kinase Inhibitors as Anticancer Therapeutics. Drug Discov. Today 2022, 27, 808–821. [Google Scholar] [CrossRef]
- Pang, K.; Wang, W.; Qin, J.X.; Shi, Z.D.; Hao, L.; Ma, Y.Y.; Xu, H.; Wu, Z.X.; Pan, D.; Chen, Z.S.; et al. Role of Protein Phosphorylation in Cell Signaling, Disease, and the Intervention Therapy. MedComm 2022, 3, e175. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-Targeted Cancer Therapies: Progress, Challenges and Future Directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Dagher, R.; Cohen, M.; Williams, G.; Rothmann, M.; Gobburu, J.; Robbie, G.; Rahman, A.; Chen, G.; Staten, A.; Griebel, D.; et al. Approval Summary: Imatinib Mesylate in the Treatment of Metastatic and/or Unresectable Malignant Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2002, 8, 3034–3038. [Google Scholar]
- Miller, M.J.; Foy, K.C.; Kaumaya, P.T.P. Cancer Immunotherapy: Present Status, Future Perspective, and a New Paradigm of Peptide Immunotherapeutics. Discov. Med. 2013, 15, 166–176. [Google Scholar]
- Ling, Y.; Liu, J.; Qian, J.; Meng, C.; Guo, J.; Gao, W.; Xiong, B.; Ling, C.; Zhang, Y. Recent Advances in Multi-Target Drugs Targeting Protein Kinases and Histone Deacetylases in Cancer Therapy. Curr. Med. Chem. 2020, 27, 7264–7288. [Google Scholar] [CrossRef]
- Zhao, Y.; Bilal, M.; Raza, A.; Khan, M.I.; Mehmood, S.; Hayat, U.; Hassan, S.T.S.; Iqbal, H.M.N. Tyrosine Kinase Inhibitors and Their Unique Therapeutic Potentialities to Combat Cancer. Int. J. Biol. Macromol. 2021, 168, 22–37. [Google Scholar] [CrossRef]
- Niederst, M.J.; Engelman, J.A. Bypass Mechanisms of Resistance to Receptor Tyrosine Kinase Inhibition in Lung Cancer. Sci. Signal. 2013, 6, re6. [Google Scholar] [CrossRef] [PubMed]
- Gurden, M.D.; Westwood, I.M.; Faisal, A.; Naud, S.; Cheung, K.M.J.; McAndrew, C.; Wood, A.; Schmitt, J.; Boxall, K.; Mak, G.; et al. Naturally Occurring Mutations in the MPS1 Gene Predispose Cells to Kinase Inhibitor Drug Resistance. Cancer Res. 2015, 75, 3340–3354. [Google Scholar] [CrossRef]
- Blay, V.; Gailiunaite, S.; Lee, C.Y.; Chang, H.Y.; Hupp, T.; Houston, D.R.; Chi, P. Comparison of ATP-Binding Pockets and Discovery of Homologous Recombination Inhibitors. Bioorg. Med. Chem. 2022, 70, 116923. [Google Scholar] [CrossRef]
- Guo, Y.; Li, X.; Xie, Y.; Wang, Y. What Influences the Activity of Degrader−Antibody Conjugates (DACs). Eur. J. Med. Chem. 2024, 268, 116216. [Google Scholar] [CrossRef]
- Veth, T.S.; Kannegieter, N.M.; de Graaf, E.L.; Ruijtenbeek, R.; Joore, J.; Ressa, A.; Altelaar, M. Innovative Strategies for Measuring Kinase Activity to Accelerate the next Wave of Novel Kinase Inhibitors. Drug Discov. Today 2024, 29, 103907. [Google Scholar] [CrossRef] [PubMed]
- Grela-Wojewoda, A.; Pacholczak-Madej, R.; Adamczyk, A.; Korman, M.; Püsküllüoğlu, M. Cardiotoxicity Induced by Protein Kinase Inhibitors in Patients with Cancer. Int. J. Mol. Sci. 2022, 23, 2815. [Google Scholar] [CrossRef] [PubMed]
- Maliepaard, M.; Faber, Y.S.; van Bussel, M.T.J. Reported Hepatotoxicity and Hepatotoxicity Guidance in the Product Information of Protein Kinase Inhibitors in Oncology Registered at the European Medicines Agency. Pharmacol. Res. Perspect. 2023, 11, e01067. [Google Scholar] [CrossRef]
- Das, A.; Sil, A.; Khan, I.A.; Bandyopadhyay, D. Dermatological Adverse Drug Reactions to Tyrosine Kinase Inhibitors: A Narrative Review. Clin. Exp. Dermatol. 2023, 48, 599–608. [Google Scholar] [CrossRef]
- Liu, C.; Amin, R.; Shatila, M.; Short, N.; Altan, M.; Shah, A.; Alhalabi, O.; Okhuysen, P.; Thomas, A.S.; Wang, Y. Clinical Characteristics and Outcomes of Tyrosine Kinase Inhibitor-Related Lower GI Adverse Effects. J. Cancer Res. Clin. Oncol. 2023, 149, 3965–3976. [Google Scholar] [CrossRef]
- Corrie, P.G. Cytotoxic Chemotherapy: Clinical Aspects Clinical Applications of Cytotoxic Chemotherapy. Medicine 2007, 36, 24–28. [Google Scholar] [CrossRef]
- WHO Collaborating Centre for Drug Statistics Methodology. ATC/ DDD Index 2024. Available online: https://atcddd.fhi.no/atc_ddd_index/ (accessed on 15 December 2024).
- Takarabe, M.; Kotera, M.; Nishimura, Y.; Goto, S.; Yamanishi, Y. Drug Target Prediction Using Adverse Event Report Systems: A Pharmacogenomic Approach. Bioinformatics 2012, 28, i611–i618. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://Www.Meddra.Org/about-Meddra/Vision (accessed on 30 August 2024).
- Böhm, R.; Von Hehn, L.; Herdegen, T.; Klein, H.J.; Bruhn, O.; Petri, H.; Höcker, J. OpenVigil FDA—Inspection of U.S. American Adverse Drug Events Pharmacovigilance Data and Novel Clinical Applications. PLoS ONE 2016, 11, e0157753. [Google Scholar] [CrossRef] [PubMed]
- Kant, A.; van Hunsel, F.; van Puijenbroek, E. Numbers of Spontaneous Reports: How to Use and Interpret? Br. J. Clin. Pharmacol. 2022, 88, 1365–1368. [Google Scholar] [CrossRef]
- Faillie, J.L. Case–Non-Case Studies: Principle, Methods, Bias and Interpretation. Therapies 2019, 74, 225–232. [Google Scholar] [CrossRef]
- Trillenberg, P.; Sprenger, A.; Machner, B. Sensitivity and Specificity in Signal Detection with the Reporting Odds Ratio and the Information Component. Pharmacoepidemiol. Drug Saf. 2023, 32, 910–917. [Google Scholar] [CrossRef] [PubMed]
- IBM Corp. SPSS Statistics for Windows, Version 26.0. Statistical Software Suite; IBM Corp.: Armonk, NY, USA, 2019. [Google Scholar]
- George, S.; Chi, P.; Heinrich, M.C.; von Mehren, M.; Jones, R.L.; Ganjoo, K.; Trent, J.; Gelderblom, H.; Razak, A.A.; Gordon, M.S.; et al. Ripretinib Intrapatient Dose Escalation after Disease Progression Provides Clinically Meaningful Outcomes in Advanced Gastrointestinal Stromal Tumour. Eur. J. Cancer 2021, 155, 236–244. [Google Scholar] [CrossRef]
- Rodriguez, Y.E.; Batra, R.; Patel, K.; Martinez, S. Capivasertib-Induced Diabetic Ketoacidosis in a Patient With Estrogen Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative (ER+/HER2-) Metastatic Breast Cancer and No Prior History of Diabetes Mellitus: A Case Report. Cureus 2024, 16, e63710. [Google Scholar] [CrossRef]
- Available online: https://Clincalc.Com/DrugStats/Top300Drugs.Aspx (accessed on 15 December 2024).
- Dhanyamraju, P.K. Drug Resistance Mechanisms in Cancers: Execution of Prosurvival Strategies. J. Biomed. Res. 2024, 38, 95–121. [Google Scholar] [CrossRef]
- Joshi, D.C.; Sharma, A.; Prasad, S.; Singh, K.; Kumar, M.; Sherawat, K.; Tuli, H.S.; Gupta, M. Novel Therapeutic Agents in Clinical Trials: Emerging Approaches in Cancer Therapy. Discov. Oncol. 2024, 15, 342. [Google Scholar] [CrossRef]
- Gao, H.; Xi, Z.; Dai, J.; Xue, J.; Guan, X.; Zhao, L.; Chen, Z.; Xing, F. Drug Resistance Mechanisms and Treatment Strategies Mediated by Ubiquitin-Specific Proteases (USPs) in Cancers: New Directions and Therapeutic Options. Mol. Cancer 2024, 23, 88. [Google Scholar] [CrossRef]
- Zhang, J.; Vokes, N.; Li, M.; Xu, J.; Bai, H.; Wang, J.; Wang, Z.; Zhang, J. Overcoming EGFR-TKI Resistance by Targeting the Tumor Microenvironment. Chin. Med. J. Pulm. Crit. Care Med. 2024, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting Cancer with Small Molecule Kinase Inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Hofheinz, R.D.; Segaert, S.; Safont, M.J.; Demonty, G.; Prenen, H. Management of Adverse Events during Treatment of Gastrointestinal Cancers with Epidermal Growth Factor Inhibitors. Crit. Rev. Oncol. Hematol. 2017, 114, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Zembower, T.R. Epidemiology of Infections in Cancer Patients. In Infectious Complications in Cancer Patients; Springer: Berlin/Heidelberg, Germany, 2014; pp. 43–89. [Google Scholar]
- Vento, S.; Cainelli, F. Infections in Patients with Cancer Undergoing Chemotherapy: Aetiology, Prevention, and Treatment. Lancet Oncol. 2003, 4, 595–604. [Google Scholar] [CrossRef]
- Holcmann, M.; Sibilia, M. Mechanisms Underlying Skin Disorders Induced by EGFR Inhibitors. Mol. Cell. Oncol. 2015, 2, e1004969. [Google Scholar] [CrossRef]
- Radulian, I.L.; Nitulescu, G.; Zanfirescu, A.; Nitulescu, G.M. Statistical and Clustering Analysis of Safety Profiles of FDA-Approved Protein Kinase Inhibitors. Appl. Sci. 2024, 14, 11757. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Range of frequency | >10% | 1–10% | 0.1–1% | 0.01–0.1% | 0.001–0.01% |
| Number of AEs values | 303 | 5733 | 21,426 | 30,101 | 6318 |
| PT | Mean of the Frequencies (%) | Number of Drugs Associated with the PT |
|---|---|---|
| death | 17.69 | 44 |
| diarrhea | 13.95 | 49 |
| off label use | 11.34 | 24 |
| fatigue | 10.92 | 40 |
| nausea | 10.72 | 28 |
| extra dose administered | 9.72 | 1 |
| blood glucose increased | 9.61 | 2 |
| product dose omission issue | 9.21 | 10 |
| rash | 8.40 | 30 |
| dyspnoea | 6.62 | 4 |
| headache | 6.60 | 3 |
| pyrexia | 6.47 | 24 |
| pain | 6.46 | 2 |
| dizziness | 6.40 | 5 |
| product use in unapproved indication | 6.27 | 9 |
| idiopathic pulmonary fibrosis | 6.07 | 1 |
| vomiting | 6.02 | 28 |
| hair colour changes | 6.02 | 6 |
| no adverse event | 5.78 | 8 |
| arthralgia | 5.63 | 7 |
| pruritus | 5.47 | 7 |
| vitamin B1 decreased | 5.27 | 1 |
| alopecia | 5.27 | 16 |
| PT | Mean nonPKI | Mean PKI | Difference |
|---|---|---|---|
| febrile neutropenia | 5.25 | 1.93 | 3.32 |
| neutropenia | 5.10 | 2.74 | 2.36 |
| thrombocytopenia | 3.87 | 2.18 | 1.69 |
| anemia | 4.09 | 2.80 | 1.30 |
| myelodysplastic syndrome | 1.25 | 0.28 | 0.97 |
| mucosal inflammation | 1.87 | 0.90 | 0.97 |
| pancytopenia | 2.13 | 1.26 | 0.87 |
| hepatic pain | 0.81 | 0.10 | 0.71 |
| bone marrow failure | 1.31 | 0.66 | 0.65 |
| haematotoxicity | 0.83 | 0.23 | 0.60 |
| respiratory failure | 1.54 | 0.95 | 0.59 |
| leukopenia | 1.39 | 0.82 | 0.57 |
| hepatic function abnormal | 0.78 | 1.36 | −0.58 |
| white blood cell count increased | 0.61 | 1.26 | −0.65 |
| metastases to central nervous system | 0.57 | 1.62 | −1.05 |
| cryptococcosis | 0.05 | 1.11 | −1.06 |
| breast cancer metastatic | 0.87 | 1.97 | −1.10 |
| decreased appetite | 3.61 | 4.74 | −1.13 |
| nail disorder | 0.19 | 1.32 | −1.13 |
| drug resistance | 0.58 | 1.93 | −1.35 |
| cognitive disorder | 0.63 | 2.18 | −1.55 |
| chronic myeloid leukemia | 0.20 | 2.20 | −1.99 |
| diarrhea | 8.36 | 13.95 | −5.59 |
| SOC | Kruskal–Wallis Test | Number of PTs -PKI | Number of PTs -nonPKI | PKI Mean | nonPKI Mean |
|---|---|---|---|---|---|
| Blood and lymphatic system disorders | p < 0.001 | 244 | 460 | 0.539 | 0.553 |
| Cardiac disorders | p < 0.001 | 160 | 247 | 0.554 | 0.308 |
| Congenital, familial and genetic disorders | p < 0.001 | 48 | 210 | 0.174 | 0.067 |
| Ear and labyrinth disorders | p = 0.204 | 34 | 58 | 0.206 | 0.146 |
| Endocrine disorders | p < 0.001 | 97 | 162 | 0.379 | 0.167 |
| Eye disorders | p < 0.001 | 146 | 319 | 0.414 | 0.118 |
| Gastrointestinal disorders | p < 0.001 | 450 | 806 | 0.310 | 0.960 |
| General disorders and administration site conditions | p < 0.001 | 203 | 435 | 2.291 | 0.878 |
| Hepatobiliary disorders | p < 0.001 | 110 | 187 | 0.364 | 0.230 |
| Immune system disorders | p < 0.001 | 101 | 258 | 0.289 | 0.254 |
| Infections and infestations | p < 0.001 | 299 | 714 | 0.399 | 0.237 |
| Injury, poisoning and procedural complications | p < 0.001 | 200 | 415 | 1.088 | 0.601 |
| Investigations | p < 0.001 | 469 | 829 | 0.537 | 0.299 |
| Metabolism and nutrition disorders | p < 0.001 | 84 | 143 | 0.541 | 0.352 |
| Musculoskeletal and connective tissue disorders | p < 0.001 | 128 | 296 | 0.600 | 0.259 |
| Neoplasms benign, malignant and unspecified (incl cysts and polyps) | p < 0.001 | 189 | 386 | 0.661 | 0.293 |
| Nervous system disorders | p < 0.001 | 172 | 379 | 0.545 | 0.276 |
| Pregnancy, puerperium and perinatal conditions | p = 0.010 | 13 | 59 | 0.061 | 0.232 |
| Product issues | p = 0.203 | 21 | 40 | 0.302 | 0.400 |
| Psychiatric disorders | p = 0.008 | 53 | 143 | 0.352 | 0.462 |
| Renal and urinary disorders | p = 0.019 | 74 | 135 | 0.518 | 0.347 |
| Reproductive system and breast disorders | p < 0.001 | 70 | 101 | 0.058 | 0.50 |
| Respiratory, thoracic and mediastinal disorders | p < 0.001 | 126 | 222 | 0.484 | 0.275 |
| Skin and subcutaneous tissue disorders | p < 0.001 | 145 | 194 | 0.936 | 0.409 |
| Social circumstances | p = 0.168 | 22 | 57 | 0.284 | 0.120 |
| Surgical and medical procedures | p < 0.001 | 189 | 277 | 0.354 | 0.144 |
| Vascular disorders | p = 0.233 | 81 | 134 | 0.467 | 0.227 |
| Cluster | Set PKI | ATC Code | Set nonPKI | ATC Code |
|---|---|---|---|---|
| 1 | Imatinib | L01EA01 | Cyclophosphamide | L01AA01 |
| Dasatinib | L01EA02 | Melphalan | L01AA03 | |
| Nilotinib | L01EA03 | Temozolomide | L01AX03 | |
| Alectinib | L01ED03 | Methotrexate | L01BA01 | |
| Ruxolitinib | L01EJ01 | Pemetrexed | L01BA04 | |
| Ibrutinib | L01EL01 | Mercaptopurine | L01BB02 | |
| Fludarabine | L01BB05 | |||
| Cytarabine | L01BC01 | |||
| Fluorouracil | L01BC02 | |||
| Gemcitabine | L01BC05 | |||
| Capecitabine | L01BC06 | |||
| Vincristine | L01CA02 | |||
| Etoposide | L01CB01 | |||
| Paclitaxel | L01CD01 | |||
| Docetaxel | L01CD02 | |||
| Irinotecan | L01CE02 | |||
| Doxorubicin | L01DB01 | |||
| Epirubicin | L01DB03 | |||
| Cisplatin | L01XA01 | |||
| Carboplatin | L01XA02 | |||
| Oxaliplatin | L01XA03 | |||
| Bortezomib | L01XG01 | |||
| Celecoxib | L01XX33 | |||
| Venetoclax | L01XX52 | |||
| 2 | Ponatinib | L01EA05 | Ifosfamide | L01AA06 |
| Gefitinib | L01EB01 | Bendamustine | L01AA09 | |
| Erlotinib | L01EB02 | Busulfan | L01AB01 | |
| Osimertinib | L01EB04 | Thiotepa | L01AC01 | |
| Vemurafenib | L01EC01 | Carmustine | L01AD01 | |
| Dabrafenib | L01EC02 | Dacarbazine | L01AX04 | |
| Crizotinib | L01ED01 | Cladribine | L01BB04 | |
| Trametinib | L01EE01 | Azacitidine | L01BC07 | |
| Palbociclib | L01EF01 | Vinblastine | L01CA01 | |
| Ribociclib | L01EF02 | Vinorelbine | L01CA04 | |
| Abemaciclib | L01EF03 | Topotecan | L01CE01 | |
| Lapatinib | L01EH01 | Daunorubicin | L01DB02 | |
| Axitinib | L01EK01 | Idarubicin | L01DB06 | |
| Acalabrutinib | L01EL02 | Mitoxantrone | L01DB07 | |
| Idelalisib | L01EM01 | Bleomycin | L01DC01 | |
| Sunitinib | L01EX01 | Mitomycin | L01DC03 | |
| Sorafenib | L01EX02 | Procarbazine | L01XB01 | |
| Pazopanib | L01EX03 | Tretinoin | L01XF01 | |
| Regorafenib | L01EX05 | Carfilzomib | L01XG02 | |
| Cabozantinib | L01EX07 | Ixazomib | L01XG03 | |
| Lenvatinib | L01EX08 | Olaparib | L01XK01 | |
| Nintedanib | L01EX09 | |||
| 3 | Bosutinib | L01EA04 | Chlorambucil | L01AA02 |
| Afatinib | L01EB03 | Lomustine | L01AD02 | |
| Encorafenib | L01EC03 | Clofarabine | L01BB06 | |
| Lorlatinib | L01ED05 | Decitabine | L01BC08 | |
| Cobimetinib | L01EE02 | Trifluridine | L01BC59 | |
| Binimetinib | L01EE03 | Cabazitaxel | L01CD04 | |
| Tucatinib | L01EH03 | Dactinomycin | L01DA01 | |
| Alpelisib | L01EM03 | Niraparib | L01XK02 | |
| Midostaurin | L01EX10 | Arsenic trioxide | L01XX27 | |
| Gilteritinib | L01EX13 | Anagrelide | L01XX35 | |
| Eribulin | L01XX41 | |||
| 4 | Ceritinib | L01ED02 | Ixabepilone | L01DC04 |
| Brigatinib | L01ED04 | Bexarotene | L01XF03 | |
| Zanubrutinib | L01EL03 | Romidepsin | L01XH02 | |
| Vandetanib | L01EX04 | Rucaparib | L01XK03 | |
| Ripretinib | L01EX19 | Vorinostat | L01XX53 | |
| Enasidenib | L01XX59 | |||
| Selinexor | L01XX66 | |||
| 5 | Mobocertinib | L01EB10 | Streptozocin | L01AD04 |
| Floxuridine | L01BC09 | |||
| Valrubicin | L01DB09 | |||
| Adagrasib | L01XX77 | |||
| 6 | Asciminib | L01EA06 | Nelarabine | L01BB07 |
| Dacomitinib | L01EB07 | Trabectedin | L01CX01 | |
| Selumetinib | L01EE04 | Aminolevulinic acid | L01XD04 | |
| Fedratinib | L01EJ02 | Alitretinoin | L01XF02 | |
| Pacritinib | L01EJ03 | Panobinostat | L01XH03 | |
| Tivozanib | L01EK03 | Vismodegib | L01XJ01 | |
| Fruquintinib | L01EK04 | Sonidegib | L01XJ02 | |
| Erdafitinib | L01EN01 | Talazoparib | L01XK04 | |
| Pemigatinib | L01EN02 | Pentostatin | L01XX08 | |
| Larotrectinib | L01EX12 | Mitotane | L01XX23 | |
| Entrectinib | L01EX14 | Ivosidenib | L01XX62 | |
| Capmatinib | L01EX17 | Lurbinectedin | L01XX69 | |
| Avapritinib | L01EX18 | |||
| 7 | Quizartinib | L01EX11 | Belinostat | L01XH04 |
| Pralsetinib | L01EX23 | Belzutifan | L01XX74 | |
| Capivasertib | L01EX27 | |||
| 8 | Neratinib | L01EH02 | Pralatrexate | L01BA05 |
| Momelotinib | L01EJ04 | Porfimer sodium | L01XD01 | |
| Copanlisib | L01EM02 | Glasdegib | L01XJ03 | |
| Duvelisib | L01EM04 | Tazemetostat | L01XX72 | |
| Pexidartinib | L01EX15 | Sotorasib | L01XX73 | |
| Tepotinib | L01EX21 | Eflornithine | L01XX79 | |
| Selpercatinib | L01EX22 | |||
| Umbralisib | L01EX25 | |||
| 9 | Pirtobrutinib | L01EL05 | Methyl aminolevulinate | L01XD03 |
| Infigratinib | L01EN03 | |||
| Futibatinib | L01EN04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Österreichische Pharmazeutische Gesellschaft. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radulian, I.L.; Nitulescu, G.; Zanfirescu, A.; Nitulescu, G.M. Comparative Analysis of Adverse Effects: Protein Kinase Inhibitors Versus Traditional Anticancer Therapies. Sci. Pharm. 2025, 93, 20. https://doi.org/10.3390/scipharm93020020
Radulian IL, Nitulescu G, Zanfirescu A, Nitulescu GM. Comparative Analysis of Adverse Effects: Protein Kinase Inhibitors Versus Traditional Anticancer Therapies. Scientia Pharmaceutica. 2025; 93(2):20. https://doi.org/10.3390/scipharm93020020
Chicago/Turabian StyleRadulian, Ioana Lavinia, Georgiana Nitulescu, Anca Zanfirescu, and George Mihai Nitulescu. 2025. "Comparative Analysis of Adverse Effects: Protein Kinase Inhibitors Versus Traditional Anticancer Therapies" Scientia Pharmaceutica 93, no. 2: 20. https://doi.org/10.3390/scipharm93020020
APA StyleRadulian, I. L., Nitulescu, G., Zanfirescu, A., & Nitulescu, G. M. (2025). Comparative Analysis of Adverse Effects: Protein Kinase Inhibitors Versus Traditional Anticancer Therapies. Scientia Pharmaceutica, 93(2), 20. https://doi.org/10.3390/scipharm93020020