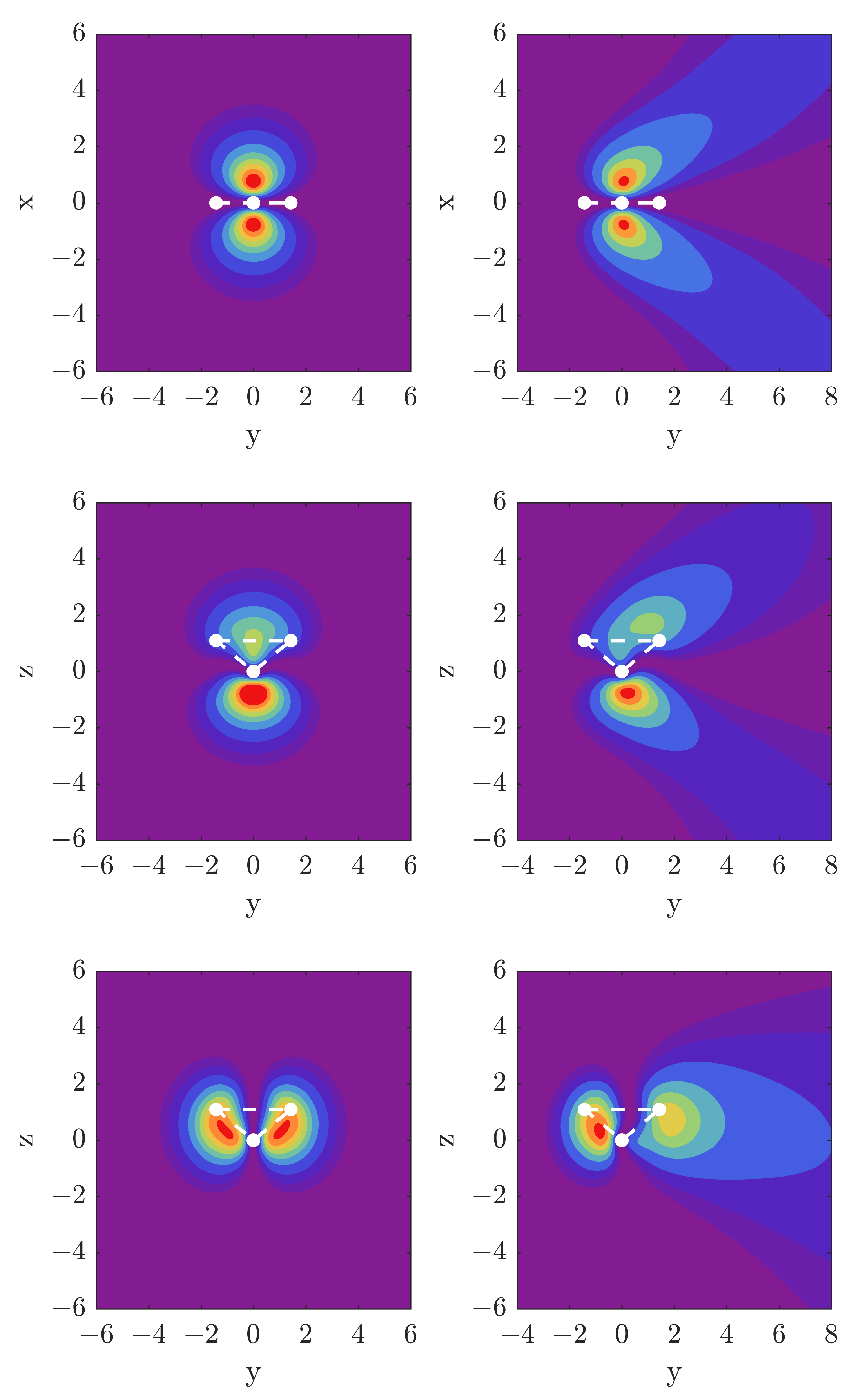
3.1. Visualization of Molecular Orbitals for x-, y-, and z-Directed Forces
In Ref. [
1], results for probability densities obtained from CAP-FEM solutions in Mathematica
® NDEigensystem were shown. For the present work, we calculated the orbitals from the ECS-FEM eigenvectors with the help of a Fortran program. The orbitals from the ECS-FEM have prescribed discontinuities at the scaling radius [
19], while the CAP-FEM solutions are smooth. Nevertheless, for radial distances below the scaling radius the shapes are consistent between these methods to the point that visual inspection alone cannot identify differences (the color schemes are similar, but not identical). For the same force orientation as presented in Ref. [
1], we now add a comparison of
.
All density plots show some peculiarities. For the force applied in the
x direction, the
MO in
Figure 1 shows a structure in the orbital shape, where the left lobe of the orbital bends down, along with the right lobe of the orbital. In other words, the electron density is not purely directed along the direction of the force. For the
MO, we observe that it is pushed toward the top right of the box, which implies that the
x-directed force leads to part of the density being pushed in the positive
z direction. For the
MO, there is a run-away of the density to both positive and negative
z along with a movement towards positive
x.
When looking at results for the
y-directed force in
Figure 2, one notices the continuation of the trend as the MOs
and
also have a run-away movement of the density away from the direction of the force. Finally, for
we also observe a structure in the right lobe downwards, away from the direction of the force. All these deformations of the MO probability densities reflect the combined influence of molecular potential and external dc electric field potential.
In
Figure 3 we display the MO densities for the case when the force is directed in the
z direction. Here we carry out a comparison between
and
. When increasing
we observe: (i) a narrowing and re-direction of the MO density of
towards the direction of the force; (ii) the same occurs for
, although there are also two maxima in the density which form at the base of the uppermost lobe; and (iii) we find that the MO
slightly changes in shape, and the lobes lengthen, in part towards the direction of the force. This is caused by the added flexibility in the partial-wave expansion.
To summarize the visualization of our results, which required added computational effort compared to the resonance parameter calculations, we note that the spatial distribution of ionized electrons even under dc field conditions is far from trivial, and would present a fertile ground for comparison with experiment. We note that in the context of ac laser field ionization, R-matrix theory was used recently to predict interesting ionization patterns for the water molecule [
20].
3.2. Dominant Field Direction for Ionization of MOs and Their dc Stark Shift
We now proceed with a presentation of the resonance parameters for the MOs, and focus on the behavior of the dc Stark shift for the MO with the largest ionization rate for a given field direction. Our main observation in this work is that the orbital with the strongest ionization rate for a given external field direction displays a non-monotonic behavior in the dc Stark shift.
Graphs of the resonance position for fields along the molecular axis
as a function of field strength were shown in Ref. [
1] to display such behavior for the
orbital for the orientation where electrons are pushed out opposite to the hydrogen atoms. The shift was shown to be initially positive, then reached a maximum, and became zero at about
a.u., in order to continue to negative values for stronger fields.
For the orbital, non-monotonic dependence of the the dc shift was observed in both directions with minima occurring at force strength of the order of a.u. and a.u., respectively. The MO is the orbital with the strongest ionization rate in that case, and the minimum in the dc shift at a.u. yields to an almost vanishing Stark shift. The bonding MO, on the other hand, showed monotonic behavior for fields along . The purpose of this section is to show that these features can be generalized to other field directions.
In
Figure 4, the three valence orbitals are shown for the case of an external field perpendicular to the molecular plane. The results are obviously symmetric with respect to reversing the field orientation. The half-widths show clearly that the MO oriented with lobes in this direction (
) is the most easily ionized valence orbital, with rates exceeding the other MOs by one or more orders of magnitude before saturation sets in.
While the MO shows non-monotonicity in the dc Stark shift with a minimum at a.u., and a vanishing shift at a.u., the other MOs simply acquire increasingly negative dc shifts. We can now ask whether this behavior is more than coincidental: is it true that for a field oriented along the direction the bonding orbital might be affected similarly to the MO’s behavior in the case of a field along ?
The answer is provided in
Figure 5 below; indeed, despite its deep binding energy for substantial field strengths, such as
a.u., this orbital is clearly the most easily ionized of the three valence orbitals. Note that this is not the case for weak fields, i.e., in the pure tunneling regime. The implication would be that strong fields along the
direction ionization with break-up of a hydrogen bond is becoming the dominant process.
To understand the dominance as far as ionization rate is concerned, geometric considerations are, of course, an important indicator: in a simplistic representation of the three orbitals, i.e., , , and , it is obvious that they respond strongly to fields aligned with , respectively, due to the occurrence of substantial dipole matrix elements from the external field.
Interestingly, the non-monotonic behavior in the dc Stark shift for the orbital occurs only for field strength a.u., with a minimum at a.u., and a vanishing shift at a.u. Thus the overall trend is comparable to the orbital , which displays very similar behaviour when under a force in the x direction. The trends of both these orbitals under their respective forces outside of a.u. are very similar to the trend in when the z force is in the negative direction.
For the case of fields along the molecular axis
that was discussed in Ref. [
1], we report some additional results here, having extended the calculations to the level of
. Compared to the field orientations along
and
we have the complication of asymmetry of the molecule along the molecular axis; thus, we separate the presentation for fields aligned such that the force pushes electrons out along this axis (
Figure 6), and in the opposite direction (
Figure 7).
We begin with electrons being pushed out towards the oxygen atom. When one considers field strengths beyond the tunneling regime (where the width turns over towards saturation), the MO emerges as the one with the highest ionization rate. Due to the asymmetry, the dc Stark shift has a non-zero slope at zero field. The resonance position decreases with increasing field and reaches a minimum at a.u. The more accurate results run parallel to those for for the dc shift. For the resonance width, the two results are not well distinguishable on a logarithmic scale except for the weakest field strengths shown.
Concerning the convergence with , we can state that the weakest bound MO () is showing the smallest discrepancy, while the bonding orbital () is affected most, because the partial-wave expansion is sampling the full potential from the hydrogenic parts only gradually as is increased. We observe, however, that the convergence (or lack thereof) does not have a strong impact on the features reported in this work, i.e., the non-monotonicity of the shifts. For fields in the opposite direction, i.e., ionization into the half-space on the hydrogenic side (away from the oxygen atom), we again notice that the dominant ionization contribution is from the MO , and that this MO displays complicated non-monotonic behavior in the dc Stark shift.
To summarize this section we observe that the conclusions are consistent for the four possible field orientations (the z direction has two possible orientations due to asymmetry of the molecule) associated with symmetry axes of the orbitals. We conclude that the non-monotonic behavior of the dc Stark shift goes hand-in-hand with the relatively strong ionization rate for the orbital of that particular symmetry.
While our results for the widths of the
orbital show an asymmetry when changing the
z force direction from positive to negative, this behavior is different from what was observed for the net molecular width in HF theory [
3], as shown in
Figure 7 of Ref. [
1].
Our own net widths are dominated by the orbital, and so a natural question to ask is whether there is a convergence issue in our partial-wave approach. The results do not deviate, however, substantially from those for . Thus, one will have to investigate further whether this difference in behavior is related to the determination of the net decay width, or whether it is the model potential approach that fails to account for self-consistent field effects in the presence of the external field.
3.3. Comparison with HF and CCSD(T) Calculations for Net Ionization
In
Figure 8 we compare with the HF and CCSD(T) total molecular Stark shifts and resonance widths given in Ref. [
3]. As explained in Ref. [
1] for our model calculation a meaningful comparison is to consider the direct sum of orbital energies. Such an analysis corresponds to total (net) ionization from the molecule, and the five MOs are counted twice to account for the spin degeneracy.
The most direct comparison for the present results should be with the mean-field single-particle approach, i.e., the HF method, as our model potential is designed to match HF orbital energies. The comparison of the dc Stark shifts for field directions along
and
shows that the present model calculation yields stronger shifts as the field strength increases, but that the overall trend agrees with the HF results of Ref. [
3].
We observe a similar trend in the decay widths: they agree at the factor-of-two level for the cases involving fields along
and
. This is an improvement compared to the results for forces along the molecular axis in the positive
z direction, while comparable to the case when the force is in the negative
z direction [
1]. We note that the additional corrections due to electronic correlation, i.e., the CCSD(T) over the HF results, are also on a similar scale, i.e., a discrepancy at the level of factors of two–three when the ionization rate is strong.
The blue dots (
•) mark the DS (2) values, i.e., the direct sum method, in which we calculate the total molecular Stark shift and width by adding the resonance parameters for every MO (as reported in
Section 3.2) assuming double occupancy due to spin degeneracy [
1]. The agreement between DS (2) and the HF results of Ref. [
3] is generally good for the Stark shifts but less so for the widths. At higher field strengths, the agreement weakens, and more so for the force in the
y direction. For the widths, DS (2) results (shown as
•) begin with improving the agreement with HF (shown as
•) as a function of the force strength. We note that the correlated CCSD(T) calculations of Ref. [
3] generally do not deviate much from the HF data, and that the present model calculations in some cases, perhaps fortuitously, agree with them.
The Stark shifts for the
x-directed force agree very well with the CCSD(T) results of Ref. [
3] with single-digit percentage deviations. It would be of interest to compare the MO resonance parameters from HF calculations with the present model potential results in order to complement the comparison of net quantities which follow from the total energy. For the purposes of eventual comparison with the experiment, and to understand the theory better, more work must be done to bridge the gap between the multi-electron solutions, HF and CCSD(T), and the present single-electron, local potential approach. For future work, it is planned to extend the current work to a potential model from density functional theory, such as the local HF potential method [
21,
22].
3.4. Interactions between
the Molecular Orbitals
A perturbative treatment of the distortion of the energy level spectrum due to an external field leads to a power series in an expansion parameter which is proportional to the external field. Nonlinear Stark shifts (or non-monotonic behavior) arise for strong perturbations. This approach of a limited-subspace diagonalization involving only neighboring levels shows distortions in the energy levels (in our case resonance positions) as a function of field strength which can be interpreted as ‘interactions’ between the unperturbed levels. We maintain the designation of the field-free MOs (), even though the symmetries of the MOs are no longer observed once the electric field is turned on.
Strong non-perturbative behavior was obtained for Rydberg states of atomic hydrogen using sophisticated numerical work [
23], leading to avoided energy level crossings at critical field strengths. Thus, it is of interest to view our results not only one MO at a time, but to show the energy levels for the three valence orbitals together.
For fields along the molecular axis, the asymmetry of the
orbital causes a marked difference in Stark shift for the two cases shown in
Figure 9. The densities in the two top rows of
Figure 1 serve to visualize how the
and
MOs are affected by the external field acting along
. First of all, we note the different energy scales for the left and right panels, and how for the external electric force pushing in the
direction larger shifts are obtained than for the case of
. For the bonding
orbital, the Stark shift is monotonic and on a relative scale decoupled from the other two valence orbitals except for some interaction with the
MO for force in the
direction. The two outer orbitals interact strongly with each other for electic force along
with an avoided crossing at
0.17 a.u.
It is interesting to consult
Figure 7 to observe what happens to the decay widths as one varies the electric field strength across the crossing point. While the decay rate for what is the
MO at zero field levels off to a (relatively large) constant value as one increases the field strength, the one for the
MO keeps increasing.
For the case of fields along
and
shown in
Figure 10, we observe a repulsion of the
and
MO energy positions for fields along
. For the other case, i.e., fields along
complicated behavior sets in between the bonding
MO with
at the strongest fields shown with a possible curve crossing at even stronger fields.
To summarize this section, we conclude that with the exception of the MO, with the electrons being pushed to positive , for which a very nonlinear Stark shift is observed as a result of an avoided curve crossing, the non-monotonic shift behavior is not obviously related to MO levels interacting very strongly with each other. Another way to state this is that the level interactions occur while the energy levels are well separated in those cases.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}