

Dynamics of Site Selectivity in Dissociative Electron Attachment in Aromatic Molecules


Abstract
1. Introduction
2. Experimental Setup
3. Results and Discussions
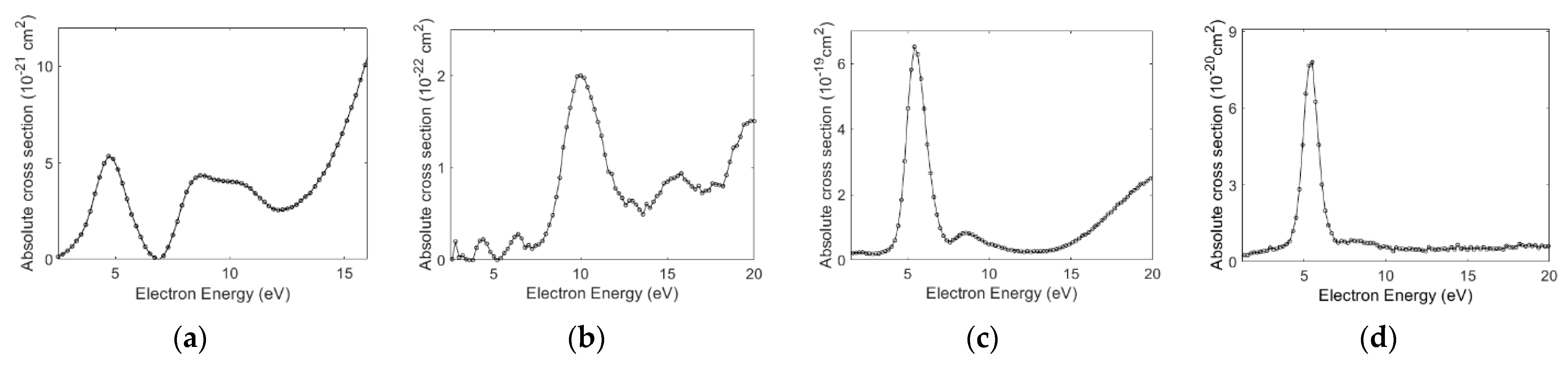
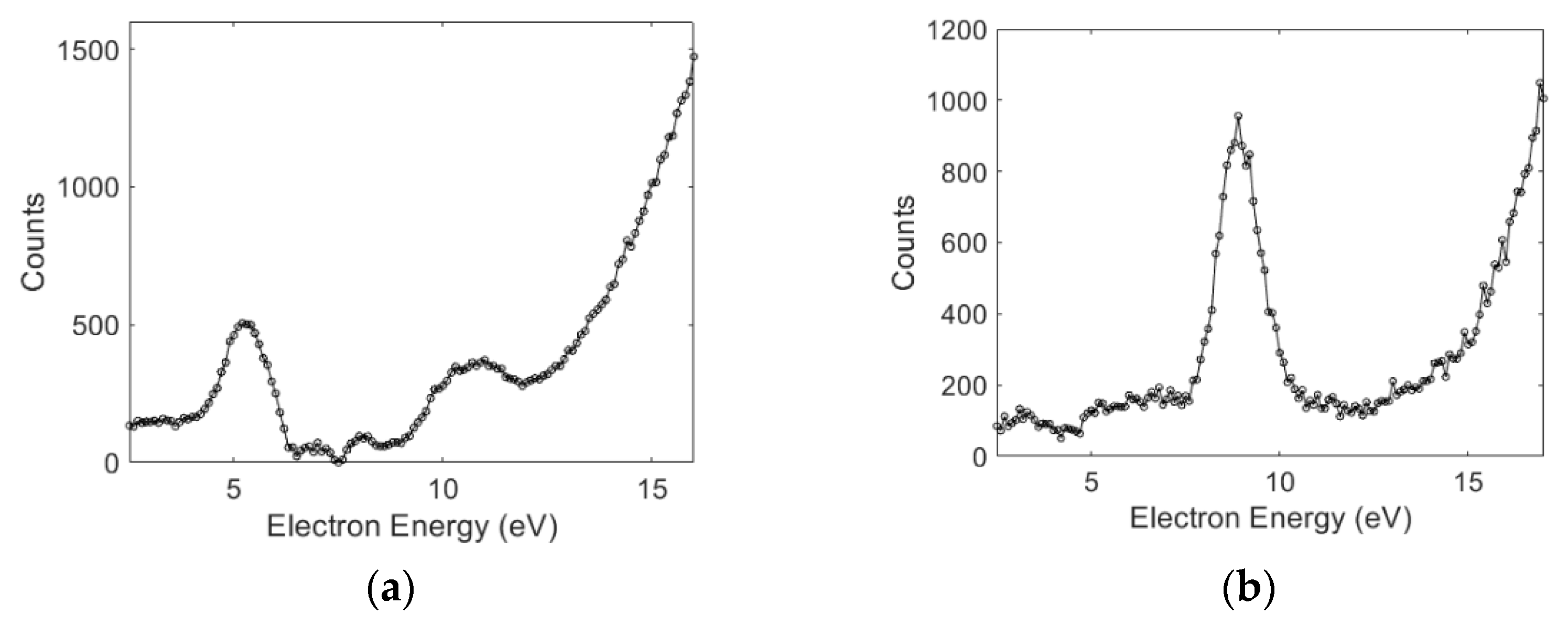
3.1. Absolute Cross-Sections
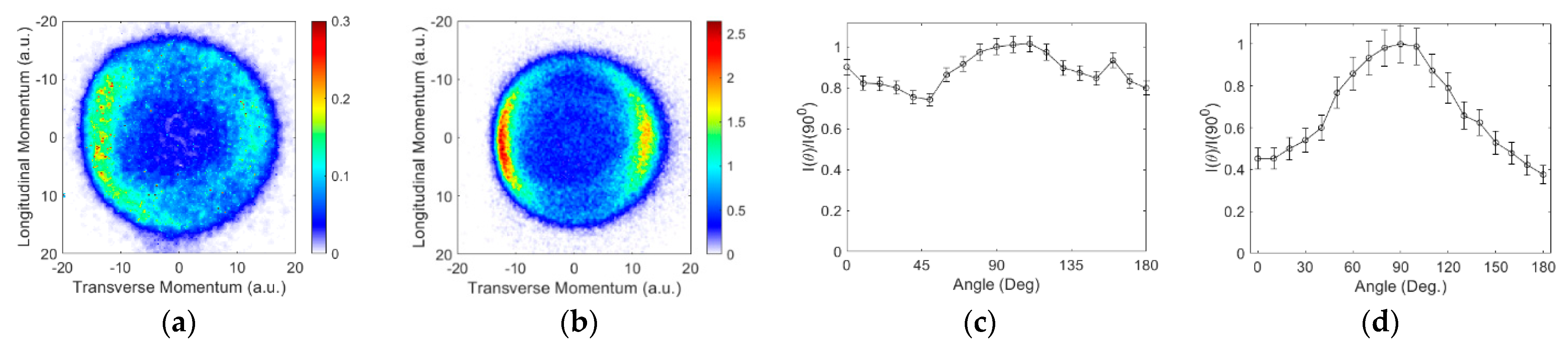
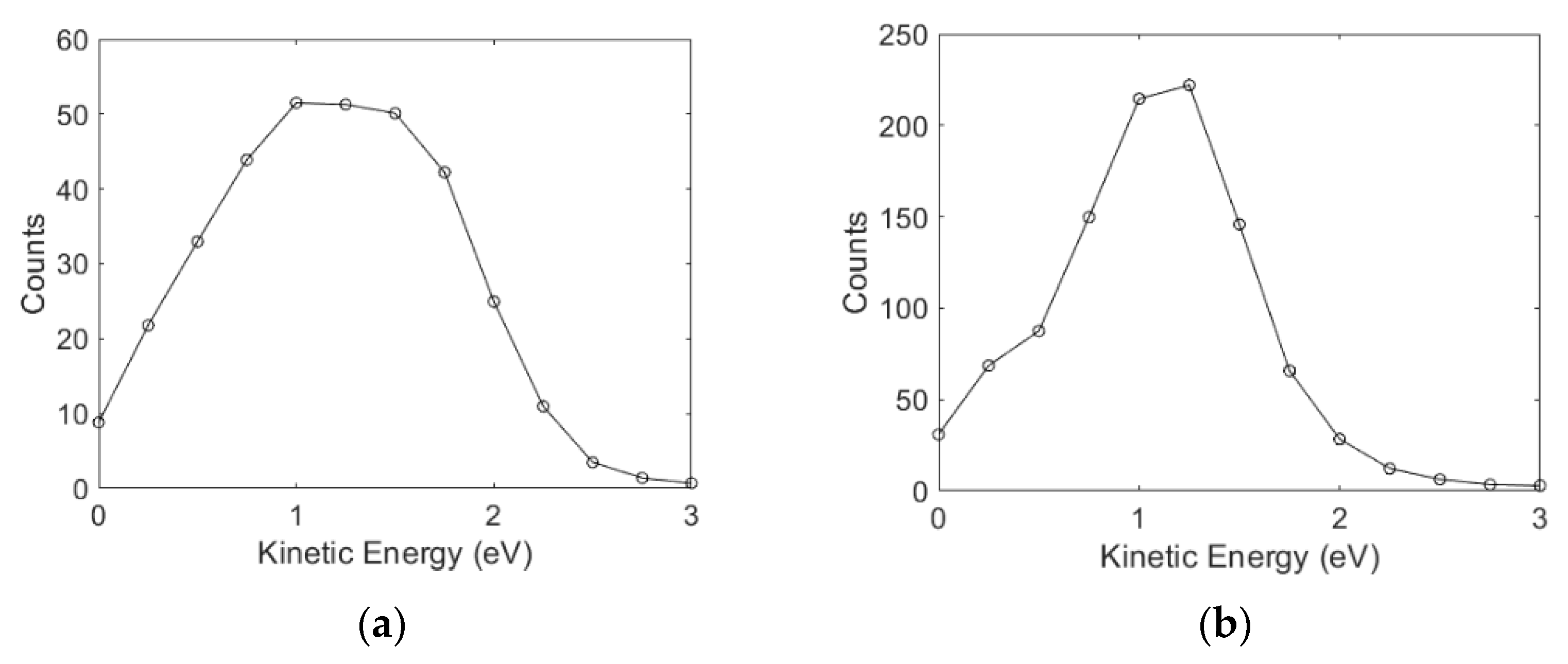
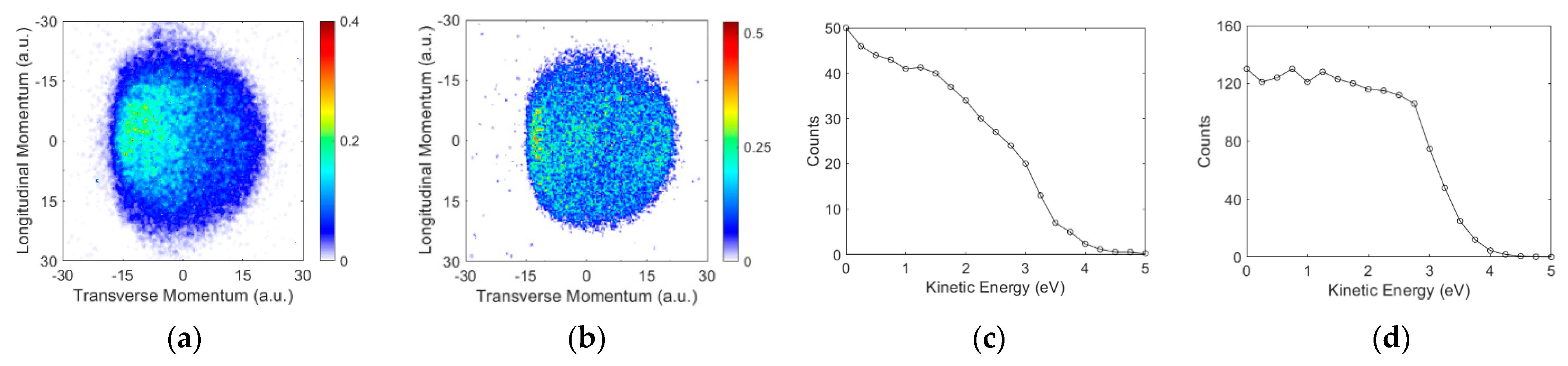
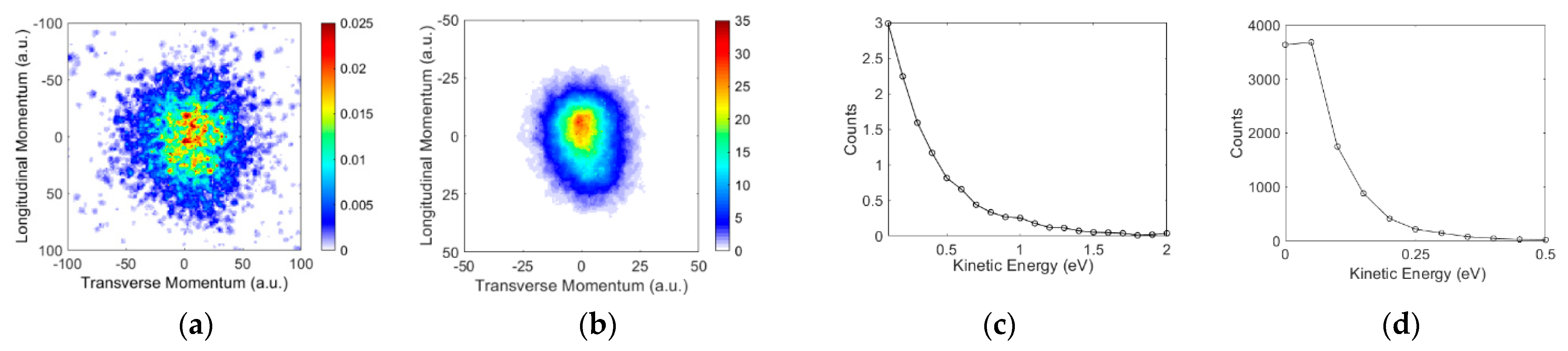
3.2. Angular and Kinetic Energy Distributions
Ions
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prabhudesai, V.S.; Kelkar, A.H.; Nandi, D.; Krishnakumar, E. Functional Group Dependent Site Specific Fragmentation of Molecules by Low Energy Electrons. Phys. Rev. Lett. 2005, 95, 143202. [Google Scholar] [CrossRef]
- Ptasińska, S.; Denifl, S.; Grill, V.; Märk, T.D.; Illenberger, E.; Scheier, P. Bond- and Site-Selective Loss of H− from Pyrimidine Bases. Phys. Rev. Lett. 2005, 95, 093201. [Google Scholar] [CrossRef] [PubMed]
- Oster, T.; Kuhn, A.; Illenberger, E. Gas phase negative ion chemistry. Int. J. Mass Spectrom. Ion Processes 1989, 89, 1–72. [Google Scholar] [CrossRef]
- Prabhudesai, V.S.; Nandi, D.; Kelkar, A.H.; Krishnakumar, E. Functional group dependent dissociative electron attachment to simple organic molecules. J. Chem. Phys. 2008, 128, 154309. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, T.N.; Trevisan, C.S.; Orel, A.E.; Slaughter, D.S.; Adaniya, H.; Belkacem, A.; Weyland, M.; Dorn, A.; McCurdy, C.W. Dynamics of dissociative electron attachment to ammonia. Phys. Rev. A 2016, 93, 052704. [Google Scholar] [CrossRef]
- Ram, N.B.; Krishnakumar, E. Dissociative Electron Attachment to Polyatomic Molecules—V: Formic Acid and Propyl Amine. arXiv 2010, arXiv:1007.5169. [Google Scholar]
- Skalicky, T.; Allan, M. The assignment of dissociative electron attachment bands in compounds containing hydroxyl and amino groups. J. Phys. B At. Mol. Opt. Phys. 2004, 37, 4849–4859. [Google Scholar] [CrossRef][Green Version]
- Sanche, L. Nanoscopic aspects of radiobiological damage: Fragmentation induced by secondary low-energy electrons. Mass Spectrom. Rev. 2002, 21, 349–369. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, S.; Illenberger, E. Probing biomolecules: Gas phase experiments and biological relevance. Europhys. News 2002, 33, 207–209. [Google Scholar] [CrossRef][Green Version]
- Gope, K.; Prabhudesai, V.S.; Mason, N.J.; Krishnakumar, E. Dissociation dynamics of transient anion formed via electron attachment to sulfur dioxide. J. Chem. Phys. 2017, 147, 054304. [Google Scholar] [CrossRef]
- Pikhtovnikov, S.V.; Mavrodiev, V.K.; Furley, I.I.; Gataullin, R.R.; Abdrakhmanov, I.B. Resonance electron capture by aniline molecules and its derivatives. High Energy Chem. 2006, 40, 224–229. [Google Scholar] [CrossRef]
- Rawat, P.; Prabhudesai, V.S.; Rahaman, M.A.; Ram, N.B.; Krishnakumar, E. Absolute cross sections for dissociative electron attachment to NH3 and CH4. Int. J. Mass Spectrom. 2008, 277, 96–102. [Google Scholar] [CrossRef]
- King, G.A.; Oliver, T.A.A.; Ashfold, M.N.R. Dynamical insights into 1πσ∗ state mediated photodissociation of aniline. J. Chem. Phys. 2010, 132, 214307. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, B.N.; Veeraiah, A.; Sunanda, K.; Jagatap, B.N. Excited states of aniline by photoabsorption spectroscopy in the 30,000–90,000 cm−1 region using synchrotron radiation. J. Chem. Phys. 2013, 139, 064303. [Google Scholar] [CrossRef] [PubMed]
- Ari, T.; Guven, H.; Ecevit, N. Electron energy-loss spectroscopy in monosubstituted benzenes. J. Electron Spectrosc. Relat. Phenom. 1995, 73, 13–23. [Google Scholar] [CrossRef]
- Fenzlaff, H.-P.; Illenberger, E. Low energy electron impact on benzene and the fluorobenzenes: Formation and dissociation of negative ions. Int. J. Mass Spectrom. Ion Processes 1984, 59, 185–202. [Google Scholar] [CrossRef]
- Ram, N.B.; Krishnakumar, E. Dissociative electron attachment resonances in ammonia: A velocity slice imaging based study. J. Chem. Phys. 2012, 136, 164308. [Google Scholar] [CrossRef] [PubMed]
- NIST Chemistry Webbook. Available online: https://webbook.nist.gov/chemistry/ (accessed on 1 September 2022).
- Bordwell, F.G.; Zhang, X.M.; Cheng, J.P. Bond dissociation energies of the nitrogen-hydrogen bonds in anilines and in the corresponding radical anions. Equilibrium acidities of aniline radical cations. J. Org. Chem. 1993, 58, 6410–6416. [Google Scholar] [CrossRef]
- Luo, Y.R. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Ram, N.B.; Prabhudesai, V.S.; Krishnakumar, E. Velocity imaging of H− from formic acid: Probing functional group dependence in dissociative electron attachment. Eur. Phys. J. D 2020, 74, 49. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Peak Position (eV) | Cross-Section (cm2) | Peak Position (eV) | Cross-Section (cm2) | Peak Position (eV) | Cross-Section (cm2) |
|---|---|---|---|---|---|---|
| Aniline | 5.0 | 5.3 × 10−21 | 10.0 | 2.0 × 10−22 | ||
| 8.7 | 4.3 × 10−21 | 15.4 | 1.0 × 10−22 | |||
| 9.9 | 4.3 × 10−21 | |||||
| Benzylamine | 5.5 | 6.5 × 10−19 | 5.5 | 6.0 × 10−20 | 5.5 | 8 × 10−20 |
| 8.4 | 9.0 × 10−20 | 8.3 | 8 × 10−21 | |||
| Ammonia 1 | 5.7 | 2.3 × 10−18 | 5.9 | 1.6 × 10−18 | ||
| 10.5 | 5.0 × 10−19 | 10.2 | 9.0 × 10−20 | |||
| n-propylamine 2 | 5.2 | 5.2 × 10−20 | ||||
| 8.8 | 1.7 × 10−20 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tadsare, V.; Das, S.; Gokhale, S.; Krishnakumar, E.; Prabhudesai, V.S.S. Dynamics of Site Selectivity in Dissociative Electron Attachment in Aromatic Molecules. Atoms 2022, 10, 98. https://doi.org/10.3390/atoms10040098
Tadsare V, Das S, Gokhale S, Krishnakumar E, Prabhudesai VSS. Dynamics of Site Selectivity in Dissociative Electron Attachment in Aromatic Molecules. Atoms. 2022; 10(4):98. https://doi.org/10.3390/atoms10040098
Chicago/Turabian StyleTadsare, Vishvesh, Sukanta Das, Samata Gokhale, E. Krishnakumar, and Vaibhav S. S. Prabhudesai. 2022. "Dynamics of Site Selectivity in Dissociative Electron Attachment in Aromatic Molecules" Atoms 10, no. 4: 98. https://doi.org/10.3390/atoms10040098
APA StyleTadsare, V., Das, S., Gokhale, S., Krishnakumar, E., & Prabhudesai, V. S. S. (2022). Dynamics of Site Selectivity in Dissociative Electron Attachment in Aromatic Molecules. Atoms, 10(4), 98. https://doi.org/10.3390/atoms10040098