5. Running the CSFs Generation Programs
5.1. First Example: Valence–Valence, Core–Valence and Core–Core for
We want to generate an expansion for the state. In this example, the CSFs are generated by SD-excitations from the MR set to an active set characterized by a maximal principal quantum number . The expansion accounts for valence–valence, core–valence and core–core correlation.
*******************************************************************************
* RUN RCSFGENERATE *
* OUTPUT FILES: rcsf.out, rcsfgenerate.log *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program generates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,*)2s(2,*)
Give configuration 2
>>1s(2,*)2p(2,*)
Give configuration 3
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>4s,4p,4d,4f
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,0
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
......
1 blocks were created
block J/P NCSF
1 0+ 361
Please note that by answering 2 for the number of excitations, we will include both single (S) and double (D) excitations. By default, the orbitals will be in the order
etc. There is also the possibility to have a reverse orbital order
, a symmetry order
or a user defined order. We will look at these options in
Section 5.9. The generated file
rcsf.out with the CSF list looks like
Core subshells:
Peel subshells:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d 4s 4p- 4p 4d- 4d 4f- 4f
CSF(s):
1s ( 2) 2s ( 2)
0+
1s ( 2) 2s ( 1) 3s ( 1)
1/2 1/2
0+
1s ( 2) 2s ( 1) 4s ( 1)
1/2 1/2
0+
1s ( 2) 2p ( 2)
0
0+
1s ( 2) 2p-( 2)
0+
..............
In addition to the file rcsf.out with the list of CSFs, the generation program produces a log-file rcsfgenerate.log that mirrors the input. The latter looks like
* ! Orbital order
0 ! Selected core
1s(2,*)2s(2,*)
1s(2,*)2p(2,*)
*
4s,4p,4d,4f
0 0 ! Lower and higher 2*J
2 ! Number of excitations
n
In practical work, it is often convenient to edit the log-file and use this as input for additional runs of rcsfgenerate.
5.2. Second Example: Valence–Valence, Core–Valence for
We want to generate expansions for . In this example, the CSFs are generated by SD-excitations from to an active set with the restrictions that is closed and that there is at most one excitation from orbitals with . The expansions account for valence–valence and core–valence correlation.
*******************************************************************************
* RUN RCSFGENERATE *
* OUTPUT FILES: rcsf.out, rcsfgenerate.log *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,1)2p(6,i)3s(1,*)3p(1,*)
Give configuration 2
>>2s(2,i)2p(6,5)3s(1,*)3p(1,*)
Give configuration 3
>>2s(2,1)2p(6,i)3p(1,*)3d(1,*)
Give configuration 4
>>2s(2,i)2p(6,5)3p(1,*)3d(1,*)
Give configuration 5
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>5s,5p,5d,5f,5g
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,4
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
......
3 blocks were created
block J/P NCSF
1 0- 1912
2 1- 5210
3 2- 7122
5.3. Third Example: Valence–Valence, Core–Valence and Intercore for
We want to generate expansions for . In this example, the CSFs are generated by SD-excitations from to an active set with the restrictions that is closed (and hence inactive) and that there is at most one excitation from and , respectively. In this case, in addition to valence–valence and core–valence correlation, also intercore correlation are accounted for through configurations of the form , where is inactive. Please note how much the number of CSFs has increased.
*******************************************************************************
* RUN RCSFGENERATE *
* OUTPUT FILES: rcsf.out, rcsfgenerate.log *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,1)2p(6,5)3s(1,*)3p(1,*)
Give configuration 2
>>2s(2,1)2p(6,5)3p(1,*)3d(1,*)
Give configuration 3
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>5s,5p,5d,5f,5g
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,4
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
...........
3 blocks were created
block J/P NCSF
1 0- 10743
2 1- 29589
3 2- 41500
5.4. Fourth Example: Valence–Valence and Core–Valence and Large Multireference
We want to generate CSF expansions that describe all 92 states with symmetries of the configurations . In this example, the CSFs are generated by SD-excitations from to an active set with the restriction that there is at most one excitation from . The expansions account for valence–valence and core–valence correlation.
*******************************************************************************
* RUN RCSFGENERATE *
* OUTPUT FILES: rcsf.out, rcsfgenerate.log *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,1)2s(2,*)2p(2,*)
Give configuration 2
>>1s(2,1)2p(4,*)
Give configuration 3
>>1s(2,1)2s(2,*)2p(1,*)3p(1,*)
Give configuration 4
>>1s(2,1)2s(1,*)2p(2,*)3s(1,*)
Give configuration 5
>>1s(2,1)2s(1,*)2p(2,*)3d(1,*)
Give configuration 6
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>5s,5p,5d,5f,5g
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,10
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
......
6 blocks were created
block J/P NCSF
1 0+ 14351
2 1+ 38928
3 2+ 53645
4 3+ 56147
5 4+ 48973
6 5+ 36562
5.5. Fifth Example: CSFs Interacting with CSFs in the MR
In this example, we show how to reduce the number of CSFs in the previous list by retaining only the CSFs that interact with the CSFs of the MR through the Dirac–Coulomb or Dirac–Coulomb–Breit Hamiltonian. We start by copying
rcsf.out from the previous run to
rcsf.inp. After that, we generate the list of CSFs for the MR. For an additional example, see
Section 6.3. Please note that the orbital order needs to be the same for the MR file and the file with CSFs that should be reduced, to ensure that this is the case it is sometimes necessary to invoke the user specified orbital ordering, see
Section 6.6.
*******************************************************************************
* COPY FILE *
*******************************************************************************
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RCSFGENERATE *
* OUTPUT FILES: rcsf.out, rcsfgenerate.log *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,i)2s(2,i)2p(2,i)
Give configuration 2
>>1s(2,i)2p(4,i)
Give configuration 3
>>1s(2,i)2s(2,i)2p(1,i)3p(1,i)
Give configuration 4
>>1s(2,i)2s(1,i)2p(2,i)3s(1,i)
Give configuration 5
>>1s(2,i)2s(1,i)2p(2,i)3d(1,i)
Give configuration 6
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>3s,3p,3d
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,10
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>n
......
6 blocks were created
block J/P NCSF
1 0+ 14
2 1+ 25
3 2+ 28
4 3+ 16
5 4+ 7
6 5+ 2
*******************************************************************************
* COPY RCSF.OUT TO RCSFMR.INP *
*******************************************************************************
>>cp rcsf.out rcsfmr.inp
*******************************************************************************
* RUN RCSFINTERACT *
* INPUT FILES: rcsf.inp, rcsfmr.inp *
* OUTPUT FILE: rcsf.out *
*******************************************************************************
>>rcsfinteract
RCSFinteract: Determines all the CSFs (rcsf.inp) that interact
with the CSFs in the multireference (rcsfmr.inp)
(C) Copyright by G. Gaigalas and Ch. F. Fischer
(Fortran 95 version) NIST (2017).
Input files: rcsfmr.inp, rcsf.inp
Output file: rcsf.out
Reduction based on Dirac-Coulomb (1) or
Dirac-Coulomb-Breit (2) Hamiltonian?
>>1
.....
There are 25 relativistic subshells;
Block MR NCSF Before NCSF After NCSF
1 14 14351 7765
2 25 38928 24492
3 28 53645 33925
4 16 56147 29299
5 7 48973 17134
6 2 36562 7542
RCSFINTERACT: Execution complete
Comparing with what we had before, we see that there is a great reduction in the number of CSFs, where the removed CSFs are relatively unimportant. The reduction based on the Dirac–Coulomb–Breit Hamiltonian gives somewhat more CSFs compared to the reduction based on the Dirac–Coulomb Hamiltonian. There is, however, not a big difference.
5.6. Sixth Example: Core–Core and Doubly Occupied Orbitals
Allowing SD-excitations from all subshells of an MR without restrictions leads to large expansions. We may impose different restrictions allowing, for example, at most one excitation from the core. The resulting expansion accounts for valence–valence and core–valence electron correlation. Another restriction is to require that all correlation orbitals are doubly occupied in the generated CSFs. This cuts down the expansion size quite substantially, but still efficiently accounts for much of the correlation.
We generate a CSF expansion that describes the states with symmetries
of the configuration
. CSFs are generated by SD-excitations from
to an active set
and symmetry
with the restriction that there is at most one excitation from
. The expansion accounts for valence–valence and core–valence correlation. In addition, there are SD-excitations from
to an active set
and symmetry
with the restriction that the correlation orbitals are doubly occupied (see
Section 5.5). This part of the expansion accounts for part of the core–core correlation.
*******************************************************************************
* RUN RCSFGENERATE *
* OUTPUT FILES: rcsf.out, rcsfgenerate.log *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,i)2p(6,5)3s(1,*)3p(1,*)
Give configuration 2
>>2s(2,1)2p(6,i)3s(1,*)3p(1,*)
Give configuration 3
>>2s(2,i)2p(6,5)3p(1,*)3d(1,*)
Give configuration 4
>>2s(2,1)2p(6,i)3p(1,*)3d(1,*)
Give configuration 5
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>8s,8p,8d,8f,8g,8h
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,4
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>y
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,*)2p(6,*)3s(1,*)3p(1,*)
Give configuration 2
>>2s(2,*)2p(6,*)3p(1,*)3d(1,*)
Give configuration 3
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>8s,8p,8d,8f,8g,8h
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,4
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>-2
Generate more lists ? (y/n)
>>n
......
3 blocks were created
block J/P NCSF
1 0- 21399
2 1- 59512
3 2- 85284
5.7. Running rcsfgenerate More Than Once
We may merge CSF expansions by running rcsfgenerate more than once. In this example, we first generate a CAS expansion for to the orbital set . This is then merged by an SD expansion to a larger orbital set.
*******************************************************************************
* RUN RCSFGENERATE *
* OUTPUT FILES: rcsf.out, rcsfgenerate.log *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,*)2p(1,*)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>5s,5p,5d,5f,5g
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,3
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>3
Generate more lists ? (y/n)
>>y
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,*)2p(1,*)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>7s,7p,7d,7f,7g,7h,7i
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,3
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
.........
2 blocks were created
block J/P NCSF
1 1/2- 2408
2 3/2- 4174
As expected, we get the same number of CSFs in the two runs. Please note that the resulting J number needs to be the same when running rcsfgenerate several times for the same parity.
5.8. Running rcsfgenerate for Even and Odd Parity
We want to generate CSFs for odd states with by allowing all SDT-excitations from and for even states with by allowing all SDT-excitations from . In both cases, the excitations are to an active set with .
*******************************************************************************
* RUN RCSFGENERATE FOR ODD AND EVEN PARITY *
* OUTPUT FILES: rcsf.out, rcsfgenerate.log *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,*)2p(1,*)
Give configuration 1
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>5s,5p,5d,5f,5g
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,3
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>3
Generate more lists ? (y/n)
>>y
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,*)2s(1,*)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>5s,5p,5d,5f,5g
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,1
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>3
Generate more lists ? (y/n)
>>n
.........
3 blocks were created
block J/P NCSF
1 1/2+ 1463
1 1/2- 1454
2 3/2- 2478
5.9. User Defined Orbital Ordering
In Ce III the ground configuration is , where is to the right of the and orbitals and a user defined orbital order is needed. To illustrate the user defined orbital ordering, we generate a list of CSFs by allowing SD-excitations from to an active orbital set (or 6s,6p,5d,5f in the notation of the rcsfgenerate program).
To generate a list of CSFs where, in the configurations, is to the right of the and orbitals, start by creating a file clist.ref with the desired orbital order; one orbital per line, left justified and with a non-relativistic notation.
1s
2s
2p
3s
3p
3d
4s
4p
4d
5s
5p
4f
5d
5f
6s
6p
Then run rcsfgenerate as usual, but select the user defined orbital order.
*******************************************************************************
* RUN RCSFGENERATE USING USER DEFINED ORBITAL ORDERING *
* INPUT FILE: clist.ref *
* OUTPUT FILES: rcsf.out, rcsfgenerate.log *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>u
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>3
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>3d(10,c)4s(2,*)4p(6,*)4d(10,*)5s(2,*)5p(6,*)4f(2,*)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>6s,6p,5d,5f
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,12
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
.........
7 blocks were created
block J/P NCSF
1 0+ 26477
2 1+ 74434
3 2+ 112054
4 3+ 133012
5 4+ 137871
6 5+ 127297
7 6+ 107194
The produced output file rcsf.out looks like this
Core subshells:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Peel subshells:
4s 4p- 4p 4d- 4d 5s 5p- 5p 4f- 4f 5d- 5d 5f- 5f 6s 6p- 6p
CSF(s):
4s ( 2) 4p-( 2) 4p ( 4) 4d-( 4) 4d ( 6) 5s ( 2) 5p ( 4) 4f ( 4)
0
0+
4s ( 2) 4p-( 2) 4p ( 4) 4d-( 4) 4d ( 6) 5s ( 2) 5p-( 1) 5p ( 3) 4f ( 4)
1/2 3/2 2; 2
2 0+
4s ( 2) 4p-( 2) 4p ( 4) 4d-( 4) 4d ( 6) 5s ( 2) 5p-( 1) 5p ( 3) 4f ( 4)
1/2 3/2 4; 2
2 0+
4s ( 2) 4p-( 2) 4p ( 4) 4d-( 4) 4d ( 6) 5s ( 2) 5p-( 2) 5p ( 2) 4f ( 4)
0 0
0+
4s ( 2) 4p-( 2) 4p ( 4) 4d-( 4) 4d ( 6) 5s ( 2) 5p-( 2) 5p ( 2) 4f ( 4)
2 2; 2
0+
4s ( 2) 4p-( 2) 4p ( 4) 4d-( 4) 4d ( 6) 5s ( 2) 5p-( 2) 5p ( 2) 4f ( 4)
2 4; 2
0+
4s ( 2) 4p-( 2) 4p ( 4) 4d-( 4) 4d ( 6) 5p-( 2) 5p ( 4) 4f ( 4)
0
0+
4s ( 2) 4p-( 2) 4p ( 4) 4d-( 4) 4d ( 6) 5s ( 2) 5p ( 4) 4f-( 1) 4f ( 3)
5/2 5/2
0+
...............
Comment: when using rcsfinteract make sure that you have the same orbital order (and core) for both rcsf.inp and rcsfmr.inp. The additional quantum numbers 2; and 4; for the 4f ( 4) subshell are the seniority quantum numbers.
5.10. Running jjgen
The
jjgen program is a more flexible generation program than
rcsfgenerate. It has several useful properties, but the input is somewhat longer and more involved. The use of
jjgen is described in detail in the original write-up [
10]. Please note that after generating a CSF list with
jjgen the list needs to be put in block form by
rcsfblock.
6. Running the Application Programs
In this section we demonstrate the use of the application programs of
grasp in six cases. The use of the tools of
grasp is described in
Section 7. All data written to the output files are shown, explained and discussed in detail in
Section 8. Scripts for example 1 are found in
grasptest/example1/script, scripts for example 2 in
grasptest/example2/script, etc. Please note that the executables must be on the path! When running the application programs and the tools, the user is encouraged to look at all the output files and use the information in
Section 8 to correctly interpret the output data.
6.1. First Example: and
in Li I
The first example is for and in Li. The example shows the computation of rmcdhf and rci wave functions, and the subsequent evaluation of hyperfine structure constants, Landé -factors, and isotope shift parameters. In addition, the biorthogonal transformation is applied, and the transition rates computed from the transformed wave functions. The example also illustrates the use of jj2lsj for labeling purposes.
Define nuclear data.
Obtain common spectroscopic orbitals for the MR set.
- (a)
Generate configuration state list containing three CSFs: .
- (b)
Perform angular integration.
- (c)
Generate initial estimates of radial orbitals.
- (d)
Perform SCF calculation on the weighted average of .
- (e)
Save output to 2s_2p_DF.
Improve even states
- (a)
Generate complete active space (CAS) expansion for .
- (b)
Perform angular integration.
- (c)
Generate initial estimates of radial orbitals.
- (d)
Perform SCF calculation on .
- (e)
Save output to 2s_3.
- (f)
Perform RCI calculation in which the transverse photon interaction (Breit) and vacuum polarization and self-energy (QED) corrections are added.
Transform from - to -coupling
Improve odd states
- (a)
Generate complete active space (CAS) expansion for .
- (b)
Perform angular integration.
- (c)
Generate initial estimates of radial orbitals.
- (d)
Perform SCF calculation on the weighted average of .
- (e)
Save output to 2p_3.
- (f)
Perform RCI calculation in which the transverse photon interaction (Breit) and vacuum polarization and self-energy (QED) corrections are added.
Transform from - to -coupling
Run rlevels to view energy separations.
Calculate properties
- (a)
Calculate hyperfine structure using the rci wave functions.
- (b)
Calculate isotope shift using the rci wave functions.
- (c)
Compute the transition rates from the rci wave functions. Calculation in two steps: biorthonormal transformation and evaluation of transition matrix elements using standard Racah algebra methods.
In the test-runs, prompt marked by >> or >>3, for example, indicates that the user should input 3 and then strike the return key. When >> is followed by blanks, just strike the return key.
*******************************************************************************
* RUN RNUCLEUS TO GENERATE NUCLEAR DATA AND DEFINE RADIAL GRID *
* IOUTPUT FILE: isodata *
*******************************************************************************
>>rnucleus
Enter the atomic number:
>>3
Enter the mass number (0 if the nucleus is to be modelled as a point source:
>>7
The default root mean squared radius is 2.4440000057220459 fm; (Angeli)
the default nuclear skin thickness is 2.2999999999999998 fm;
Revise these values?
>>n
Enter the mass of the neutral atom (in amu) (0 if the nucleus is to be static):
>>6.941
Enter the nuclear spin quantum number (I) (in units of h / 2 pi):
>>1.5
Enter the nuclear dipole moment (in nuclear magnetons):
>>3.2564268
Enter the nuclear quadrupole moment (in barns):
>>-0.040
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE LIST OF CSFs FOR 2S *
* AND 2P WITH THREE CSFs: 1s(2)2s J=1/2, 1s(2)2p- J=1/2, *
* 1s(2)2p J=3/2 *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program generates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
OUTPUT FILES: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,i)2s(1,i)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>2s
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,1
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>y
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*).
Give configuration 1
>>1s(2,i)2p(1,i)
Give configuration 2
>>
Give set of active orbitals in a comma delimited list ordered by l-symmetry, e.g., 5s,4p,3d
>>1s,2p
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,3
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>n
.........
3 blocks were created
block J/P NCSF
1 1/2+ 1
2 1/2- 1
3 3/2- 1
*******************************************************************************
* COPY FILES *
* IT IS ADVISABLE TO SAVE THE rcsfgenerate.log FILE TO HAVE A *
* RECORD ON HOW THE LIST OF CSFs WAS CREATED *
*******************************************************************************
>>cp rcsfgenerate.log 2s_2p_DF.exc
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.log, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
.....
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* * MEANS ALL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp, rwfnestimate.log *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 4 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.2476D+01 0.9246D+01 0.1000D+01 0.5691D+00 332 T-F
2s 0.2895D+00 0.2308D+01 0.1000D+01 0.3010D+01 355 T-F
2p- 0.2173D+00 0.1444D-03 0.1000D+01 0.3019D+01 358 T-F
2p 0.2173D+00 0.1204D+01 0.2000D+01 0.3020D+01 358 T-F
RWFNESTIMATE: Execution complete
Comment: <r> is the mean orbital radius in a.u. (). MTP is the extension of the orbitals on the grid, for which the upper limit in the default installation is 590 points. SRC is the source of the estimate, in this case T-F (Thomas-Fermi).
*******************************************************************************
* RUN RMCDHF_MEM TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: ORBITALS BUILDING REFERENCE STATES ARE REQUIRED TO HAVE *
* THE CORRECT NUMBER OF NODES. THEY ARE REFERRED TO AS SPECTROSCOPIC *
* ORBITALS. IN THIS RUN WE VARY 1s, 2s, 2p AND THEY ARE ALL *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* * MEANS ALL ORBITALS *
*******************************************************************************
>>rmcdhf_mem
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 4 relativistic subshells;
Loading CSF File for ALL blocks
There are 3 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 3 blocks (block J/Parity NCF):
1 1/2+ 1 2 1/2- 1 3 3/2- 1
Enter ASF serial numbers for each block
Block 1 ncf = 1 id = 1/2+
>>1
Block 2 ncf = 1 id = 1/2-
>>1
Block 3 ncf = 1 id = 3/2-
>>1
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p
Enter orbitals to be varied (Updating order)
>>*
Which of these are spectroscopic orbitals?
>>*
Enter the maximum number of SCF cycles:
>>100
..........
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog, name.log *
*******************************************************************************
>>rsave 2s_2p_DF
Created 2s_2p_DF.w, 2s_2p_DF.c, 2s_2p_DF.m, 2s_2p_DF.sum, 2s_2p_DF.alog and 2s_2p_DF.log
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE n = 3 CAS LIST *
* OF CSFs FOR 2S *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfile: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,*)2s(1,*)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>3s,3p,3d
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,1
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>3
Generate more lists ? (y/n)
>>n
.........
1 blocks were created
block J/P NCSF
1 1/2+ 79
*******************************************************************************
* COPY FILES *
* IT IS ADVISABLE TO SAVE THE rcsfgenerate.log FILE TO HAVE A *
* RECORD ON HOW THE LIST OF CSFs WAS CREATED *
*******************************************************************************
>>cp rcsfgenerate.log 2s_3.exc
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.log, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
...........
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>1
Enter the file name (Null then "rwfn.out")
>>
Enter the list of relativistic subshells:
>>*
The following subshell radial wavefunctions remain to be estimated:
3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.2518D+01 0.9280D+01 0.1000D+01 0.5732D+00 355 rwf
2s 0.1963D+00 0.1452D+01 0.1000D+01 0.3873D+01 361 rwf
2p- 0.1287D+00 0.5116D-04 0.1000D+01 0.4796D+01 366 rwf
2p 0.1287D+00 0.4265D+00 0.2000D+01 0.4796D+01 366 rwf
3s 0.9128D-01 0.9783D+00 0.1000D+01 0.8483D+01 369 T-F
3p- 0.7531D-01 0.6591D-04 0.1000D+01 0.9267D+01 371 T-F
3p 0.7531D-01 0.5494D+00 0.2000D+01 0.9267D+01 371 T-F
3d- 0.6228D-01 0.3234D-05 0.2000D+01 0.9127D+01 373 T-F
3d 0.6228D-01 0.3237D-01 0.3000D+01 0.9128D+01 373 T-F
RWFNESTIMATE: Execution complete.
Comment: please note how we used the wild card
* twice. We start by reading the orbitals from a
grasp file (previous run
rwfn.out). Using the wild card
* the program reads as many orbitals as possible, i.e.,
,
,
-,
. The orbitals
,
-,
,
-,
then remain to be estimated, and we use Thomas-Fermi estimates. By again using the wild card
* all the remaining orbitals will be Thomas-Fermi estimates. Instead of Thomas-Fermi estimates, we could have used option 4, screened hydrogenic with custom
Z and adjusted the charge until the radii
<r> of the estimated orbitals overlapped the radii
<r> of the
and
spectroscopic orbitals, see
Section 6.8 for an example of the use of option 4.
*******************************************************************************
* RUN RMCDHF_MEM TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: FOR CORRELATION ORBITALS THERE ARE NO RESTRICTIONS ON THE *
* NUMBER OF NODES, I.E. THEY ARE NOT SPECTROSCOPIC. IN THIS RUN WE *
* VARY THE CORRELATION ORBITALS 3s,3p, 3d. NONE OF THESE ARE *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* 3* MEANS 3s, 3p-, 3p, 3d-, 3d *
*******************************************************************************
>>rmcdhf_mem
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-consistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Loading CSF File for ALL blocks
There are 79 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 1 blocks (block J/Parity NCF):
1 1/2+ 79
Enter ASF serial numbers for each block
Block 1 ncf = 79 id = 1/2+
>>1
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Enter orbitals to be varied (Updating order)
>>3*
Which of these are spectroscopic orbitals?
>>
Enter the maximum number of SCF cycles:
>>100
..........
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog, name.log *
*******************************************************************************
>>rsave 2s_3
Created 2s_3.w, 2s_3.c, 2s_3.m, 2s_3.sum, 2s_3.alog and 2s_3.log
*******************************************************************************
* RUN RCI TO INCLUDE TRANSVERSE PHOTON INTERACTION AND QED EFFECTS *
* INPUT FILES : isodata, 2s_3.c, 2s_3.w *
* OUTPUT FILES: 2s_3.cm, 2s_3.csum, 2s_3.clog, rci.res *
* *
* THE TRANSVERSE PHOTON FREQUENCIES CAN BE SET TO THE LOW FREQUENCY *
* LIMIT. RECOMMENDED IN CASES WHERE YOU HAVE CORRELATION ORBITALS *
* THE SELF ENERGY CORRECTION MAY FAIL FOR CORRELATION ORBITALS WITH *
* HIGH N. *
*******************************************************************************
>>rci
RCI
This is the configuration interaction program
Input file: isodata, name.c, name.w
Outputfiles: name.cm, name.csum, name.clog, rci.res
Default settings?
>>y
Name of state:
>>2s_3
Block 1 , ncf = 79
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Include contribution of H (Transverse)?
>>y
Modify all transverse photon frequencies?
>>y
Enter the scale factor:
>>1.d-6
Include H (Vacuum Polarisation)?
>>y
Include H (Normal Mass Shift)?
>>n
Include H (Specific Mass Shift)?
>>n
Estimate self-energy?
>>y
Largest n quantum number for including self-energy for orbital
n should be less or equal 8
>>3
Loading Radial WaveFunction File ...
There are 1 blocks (block J/Parity NCF):
1 1/2+ 79
Enter ASF serial numbers for each block
Block 1 ncf = 79 id = 1/2+
>>1
......
RCI: Execution complete.
*******************************************************************************
* RUN JJ2LSJ TO TRANSFORM FROM JJ- TO LSJ-COUPLING *
* INPUT FILES: 2s_3.c, 2s_3.cm *
* OUTPUT FILE: 2s_3.lsj.lbl, 2s_3.uni.lsj.lbl *
*******************************************************************************
>>jj2lsj
jj2lsj: Transformation of ASFs from a jj-coupled CSF basis
into an LSJ-coupled CSF basis (Fortran 95 version)
(C) Copyright by G. Gaigalas and Ch. F. Fischer,
(2021).
Input files: name.c, name.(c)m
Output files: name.lsj.lbl
(optional) name.lsj.c, name.lsj.j,
name.uni.lsj.lbl, name.uni.lsj.sum
Name of state
>>2s_3
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 79 relativistic CSFs;
... load complete;
Mixing coefficients from a CI calc.?
>>y
Do you need a unique labeling? (y/n)
>>y
nelec = 3
ncftot = 79
nw = 9
nblock = 1
block ncf nev 2j+1 parity
1 79 1 2 1
Default settings? (y/n)
>>y
....
jj2lsj: Execution Complete
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE n = 3 CAS LIST *
* OF CSFs FOR 2P *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,*)2p(1,*)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>3s,3p,3d
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,3
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>3
Generate more lists ? (y/n)
>>n
....
2 blocks were created
block J/P NCSF
1 1/2- 76
2 3/2- 110
*******************************************************************************
* COPY FILES *
*******************************************************************************
>>cp rcsfgenerate.log 2p_3.exc
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.log, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
....
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* WE CAN USE WILD CARDS * TO SPECIFY ORBITALS *
* * MEANS ALL ORBITALS *
* WE TAKE THE SPECTROSCOPIC ORBITALS FROM OUR DF CALCULATION *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>1
Enter the file name (Null then "rwfn.out")
>>2s_2p_DF.w
Enter the list of relativistic subshells:
>>*
The following subshell radial wavefunctions remain to be estimated:
3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.2518D+01 0.9280D+01 0.1000D+01 0.5732D+00 355 2s_
2s 0.1963D+00 0.1452D+01 0.1000D+01 0.3873D+01 361 2s_
2p- 0.1287D+00 0.5116D-04 0.1000D+01 0.4796D+01 366 2s_
2p 0.1287D+00 0.4265D+00 0.2000D+01 0.4796D+01 366 2s_
3s 0.9128D-01 0.9783D+00 0.1000D+01 0.8483D+01 369 T-F
3p- 0.7531D-01 0.6591D-04 0.1000D+01 0.9267D+01 371 T-F
3p 0.7531D-01 0.5494D+00 0.2000D+01 0.9267D+01 371 T-F
3d- 0.6228D-01 0.3234D-05 0.2000D+01 0.9127D+01 373 T-F
3d 0.6228D-01 0.3237D-01 0.3000D+01 0.9128D+01 373 T-F
RWFNESTIMATE: Execution complete.
*******************************************************************************
* RUN RMCDHF_MEM TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: FOR CORRELATION ORBITALS THERE ARE NO RESTRICTIONS ON THE *
* NUMBER OF NODES, I.E. THEY ARE NOT SPECTROSCOPIC. IN THIS RUN WE *
* VARY THE CORRELATION ORBITALS 3s,3p, 3d. NONE OF THESE ARE *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* 3* MEANS 3s, 3p-, 3p, 3d-, 3d *
*******************************************************************************
>>rmcdhf_mem
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Loading CSF File for ALL blocks
There are 186 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 2 blocks (block J/Parity NCF):
1 1/2- 76 2 3/2- 110
Enter ASF serial numbers for each block
Block 1 ncf = 76 id = 1/2-
>>1
Block 2 ncf = 110 id = 3/2-
>>1
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Enter orbitals to be varied (Updating order)
>>3*
Which of these are spectroscopic orbitals?
>>
Enter the maximum number of SCF cycles:
>>100
......
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES *
*******************************************************************************
>>rsave 2p_3
Created 2p_3.w, 2p_3.c, 2p_3.m, 2p_3.sum, 2p_3.alog and 2p_3.log
*******************************************************************************
* RUN RCI TO INCLUDE TRANSVERSE PHOTON INTERACTION AND QED EFFECTS *
* INPUT FILES : isodata, 2p_3.c, 2p_3.w *
* OUTPUT FILES: 2p_3.cm, 2p_3.csum, 2p_3.clog, rci.res *
* *
* THE TRANSVERSE PHOTON FREQUENCIES CAN BE SET TO THE LOW FREQUENCY *
* LIMIT. RECOMMENDED IN CASES WHERE YOU HAVE CORRELATION ORBITALS *
* THE SELF ENERGY CORRECTION MAY FAIL FOR CORRELATION ORBITALS WITH *
* HIGH N. *
*******************************************************************************
>>rci
RCI
This is the configuration interaction program
Input file: isodata, name.c, name.w
Outputfiles: name.cm, name.csum, name.clog, rci.res
Default settings?
>>y
Name of state:
>>2p_3
Block 1 , ncf = 76
Block 2 , ncf = 110
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Include contribution of H (Transverse)?
>>y
Modify all transverse photon frequencies?
>>y
Enter the scale factor:
>>1.d-6
Include H (Vacuum Polarisation)?
>>y
Include H (Normal Mass Shift)?
>>n
Include H (Specific Mass Shift)?
>>n
Estimate self-energy?
>>y
Largest n quantum number for including self-energy for orbital
n should be less or equal 8
>>3
Loading Radial WaveFunction File ...
There are 2 blocks (block J/Parity NCF):
1 1/2- 76 2 3/2- 110
Enter ASF serial numbers for each block
Block 1 ncf = 76 id = 1/2-
>>1
Block 2 ncf = 110 id = 3/2-
>>1
....
RCI: Execution complete.
*******************************************************************************
* RUN JJ2LSJ TO TRANSFORM FROM JJ- TO LSJ-COUPLING *
* INPUT FILES: 2p_3.c, 2p_3.cm *
* OUTPUT FILE: 2p_3.lsj.lbl, 2p_3.uni.lsj.lbl *
*******************************************************************************
>>jj2lsj
jj2lsj: Transformation of ASFs from a jj-coupled CSF basis
into an LSJ-coupled CSF basis (Fortran 95 version)
(C) Copyright by G. Gaigalas and Ch. F. Fischer,
(2021).
Input files: name.c, name.(c)m
Output files: name.lsj.lbl
(optional) name.lsj.c, name.lsj.j,
name.uni.lsj.lbl, name.uni.lsj.sum
Name of state
>>2p_3
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 186 relativistic CSFs;
... load complete;
Mixing coefficients from a CI calc.?
>>y
Do you need a unique labeling? (y/n)
>>y
nelec = 3
ncftot = 186
nw = 9
nblock = 2
block ncf nev 2j+1 parity
1 76 1 2 -1
2 110 1 4 -1
Default settings? (y/n)
>>y
...
jj2lsj: Execution Complete
*******************************************************************************
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS. *
* IF DESIRED WE CAN INSTEAD RUN RLEVELSEV TO GET THE SEPARATION IN EV *
*******************************************************************************
>> rlevels 2s_3.cm 2p_3.cm
nblock = 1 ncftot = 79 nw = 9 nelec = 3
nblock = 2 ncftot = 186 nw = 9 nelec = 3
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbour
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 1/2 + -7.4719740 0.00 0.00 1s(2).2s_2S
2 1 1/2 - -7.4042610 14861.28 14861.28 1s(2).2p_2P
3 1 3/2 - -7.4042597 14861.57 0.29 1s(2).2p_2P
------------------------------------------------------------------------------------------
*******************************************************************************
* RUN RHFS FOR 2s_3 *
* INPUT FILES: isodata, 2s_3.c, 2s_3.w, 2s_3.cm *
* OUTPUT FILE: 2s_3.ch, 2s_3.choffd *
*******************************************************************************
>>rhfs
RHFS
This is the hyperfine structure program
Input files: isodata, name.c, name.(c)m, name.w
Output files: name.(c)h, name.(c)hoffd
Default settings?
>>y
Name of state
>>2s_3
Mixing coefficients from a CI calc.?
>>y
....
RHFS: Execution complete.
*******************************************************************************
* VIEW DIAGONAL HFS CONSTANTS AND GJ FACTORS *
* OUTPUT SLIGHTLY EDITED TO DISPLAY ONLY THE TOTAL GJ *
*******************************************************************************
>> more 2s_3.ch
Nuclear spin 1.500000000000000D+00 au
Nuclear magnetic dipole moment 3.256426800000000D+00 n.m.
Nuclear electric quadrupole moment -4.000000000000000D-02 barns
Interaction constants:
Level1 J Parity A (MHz) B (MHz) total g_J
1 1/2 + 3.8844184122D+02 -0.0000000000D+00 2.0023047262D+00
*******************************************************************************
* RUN RHFS FOR 2p_3 *
* INPUT FILES: isodata, 2p_3.c, 2p_3.w, 2p_3.cm *
* OUTPUT FILE: 2p_3.ch, 2p_3.choffd *
*******************************************************************************
>>rhfs
RHFS
This is the hyperfine structure program
Input files: isodata, name.c, name.(c)m, name.w
Output files: name.(c)h, name.(c)hoffd
Default settings?
>>y
Name of state
>>2p_3
Mixing coefficients from a CI calc.?
>>y
.....
RHFS: Execution complete.
*******************************************************************************
* VIEW DIAGONAL HFS CONSTANTS AND GJ FACTORS *
* OUTPUT SLIGHTLY EDITED TO DISPLAY ONLY THE TOTAL GJ *
*******************************************************************************
>> more 2p_3.ch
Nuclear spin 1.500000000000000D+00 au
Nuclear magnetic dipole moment 3.256426800000000D+00 n.m.
Nuclear electric quadrupole moment -4.000000000000000D-02 barns
Interaction constants:
Level1 J Parity A (MHz) B (MHz) total g_J
1 1/2 - 4.4821853986D+01 -0.0000000000D+00 6.6588395646D-01
1 3/2 - -3.5378452915D+00 -1.7729096327D-01 1.3340987050D+00
Please note that
rhfs computes both diagonal and off-diagonal hyperfine interaction constants. The latter are available in the
name.choffd file. The off-diagonal parameters are sometimes available from experiment. For Li I, the
interaction constant is for example measured from level-crossing spectroscopy [
38]. For systems with small fine-structure separations, the off-diagonal hyperfine parameters are of crucial importance in order to model the observed hyperfine line profiles [
39]. For systems with large fine structure separations, the off-diagonal hyperfine constants may be neglected.
*******************************************************************************
* RUN RIS4 FOR 2s_3 *
* INPUT FILES: isodata, 2s_3.c, 2s_3.w, 2s_3.cm *
* OUTPUT FILES: 2s_3.ci *
* 2s_3.IOB, 2s_3.ITB (angular files) *
*******************************************************************************
>>ris4
RIS: Execution begins ...
Default settings?
>>y
Name of state
>>2s_3
Mixing coefficients from a CI calc.?
>>y
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 79 relativistic CSFs;
... load complete;
Loading Radial WaveFunction File ...
nelec = 3
ncftot = 79
nw = 9
nblock = 1
block ncf nev 2j+1 parity
1 79 1 2 1
-------------------------------
RIS_CAL: Execution Begins ...
-------------------------------
NRNUC: 91
Compute higher order field shift electronic factors?
>>y
One-body angular file not available
Two-body angular file not available
Save ang. coefficients of one- and two-body op.?
>>y
.....
RIS: Execution complete.
*******************************************************************************
* VIEW SPECIFIC MASS SHIFT AND FIELD SHIFT PARAMETERS *
* OUTPUT EDITED TO FIT THE PAGE *
*******************************************************************************
>> more 2s_3.ci
Number of eigenvalues: 1
Level J Parity Energy
1 1/2 + -0.7471973983D+01 (a.u.)
Level J Parity Normal mass shift parameter
<K^1> <K^2+K^3> <K^1+K^2+K^3>
1 1/2 + 0.7475765524D+01 -0.6760181109D-02 0.7469005343D+01 (a.u.)
0.2698364414D+05 -0.2440075478D+02 0.2695924338D+05 (GHz u)
Level J Parity Specific mass shift parameter
<K^1> <K^2+K^3> <K^1+K^2+K^3>
1 1/2 + 0.3072684862D+00 -0.2114198685D-03 0.3070570663D+00 (a.u.)
0.1109080195D+04 -0.7631162959D+00 0.1108317079D+04 (GHz u)
Level J Parity Electron density in atomic units
Dens. (a.u.)
1 1/2 + 0.1388454525D+02
Level J Parity Field shift electronic factors and average point discrepancy in fit
F0 (GHz/fm^2) F2 (GHz/fm^4) F4 (GHz/fm^6)
1 1/2 + 0.2049813242D+00 -0.3342886617D-05 0.5289532830D-07
F6 (GHz/fm^8) Disc. (per mille)
1 1/2 + -0.7068539282D-09 0.0000
Level J Parity Field shift electronic factors (corrected for varying density inside nucleus)
F0VED0 (GHz/fm^2) F0VED1 (GHz/fm^4)
1 1/2 + 0.2049364945D+00 -0.2839055991D-05
The normal and specific mass shift parameters are those of the three terms defined in TP Section 3.3, Equations (73) and (74). The field shift electronic factors are the ones defined in TP Section 3.3, Equation (79). and are the parameters defined in TP Section 3.3, Equation (83).
*******************************************************************************
* RUN RIS4 FOR 2p_3 *
* INPUT FILES: isodata, 2p_3.c, 2p_3.w, 2p_3.cm *
* OUTPUT FILES: 2p_3.ci *
* 2p_3.IOB, 2p_3.ITB (angular files) *
*******************************************************************************
>>ris4
RIS: Execution begins ...
Default settings?
>>y
Name of state
>>2p_3
Mixing coefficients from a CI calc.?
>>y
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 186 relativistic CSFs;
... load complete;
Loading Radial WaveFunction File ...
nelec = 3
ncftot = 186
nw = 9
nblock = 2
block ncf nev 2j+1 parity
1 76 1 2 -1
2 110 1 4 -1
-------------------------------
RIS_CAL: Execution Begins ...
-------------------------------
NRNUC: 91
Compute higher order field shift electronic factors?
>>y
One-body angular file not available
Two-body angular file not available
Save ang. coefficients of one- and two-body op.?
>>y
Column 100 complete;
Column 100 complete;
....
RIS: Execution complete.
*******************************************************************************
* VIEW SPECIFIC MASS SHIFT AND FIELD SHIFT PARAMETERS *
* OUTPUT EDITED TO FIT THE PAGE *
*******************************************************************************
>> more 2p_3.ci
Number of eigenvalues: 2
Level J Parity Energy
1 1/2 - -0.7404260995D+01 (a.u.)
1 3/2 - -0.7404259683D+01 (a.u.)
Level J Parity Normal mass shift parameter
<K^1> <K^2+K^3> <K^1+K^2+K^3>
1 1/2 - 0.7409611828D+01 -0.6671237484D-02 0.7402940590D+01 (a.u.)
0.2674486353D+05 -0.2407971433D+02 0.2672078382D+05 (GHz u)
<K^1> <K^2+K^3> <K^1+K^2+K^3>
1 3/2 - 0.7409602908D+01 -0.6657064450D-02 0.7402945843D+01 (a.u.)
0.2674483134D+05 -0.2402855701D+02 0.2672080278D+05 (GHz u)
Level J Parity Specific mass shift parameter
<K^1> <K^2+K^3> <K^1+K^2+K^3>
1 1/2 - 0.2425644688D+00 -0.1746264308D-03 0.2423898424D+00 (a.u.)
0.8755321826D+03 -0.6303110296D+00 0.8749018716D+03 (GHz u)
<K^1> <K^2+K^3> <K^1+K^2+K^3>
1 3/2 - 0.2425741100D+00 -0.1915018511D-03 0.2423826081D+00 (a.u.)
0.8755669823D+03 -0.6912225626D+00 0.8748757597D+03 (GHz u)
Level J Parity Electron density in atomic units
Dens. (a.u.)
1 1/2 - 0.1372240739D+02
1 3/2 - 0.1372240990D+02
Level J Parity Field shift electronic factors and average point discrepancy in fit
F0 (GHz/fm^2) F2 (GHz/fm^4) F4 (GHz/fm^6)
1 1/2 - 0.2025876387D+00 -0.3303847114D-05 0.5227748000D-07
1 3/2 - 0.2025876757D+00 -0.3303847831D-05 0.5227749057D-07
F6 (GHz/fm^8) Disc. (per mille)
1 1/2 - -0.6985943239D-09 0.0000
1 3/2 - -0.6985944586D-09 0.0000
Level J Parity Field shift electronic factors (corrected for varying density inside nucleus)
F0VED0 (GHz/fm^2) F0VED1 (GHz/fm^4)
1 1/2 - 0.2025433326D+00 -0.2805899138D-05
1 3/2 - 0.2025433696D+00 -0.2805899756D-05
Comment: Given the information in
2s_3.ci and
2p_3.ci together with isotopic data, the frequency isotope shift can be computed using the
fical program, see
Section 12.2.
*******************************************************************************
* RUN RBIOTRANSFORM FOR 2s_3 AND 2p_3 TO TRANSFORM WAVE FUNCTIONS *
* INPUT FILES: isodata, 2s_3.c, 2s_3.w, 2s_3.cm, *
* 2p_3.c, 2p_3.w, 2p_3.cm *
* OUTPUT FILES: 2s_3.cbm, 2s_3.bw, 2p_3.cbm, 2p_3.bw *
* 2s_3.TB, 2p_3.TB (angular files) *
* NOTE THAT THE ORDER OF INITIAL AND FINAL STATE DOES NOT MATTER *
*******************************************************************************
>>rbiotransform
RBIOTRANSFORM
This program transforms the initial and final wave
functions so that standard tensor albegra can be
used in evaluation of the transition parameters
Input files: isodata, name1.c, name1.w, name1.(c)m
name2.c, name2.w, name2.(c)m
name1.TB, name2.TB (optional angular files)
Output files: name1.bw, name1.(c)bm,
name2.bw, name2.(c)bm
name1.TB, name2.TB (angular files)
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>2s_3
Name of the Final state
>>2p_3
Transformation of all J symmetries?
>>y
....
BIOTRANSFORM: Execution complete.
*******************************************************************************
* RUN RTRANSITION FOR 2s_3 and 2p_3 TO COMPUTE TRANSITION PARAMETERS *
* INPUT FILES: isodata, 2s_3.c, 2s_3.bw, 2s_3.cbm *
* 2p_3.c, 2p_3.bw, 2p_3.cbm *
* OUTPUT FILES: 2s_3.2p_3.ct *
* 2s_3.2p_3.-1T (angular file) *
* NOTE THAT THE ORDER OF INITIAL AND FINAL STATE DOES NOT MATTER *
*******************************************************************************
>>rtransition
RTRANSITION
This program computes transition parameters from
transformed wave functions
Input files: isodata, name1.c, name1.bw, name1.(c)bm
name2.c, name2.bw, name2.(c)bm
optional, name1.lsj.lbl, name2.lsj.lbl
name1.name2.KT (optional angular files)
Output files: name1.name2.(c)t
optional, name1.name2.(c)t.lsj
name1.name2.KT (angular files)
Here K is parity and rank of transition: -1,+1 etc
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>2s_3
Name of the Final state
>>2p_3
MRGCSL: Execution begins ...
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 79 relativistic CSFs;
... load complete;
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 186 relativistic CSFs;
... load complete;
1 s
2 s
2 p-
2 p
3 s
3 p-
3 p
3 d-
3 d
1
79
2
76 186
Loading Configuration Symmetry List File ...
there are 9 relativistic subshells;
there are 265 relativistic CSFs;
... load complete;
Enter the list of transition specifications
e.g., E1,M2 or E1 M2 or E1;M2 :
>>E1
.....
RTRANSITION: Execution complete.
*******************************************************************************
* VIEW COMPUTED TRANSITION PARAMETERS *
*******************************************************************************
>>more 2s_3.2p_3.ct
Transition between files:
f1 = 2s_3
f2 = 2p_3
Electric 2**( 1)-pole transitions
=================================
Upper Lower
Lev J P Lev J P E (Kays) A (s-1) gf S
f2 1 1/2 - f1 1 1/2 + 14861.28 C 3.81311D+07 5.17671D-01 1.14676D+01
B 3.74756D+07 5.08773D-01 1.12705D+01
f2 1 3/2 - f1 1 1/2 + 14861.57 C 3.81334D+07 1.03537D+00 2.29353D+01
B 3.74782D+07 1.01758D+00 2.25413D+01
*******************************************************************************
* VIEW COMPUTED TRANSITION PARAMETERS IN LSJ COUPLING *
*******************************************************************************
>>more 2s_3.2p_3.ct.lsj
Transition between files:
2s_3
2p_3
1 -7.47197398 1s(2).2s_2S
1 -7.40426099 1s(2).2p_2P
14861.28 CM-1 6728.89 ANGS(VAC) 6728.20 ANGS(AIR)
E1 S = 1.12705D+01 GF = 5.08773D-01 AKI = 3.74756D+07 dT = 0.01719
1.14676D+01 5.17671D-01 3.81311D+07
1 -7.47197398 1s(2).2s_2S
3 -7.40425968 1s(2).2p_2P
14861.57 CM-1 6728.76 ANGS(VAC) 6728.06 ANGS(AIR)
E1 S = 2.25413D+01 GF = 1.01758D+00 AKI = 3.74782D+07 dT = 0.01718
2.29353D+01 1.03537D+00 3.81334D+07
Comment: the values in Babushkin gauge are now shown on the first line. In addition, the uncertainty parameter
is given, see TP Section 3.5.
6.2. Second Example: for B II in Different Coupling Schemes – HF Initial Estimates
The second example is for B II in different coupling schemes and aims to illustrate the use of the Coupling program. In this example, we also illustrate how we can use converted HF wave function as starting estimates for the radial orbitals.
Define nuclear data
Obtain common spectroscopic orbitals for the MR set
- (a)
Generate configuration list containing 4 CSFs belonging to
- (b)
Perform angular integration
- (c)
Perform HF calculation
- (d)
Convert HF orbitals to relativistic orbitals. We do not need to run rwfnestimate since all orbitals have been estimated
- (e)
Perform SCF calculation on the weighted average on the state belonging to
- (f)
Save output to 2s2p_DF
Transform from - to -coupling
Run rlevels to view energy separations.
Run jj2lsj, Coupling, and rlevels to define energy spectra in different coupling scheme.
In the test-runs, prompt marked by >> or >>3, for example, indicates that the user should input 3 and then strike the return key. When >> is followed by blanks, just strike the return key.
*******************************************************************************
* RUN RNUCLEUS TO GENERATE NUCLEAR DATA AND DEFINE RADIAL GRID *
* OUTPUT FILE: isodata *
*******************************************************************************
>>rnucleus
RNUCLEUS
This program defines nuclear data and the radial grid
Outputfile: isodata
Enter the atomic number:
>>5
Enter the mass number (0 if the nucleus is to be modelled as a point source:
>>11
The default root mean squared radius is 2.4059998989105225 fm; (Angeli)
the default nuclear skin thickness is 2.2999999999999998 fm;
Revise these values?
>>n
Enter the mass of the neutral atom (in amu) (0 if the nucleus is to be static):
>>10.81
Enter the nuclear spin quantum number (I) (in units of h / 2 pi):
>>1.5
Enter the nuclear dipole moment (in nuclear magnetons):
>>2.6886489
Enter the nuclear quadrupole moment (in barns):
>>1
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE LIST FOR *
* 1P_1 AND 3P_0,1,2 WITH FOUR CSFs: 2s2p- J=0, 2s2p- J=1, *
* 2s2p J=1, 2s2p J = 2 *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,i)2s(1,i)2p(1,i)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>2s,2p
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,4
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>n
.........
3 blocks were created
block J/P NCSF
1 0- 1
2 1- 2
3 2- 1
*******************************************************************************
* COPY FILES *
* IT IS ADVISABLE TO SAVE THE rcsfgenerate.log FILE TO HAVE A *
* RECORD ON HOW THE LIST OF CSFs WAS CREATED *
*******************************************************************************
>>cp rcsfgenerate.log 2s2p_DF.exc
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.alog, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
....
RANGULAR: Execution complete.
*******************************************************************************
* RUN HF PROGRAM TO GENERATE NON-RELATIVISTIC RADIAL ORBITALS *
* THAT CAN BE CONVERTED TO RELATIVISTIC ORBITALS *
* OUTPUT FILE: wfn.out *
*******************************************************************************
>>hf
=============================
H A R T R E E - F O C K . 96
=============================
THE DIMENSIONS FOR THE CURRENT VERSION ARE:
NWF= 20 NO=220
START OF CASE
=============
Enter ATOM,TERM,Z
Examples: O,3P,8. or Oxygen,AV,8.
>>B,AV,5.
List the CLOSED shells in the fields indicated (blank line if none)
... ... ... ... ... ... ... ... etc.
>> 1s (Please note that the closed shells should be entered right-justified with
respect to the dots on the line above!!!)
Enter electrons outside CLOSED shells (blank line if none)
Example: 2s(1)2p(3)
>>2s(1)2p(1)
There are 3 orbitals as follows:
1s 2s 2p
Orbitals to be varied: ALL/NONE/=i (last i)/comma delimited list/H
>>all
Default electron parameters ? (Y/N/H)
>>y
Default values for remaining parameters? (Y/N/H)
>>y
WEAK ORTHOGONALIZATION DURING THE SCF CYCLE= T
SCF CONVERGENCE TOLERANCE (FUNCTIONS) = 1.00D-08
NUMBER OF POINTS IN THE MAXIMUM RANGE = 220
ITERATION NUMBER 1
----------------
................
ITERATION NUMBER 6
----------------
SCF CONVERGENCE CRITERIA (SCFTOL*SQRT(Z*NWF)) = 1.2D-06
C( 1s 2s) = 0.00000 V( 1s 2s) = -7.06535 EPS = 0.000000
E( 2s 1s) = 0.02654 E( 1s 2s) = 0.01327
EL ED AZ NORM DPM
1s 16.3418222 20.8332819 1.0000000 1.93D-08
2s 1.8579695 4.7336947 1.0000000 1.38D-08
2p 1.4015370 4.0799511 1.0000000 1.74D-08
< 1s| 2s>= 8.0D-09
TOTAL ENERGY (a.u.)
----- ------
Non-Relativistic -24.06678870 Kinetic 24.06678852
Relativistic Shift -0.00587815 Potential -48.13357722
Relativistic -24.07266685 Ratio -2.000000008
Additional parameters ? (Y/N/H)
>>n
Do you wish to continue along the sequence ?
>>n
END OF CASE
===========
*******************************************************************************
* COPY FILES *
*******************************************************************************
>>cp wfn.out wfn.inp
*******************************************************************************
* RUN RWFNMCHFMCDF TO CONVERT NON-RELATIVISTIC RADIAL ORBITALS TO *
* RELATIVISTIC ONES *
* INPUT FILE: wfn.inp *
* OUTPUT FILE: rwfn.out *
*******************************************************************************
>>rwfnmchmcdf
RWFNMCHFMCDF
This program converts non-relativistic radial
orbitals to relativistic ones in GRASP format
Input file: wfn.inp
Output file: rwfn.out
*******************************************************************************
* COPY FILES *
* WE DONT NEED TO INVOKE RWFNESTIMATE SINCE ALL ORBITALS HAVE *
* BEEN ESTIMATED THROUGH THE MCHF MCDF CONVERSION *
*******************************************************************************
>>cp rwfn.out rwfn.inp
*******************************************************************************
* RUN RMCDHF_MEM TO OBTAIN SELF CONSISTENT SOLUTIONS. *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: ORBITALS BUILDING REFERENCE STATES ARE REQUIRED TO HAVE *
* THE CORRECT NUMBER OF NODES. THEY ARE REFERRED TO AS SPECTROSCOPIC *
* ORBITALS. IN THIS RUN WE VARY 1s, 2s, 2p AND THEY ARE ALL *
* SPECTROSCOPIC. WE CAN USE WILD CARDS FOR SPECIFYING ORBITALS *
*******************************************************************************
>>rmcdhf_mem
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 4 relativistic subshells;
Loading CSF File for ALL blocks
There are 4 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 3 blocks (block J/Parity NCF):
1 0- 1 2 1- 2 3 2- 1
Enter ASF serial numbers for each block
Block 1 ncf = 1 id = 0-
>>1
Block 2 ncf = 2 id = 1-
>>1,2
Block 3 ncf = 1 id = 2-
>>1
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p
Enter orbitals to be varied (Updating order)
>>*
Which of these are spectroscopic orbitals?
>>*
Enter the maximum number of SCF cycles:
>>100
.....
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES *
*******************************************************************************
>>rsave 2s2p_DF
Created 2s2p_DF.w, 2s2p_DF.c, 2s2p_DF.m, 2s2p_DF.sum, 2s2p_DF.alog and 2s2p_DF.log
*******************************************************************************
* RUN JJ2LSJ TO GET THE LSJ-COMPOSITION *
* INPUT FILE: 2s2p_DF.c, 2s2p_DF.m *
* OUTPUT FILE: 2s2p_DF.lsj.lbl, 2s2p_DF.uni.lsj.lbl *
*******************************************************************************
>>jj2lsj
jj2lsj: Transformation of ASFs from a jj-coupled CSF basis
into an LSJ-coupled CSF basis (Fortran 95 version)
(C) Copyright by G. Gaigalas and Ch. F. Fischer,
(2021).
Input files: name.c, name.(c)m
Output files: name.lsj.lbl
(optional) name.lsj.c, name.lsj.j,
name.uni.lsj.lbl, name.uni.lsj.sum
Name of state
>>2s2p_DF
Loading Configuration Symmetry List File ...
There are 4 relativistic subshells;
There are 4 relativistic CSFs;
... load complete;
Mixing coefficients from a CI calc.?
>>n
Do you need a unique labeling? (y/n)
>>y
nelec = 4
ncftot = 4
nw = 4
nblock = 3
block ncf nev 2j+1 parity
1 1 1 1 -1
2 2 2 3 -1
3 1 1 5 -1
Default settings? (y/n)
>>y
....
jj2lsj: Execution complete.
*******************************************************************************
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* NOTE: SINCE LSJ-INFORMATION NOW IS AVAILABLE OUTPUT LABELS *
* WILL BE IN LSJ-COUPLING *
* IF DESIRED WE CAN INSTEAD RUN RLEVELSEV TO GET THE SEPARATION IN EV *
*******************************************************************************
>> rlevels 2s2p_DF.m
nblock = 3 ncftot = 4 nw = 4 nelec = 4
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 0 - -24.1270878 0.00 0.00 1s(2).2s_2S.2p_3P
2 1 1 - -24.1270404 10.39 10.39 1s(2).2s_2S.2p_3P
3 1 2 - -24.1269457 31.17 20.79 1s(2).2s_2S.2p_3P
4 2 1 - -23.9154061 46458.75 46427.58 1s(2).2s_2S.2p_1P
------------------------------------------------------------------------------------------
For interpretation of
-coupling notation produced by
jj2lsj see
Section 8.2, where we discuss in detail the transformation from
- to
-coupling for the
and
configurations in Si VIII.
*******************************************************************************
* RUN JJ2LSJ TO GET THE INPUT FOR COUPLING PROGRAM *
* INPUT FILES: 2s2p_DF.c, 2s2p_DF.m *
* OUPUT FILES: 2s2p_DF.lsj.c, 2s2p_DF.lsj.j, 2s2p_DF.lsj.lbl *
*******************************************************************************
>>jj2lsj
jj2lsj: Transformation of ASFs from a jj-coupled CSF basis
into an LS-coupled CSF basis (Fortran 95 version)
(C) Copyright by G. Gaigalas and Ch. F. Fischer,
(2021).
Input files: name.c, name.(c)m
Ouput files: name.lsj.lbl,
(optional) name.lsj.c, name.lsj.j,
name.uni.lsj.lbl, name.uni.lsj.sum
Name of state
>>2s2p_DF
Loading Configuration Symmetry List File ...
There are 4 relativistic subshells;
There are 4 relativistic CSFs;
... load complete;
Mixing coefficients from a CI calc.?
>>n
Do you need a unique labeling? (y/n)
>>n
nelec = 4
ncftot = 4
nw = 4
nblock = 3
block ncf nev 2j+1 parity
1 1 1 1 -1
2 2 2 3 -1
3 1 1 5 -1
Default settings? (y/n)
>>n
All levels (Y/N)
>>y
Maximum % of omitted composition
>>0
What is the value below which an eigenvector composition
is to be neglected for printing?
>>0.01
jj2lsj: Execution complete.
*******************************************************************************
* RUN COUPLING TO GET THE IDENTIFICATION STATES IN DIFFERENT *
* COUPLING SCHEMES *
* INPUT FILES: 2s2p_DF.lsj.c, 2s2p_DF.lsj.j *
* OUPUT FILES: 2s2p_DF.coup3.LK3.lbl, 2s2p_DF.coup3.JK3.lbl *
* 2s2p_DF.coup3.LS.lbl, 2s2p_DF.coup3.LS3.lbl *
* 2s2p_DF.coup3.LSJ3.lbl, 2s2p_DF.coup3.jj.lbl *
* 2s2p_DF.coup3.cLSJ3.lbl, 2s2p_DF.coup3.sum *
*******************************************************************************
>>Coupling
Coupling: Transformation of ASFs from a LS-coupled CSF basis
into differete coupled CSF bases (Fortran 95)
(C) (2022) G. Gaigalas, A. Kramida.
Input files: *.lsj.c, *.lsj.j (ATSP (CPC) or GRASP2K types)
Output files: *.coup*.*.lbl, *.coup*.sum
Name of state
>>2s2p_DF
Default settings ? (Y/N)
>>y
Specify the number of coupled shells for evaluation (1,2 or 3):
>>3
3
What is the value below which an eigenvector composition
is to be neglected for printing?
>>0
0.0000000000000000
Specify shells for recoupling (no more than 12)
>>1s,2s,2p
All transformations completed
There is one-to-one classification for LS coupling
There is one-to-one classification for LS3 coupling
There is one-to-one classification for LSJ3 coupling
There is one-to-one classification for LK3 coupling
There is one-to-one classification for JK3 coupling
There is one-to-one classification for cLSJ3 coupling
There is one-to-one classification for jj3 coupling
end subroutine generate_classification_data
Coupling: Execution complete.
*******************************************************************************
* COPY 2s2p_DF.coup3.LK3.lbl TO 2s2p_DF.lsj.lbl. *
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* IN LK3-COUPLING. COMMENT: RLEVELS TAKES <name.lsj.lbl> *
* FOR THIS REASON WE COPY <name.coup3.LK3.lbl> TO <name.lsj.lbl> *
*******************************************************************************
>>cp 2s2p_DF.coup3.LK3.lbl 2s2p_DF.lsj.lbl
>>rlevels 2s2p_DF.m
nblock = 3 ncftot = 4 nw = 4 nelec = 4
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 0 - -24.1270878 0.00 0.00 1s2_ 2s_2p_(3P) P_3[1]<0>
2 1 1 - -24.1270404 10.39 10.39 1s2_ 2s_2p_(3P) P_3[1]<1>
3 1 2 - -24.1269457 31.17 20.79 1s2_ 2s_2p_(3P) P_3[1]<2>
4 2 1 - -23.9154061 46458.75 46427.58 1s2_ 2s_2p_(1P) P_1[1]<1>
------------------------------------------------------------------------------------------
*******************************************************************************
* COPY 2s2p_DF.coup3.JK3.lbl TO 2s2p_DF.lsj.lbl. *
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* IN JK3-COUPLING *
*******************************************************************************
>>cp 2s2p_DF.coup3.JK3.lbl 2s2p_DF.lsj.lbl
>>rlevels 2s2p_DF.m
nblock = 3 ncftot = 4 nw = 4 nelec = 4
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
---------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
---------------------------------------------------------------------------------------------
1 1 0 - -24.1270877 0.00 0.00 1s2_<0>2s_2p_(3P) 3[1]<0>
2 1 1 - -24.1270404 10.39 10.39 1s2_<0>2s_2p_(3P) 3[1]<1>
3 1 2 - -24.1269457 31.17 20.79 1s2_<0>2s_2p_(3P) 3[1]<2>
4 2 1 - -23.9154061 46458.75 46427.58 1s2_<0>2s_2p_(1P) 1[1]<1>
---------------------------------------------------------------------------------------------
*******************************************************************************
* COPY 2s2p_DF.coup3.LS3.lbl TO 2s2p_DF.lsj.lbl. *
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* IN LS3-COUPLING *
*******************************************************************************
>>cp 2s2p_DF.coup3.LS3.lbl 2s2p_DF.lsj.lbl
>>rlevels 2s2p_DF.m
nblock = 3 ncftot = 4 nw = 4 nelec = 4
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
---------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 0 - -24.1270877 0.00 0.00 1s2_ 2s_2p_(3P) 3P<0>
2 1 1 - -24.1270404 10.39 10.39 1s2_ 2s_2p_(3P) 3P<1>
3 1 2 - -24.1269457 31.17 20.79 1s2_ 2s_2p_(3P) 3P<2>
4 2 1 - -23.9154061 46458.75 46427.58 1s2_ 2s_2p_(1P) 1P<1>
---------------------------------------------------------------------------------------------
*******************************************************************************
* COPY 2s2p_DF.coup3.LSJ3.lbl TO 2s2p_DF.lsj.lbl. *
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* IN LSJ3-COUPLING *
*******************************************************************************
>>cp 2s2p_DF.coup3.LSJ3.lbl: 2s2p_DF.lsj.lbl
>>rlevels 2s2p_DF.m
nblock = 3 ncftot = 4 nw = 4 nelec = 4
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 0 - -24.1270877 0.00 0.00 1s2_ 2s_2p_(3P) (0,0)<0>
2 1 1 - -24.1270404 10.39 10.39 1s2_ 2s_2p_(3P) (0,1)<1>
3 1 2 - -24.1269457 31.17 20.79 1s2_ 2s_2p_(3P) (0,2)<2>
4 2 1 - -23.9154061 46458.75 46427.58 1s2_ 2s_2p_(1P) (0,1)<1>
------------------------------------------------------------------------------------------
*******************************************************************************
* COPY 2s2p_DF.coup3.jj.lbl TO 2s2p_DF.lsj.lbl. *
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* IN jj-COUPLING *
*******************************************************************************
>>cp 2s2p_DF.coup3.jj.lbl 2s2p_DF.lsj.lbl
>>rlevels 2s2p_DF.m
nblock = 3 ncftot = 4 nw = 4 nelec = 4
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 0 - -24.1270877 0.00 0.00 1s+2_2s+_<1/2>.2p-_(1/2) <0>
2 1 1 - -24.1270404 10.39 10.39 1s+2_2s+_<1/2>.2p-_(1/2) <1>
3 1 2 - -24.1269457 31.17 20.79 1s+2_2s+_<1/2>.2p+_ <2>
4 2 1 - -23.9154061 46458.75 46427.58 1s+2_2s+_<1/2>.2p+_(3/2) <1>
------------------------------------------------------------------------------------------
*******************************************************************************
* COPY 2s2p_DF.coup3.cLSJ3.lbl TO 2s2p_DF.lsj.lbl. *
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* IN cLSJ3-COUPLING *
*******************************************************************************
>>cp 2s2p_DF.coup3.cLSJ3.lbl 2s2p_DF.lsj.lbl
>>rlevels 2s2p_DF.m
nblock = 3 ncftot = 4 nw = 4 nelec = 4
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
----------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
----------------------------------------------------------------------------------------------
1 1 0 - -24.1270877 0.00 0.00 1s+2_ (0,0)<0> 2s_2p_(3P)<0> (0,0)<0>
2 1 1 - -24.1270404 10.39 10.39 1s+2_ (0,0)<0> 2s_2p_(3P)<1> (0,1)<1>
3 1 2 - -24.1269457 31.17 20.79 1s+2_ (0,0)<0> 2s_2p_(3P)<2> (0,2)<2>
4 2 1 - -23.9154061 46458.75 46427.58 1s+2_ (0,0)<0> 2s_2p_(1P)<1> (0,1)<1>
----------------------------------------------------------------------------------------------
For definition of different coupling schemes see in [
14] and for interpretation of different coupling schemes notation produced by
Coupling see
Section 8.2.
6.3. Third Example: and
for Si VIII in Different Coupling Schemes–Condensing the CSF List
The third example is and in Si VIII, where we compute M1 transition rates and give the transition data in different coupling schemes. This example also illustrates the use of the rcsfinteract program to reduce the expansion sizes by retaining only the CSFs that interact with the CSFs in the MR.
Define nuclear data
Obtain common spectroscopic orbitals for the MR set
- (a)
Generate configuration list belonging to and
- (b)
Perform angular integration
- (c)
Generate initial estimates of radial orbitals
- (d)
Perform SCF calculation on the weighted average of all states belonging to and (there are two states with , four states with and one state with , see NIST Tables)
- (e)
Save output to 2s22p3_2p5_DF
Improve states
- (a)
Generate CSF list from SD-excitations from and to
- (b)
Run rcsfinteract to extract CSFs that interact with CSFs belonging to or
- (c)
Perform angular integration
- (d)
Generate initial estimates of radial orbitals
- (e)
Perform SCF calculation on the weighted average of all states belonging to and
- (f)
Save output to 2s22p3_2p5_3
- (g)
Perform rci calculation in which Breit and QED effects are added.
Transform from - to -coupling
Run rlevels to view energy separations.
Run jj2lsj, Coupling, and rlevels to define energy spectra in different coupling schemes for those levels which have , , shells in identification.
Calculate properties
- (a)
Compute the M1 transition rates from the rci wave functions. Biorthonormal transformation not needed in this case since the states are described using the same orthonormal orbital set. Copy files and run the transition program.
- (b)
Compute the M1 transition rates in different coupling schemes for those levels which have , , shells in identification. Display the transition file.
*******************************************************************************
* RUN RNUCLEUS TO GENERATE NUCLEAR DATA AND DEFINE RADIAL GRID *
* OUTPUT FILE: isodata *
*******************************************************************************
>>rnucleus
RNUCLEUS
This program defines nuclear data and the radial grid
Outputfile: isodata
Enter the atomic number:
>>14
Enter the mass number (0 if the nucleus is to be modelled as a point source:
>>28
The default root mean squared radius is 3.1224000453948975 fm; (Angeli)
the default nuclear skin thickness is 2.2999999999999998 fm;
Revise these values?
>>n
Enter the mass of the neutral atom (in amu) (0 if the nucleus is to be static):
>>27.9769271
Enter the nuclear spin quantum number (I) (in units of h / 2 pi):
>>1
Enter the nuclear dipole moment (in nuclear magnetons):
>>1
Enter the nuclear quadrupole moment (in barns):
>>1
Comment: if we are not interested in the hyperfine structure constants we may just set nuclear spin and electromagnetic moments (magnetic dipole and electric quadrupole) to 1.
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE LIST FOR ALL *
* STATES OF 2s(2)2p(3) + 2p(5) *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,i)2s(2,i)2p(3,i)
Give configuration 2
>>1s(2,i)2p(5,i)
Give configuration 3
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>2s,2p
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,5
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>n
.........
3 blocks were created
block J/P NCSF
1 1/2- 2
2 3/2- 4
3 5/2- 1
*******************************************************************************
* COPY FILES *
* NOTE THAT WE COPY THE FILE TO RCSFMR.INP FOR FUTURE USE *
* TOGETHER WITH RCSFINTERACT *
*******************************************************************************
>>cp rcsfgenerate.log 2s22p3_2p5_DF.exc
>>cp rcsf.out rcsf.inp
>>cp rcsf.out rcsfmr.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.alog, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
........
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp, rwfnestimate.log *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 4 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>3
Enter the list of relativistic subshells:
>>*
Orbital Z_eff for hydrogenic orbitals
1s 14.00
2s 14.00
2p- 14.00
2p 14.00
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.9826D+02 0.1033D+03 0.1000D+01 0.1068D+00 328 Hyd
2s 0.2458D+02 0.3670D+02 0.1000D+01 0.4269D+00 344 Hyd
2p- 0.2458D+02 0.8338D-01 0.1000D+01 0.3555D+00 343 Hyd
2p 0.2452D+02 0.1492D+03 0.2000D+01 0.3568D+00 343 Hyd
RWFNESTIMATE: Execution complete.
*******************************************************************************
* RUN RMCDHF_MEM TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: ORBITALS BUILDING REFERENCE STATES ARE REQUIRED TO HAVE *
* THE CORRECT NUMBER OF NODES. THEY ARE REFERRED TO AS SPECTROSCOPIC *
* ORBITALS. IN THIS RUN WE VARY 1s, 2s, 2p AND THEY ARE ALL *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* *
* NOTE: INSTEAD OF SAYING THAT WE WILL OPTIMIZE ON, FOR EXAMPLE, *
* STATES 1,2,3,4 WE CAN WRITE 1-4 MEANING THE SAME THING *
*******************************************************************************
>>rmcdhf_mem
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 4 relativistic subshells;
Loading CSF File for ALL blocks
There are 7 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 3 blocks (block J/Parity NCF):
1 1/2- 2 2 3/2- 4 3 5/2- 1
Enter ASF serial numbers for each block
Block 1 ncf = 2 id = 1/2-
>>1-2
Block 2 ncf = 4 id = 3/2-
>>1-4
Block 3 ncf = 1 id = 5/2-
>>1
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p
Enter orbitals to be varied (Updating order)
>>*
Which of these are spectroscopic orbitals?
>>*
Enter the maximum number of SCF cycles:
>>100
......
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES *
*******************************************************************************
>>rsave 2s22p3_2p5_DF
Created 2s22p3_2p5_DF.w, 2s22p3_2p5_DF.c, 2s22p3_2p5_DF.m, 2s2p3_2p5_DF.sum,
2s2p3_2p5_DF.alog and 2s22p3_2p5_DF.log
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE LIST OBTAINED BY *
* SD-EXCITATIONS FROM 1s(2)2s(2)2p(3) + 1s(2)2p(5) TO n = 3 *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,*)2s(2,*)2p(3,*)
Give configuration 2
>>1s(2,*)2p(5,*)
Give configuration 3
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>3s,3p,3d
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,5
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
.........
3 blocks were created
block J/P NCSF
1 1/2- 595
2 3/2- 914
3 5/2- 847
*******************************************************************************
* COPY FILES *
*******************************************************************************
>>cp rcsfgenerate.log 2s22p3_2p5_3.exc
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RCSFINTERACT PROGRAM TO DETERMINE WHICH OF THE CSFs IN THE *
* rcsf.inp LIST INTERACTS WITH THE CSFs IN rcsfmr.inp *
* THE INTERACTING CSFs ARE WRITTEN TO rcsf.out *
* INPUT FILES: rcsfmr.inp, rcsf.inp *
* OUTPUT FILE: rcsf.out *
*******************************************************************************
>>rcsfinteract
RCSFinteract: Determines all the CSFs (rcsf.inp) that interact
with the CSFs in the multireference (rcsfmr.inp)
(C) Copyright by G. Gaigalas and Ch. F. Fischer
(Fortran 95 version) NIST (2017).
Input files: rcsfmr.inp, rcsf.inp
Output file: rcsf.out
Reduction based on Dirac-Coulomb (1) or
Dirac-Coulomb-Breit (2) Hamiltonian?
>>1
....
There are 9 relativistic subshells;
Block MR NCSF Before NCSF After NCSF
1 2 595 274
2 4 914 591
3 1 847 300
RCSFINTERACT: Execution complete
Please note that the orbital orders in
rcsfmr.inp and
rcsf.inp are required to be the same. In the case above, this requirement was fulfilled. In more complex cases, to meet the above requirement, one needs to prescribe the orbital order in the
clist.ref file that is used when generating the
rcsf.inp list, see
Section 6.6.
*******************************************************************************
* COPY FILES *
*******************************************************************************
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.alog, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
....
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp, rwfnestimate.log *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>1
Enter the file name (Null then "rwfn.out")
>>
Enter the list of relativistic subshells:
>>*
The following subshell radial wavefunctions remain to be estimated:
3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>3
Enter the list of relativistic subshells:
>>*
Orbital Z_eff for hydrogenic orbitals
3s 14.00
3p- 14.00
3p 14.00
3d- 14.00
3d 14.00
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.7698D+02 0.1056D+03 0.1000D+01 0.1109D+00 347 rwf
2s 0.1236D+02 0.3088D+02 0.1000D+01 0.5172D+00 351 rwf
2p- 0.1089D+02 0.5761D-01 0.1000D+01 0.4660D+00 352 rwf
2p 0.1086D+02 0.1007D+03 0.2000D+01 0.4675D+00 352 rwf
3s 0.1092D+02 0.1998D+02 0.1000D+01 0.9615D+00 354 Hyd
3p- 0.1092D+02 0.4942D-01 0.1000D+01 0.8901D+00 354 Hyd
3p 0.1090D+02 0.8855D+02 0.2000D+01 0.8918D+00 354 Hyd
3d- 0.1090D+02 0.4311D-01 0.2000D+01 0.7490D+00 353 Hyd
3d 0.1089D+02 0.9250D+02 0.3000D+01 0.7496D+00 353 Hyd
RWFNESTIMATE: Execution complete.
*******************************************************************************
* RUN RMCDHF_MEM TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: FOR CORRELATION ORBITALS THERE ARE NO RESTRICTIONS ON THE *
* NUMBER OF NODES, I.E. THEY ARE NOT SPECTROSCOPIC. IN THIS RUN WE *
* VARY THE CORRELATION ORBITALS 3s,3p, 3d. NONE OF THESE ARE *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* *
* NOTE: INSTEAD OF SAYING THAT WE WILL OPTIMIZE ON, FOR EXAMPLE, *
* STATES 1,2,3,4 WE CAN WRITE 1-4 MEANING THE SAME THING *
*******************************************************************************
>>rmcdhf_mem
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Loading CSF File for ALL blocks
There are 1164 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 3 blocks (block J/Parity NCF):
1 1/2- 274 2 3/2- 590 3 5/2- 300
Enter ASF serial numbers for each block
Block 1 ncf = 274 id = 1/2-
>>1-2
Block 2 ncf = 590 id = 3/2-
>>1-4
Block 3 ncf = 300 id = 5/2-
>>1
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Enter orbitals to be varied (Updating order)
>>3*
Which of these are spectroscopic orbitals?
>>
Enter the maximum number of SCF cycles:
>>100
.....
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES *
*******************************************************************************
>>rsave 2s22p3_2p5_3
Created 2s22p3_2p5_3.w, 2s22p3_2p5_3.c, 2s22p3_2p5_3.m, 2s22p3_2p5_3.sum,
2s22p3_2p5_3.alog and 2s22p3_2p5_3.log
*******************************************************************************
* RUN RCI TO INCLUDE TRANSVERSE PHOTON INTERACTION AND QED EFFECTS *
* INPUT FILES: isodata, 2s22p3_2p5_3.c, 2s22p3_2p5_3.w *
* OUTPUT FILES: 2s22p3_2p5_3.cm, 2s22p3_2p5_3.csum, 2s22p3_2p5_3.clog *
* rci.res *
* *
* THE TRANSVERSE PHOTON FREQUENCIES CAN BE SET TO THE LOW FREQUENCY *
* LIMIT. RECOMMENDED IN CASES WHERE YOU HAVE CORRELATION ORBITALS *
* THE SELF ENERGY CORRECTION MAY FAIL FOR CORRELATION ORBITALS WITH *
* HIGH N. *
* *
* NOTE: INSTEAD OF SAYING THAT WE WILL COMPUTE EIGENVALUES FOR *
* STATES 1,2,3,4 WE CAN WRITE 1-4 MEANING THE SAME THING *
*******************************************************************************
>>rci
RCI
This is the configuration interaction program
Input file: isodata, name.c, name.w
Outputfiles: name.cm, name.csum, name.clog. rci.res
Default settings?
>>y
Name of state:
>>2s22p3_2p5_3
Block 1 , ncf = 274
Block 2 , ncf = 590
Block 3 , ncf = 300
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Include contribution of H (Transverse)?
>>y
Modify all transverse photon frequencies?
>>y
Enter the scale factor:
>>1.d-6
Include H (Vacuum Polarisation)?
>>y
Include H (Normal Mass Shift)?
>>n
Include H (Specific Mass Shift)?
>>n
Estimate self-energy?
>>y
Largest n quantum number for including self-energy for orbital
n should be less or equal 8
>>3
Loading Radial WaveFunction File ...
There are 3 blocks (block J/Parity NCF):
1 1/2- 274 2 3/2- 590 3 5/2- 300
Enter ASF serial numbers for each block
Block 1 ncf = 274 id = 1/2-
>>1-2
Block 2 ncf = 590 id = 3/2-
>>1-4
Block 3 ncf = 300 id = 5/2-
>>1
....
RCI: Execution complete.
*******************************************************************************
* RUN JJ2LSJ TO GET THE LSJ-COMPOSITION *
* INPUT FILE: 2s22p3_2p5_3.c, 2s22p3_2p5_3.cm *
* OUTPUT FILE: 2s22p3_2p5_3.lsj.lbl, 2s22p3_2p5_3.uni.lsj.lbl *
*******************************************************************************
>>jj2lsj
jj2lsj: Transformation of ASFs from a jj-coupled CSF basis
into an LSJ-coupled CSF basis (Fortran 95 version)
(C) Copyright by G. Gaigalas and Ch. F. Fischer,
(2017).
Input files: name.c, name.(c)m
Output files: name.lsj.lbl
(optional) name.lsj.c, name.lsj.j,
name.uni.lsj.lbl, name.uni.lsj.sum
Name of state
>>2s22p3_2p5_3
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
Mixing coefficients from a CI calc.?
>>y
Do you need a unique labeling? (y/n)
>>y
nelec = 7
ncftot = 1164
nw = 9
nblock = 3
block ncf nev 2j+1 parity
1 274 2 2 -1
2 591 4 4 -1
3 300 1 6 -1
Default settings? (y/n)
>>y
...........
jj2lsj: Execution complete.
*******************************************************************************
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* NOTE: SINCE LSJ-INFORMATION NOW IS AVAILABLE OUTPUT LABELS *
* WILL BE IN LSJ-COUPLING *
* IF DESIRED WE CAN INSTEAD RUN RLEVELSEV TO GET THE SEPARATION IN EV *
*******************************************************************************
>>rlevels 2s22p3_2p5_3.cm
nblock = 3 ncftot = 1165 nw = 9 nelec = 7
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 3/2 - -263.2797841 0.00 0.00 1s(2).2s(2).2p(3)4S3_4S
2 2 3/2 - -262.9550555 71269.67 71269.67 1s(2).2s(2).2p(3)2D3_2D
3 1 5/2 - -262.9538206 71540.71 271.04 1s(2).2s(2).2p(3)2D3_2D
4 1 1/2 - -262.7906339 107356.06 35815.34 1s(2).2s(2).2p(3)2P1_2P
5 3 3/2 - -262.7882742 107873.94 517.88 1s(2).2s(2).2p(3)2P1_2P
6 4 3/2 - -259.5241179 824273.45 716399.51 1s(2).2p(5)_2P
7 2 1/2 - -259.4979399 830018.86 5745.41 1s(2).2p(5)_2P
------------------------------------------------------------------------------------------
To interpret the
-coupling notation produced by
jj2lsj, see
Section 8.2.
*******************************************************************************
* THE ABOVE JJ2LJS RUN TRANSFORMED ALL LEVELS TO LSJ COUPLING. *
* BELOW WE WILL TRANSFORM A SUBSET OF THE LEVELS TO OTHER COUPLING *
* SCHEMES. FOR TECHNICAL REASONS WE HAVE TO ADD INFORMATION ALSO *
* FOR THE UNTRANSFORMED LEVELS IN ORDER FOR THE PROGRAMS TO WORK *
* THE LABELS FOR THE UNTRANSFORMED LEVELS WILL BE THOSE FROM THE *
* ABOVE RUN. FOR THIS REASON WE HAVE TO SAVE A COPY OF THE *
* 2s22p3_2p5_3.lsj.lbl LABEL FILE *
*******************************************************************************
>>cp 2s22p3_2p5_3.lsj.lbl 2s22p3_2p5_3.lsj.lbl_SAVE
*******************************************************************************
* RUN JJ2LSJ TO GET THE INPUT FOR COUPLING PROGRAM FOR THOSE *
* LEVELS WHICH HAVE 1s, 2s, AND 2p SHELLS IN IDENTIFICATION *
* INPUT FILES: 2s22p3_2p5_3.c, 2s22p3_2p5_3.cm *
* OUPUT FILES: 2s22p3_2p5_3.lsj.c, 2s22p3_2p5_3.lsj.j, *
* 2s22p3_2p5_3.lsj.lbl *
* *
* THE LEVELS WE ARE INTERESTED IN ARE *
* BLOCK 1, J = 1/2, LEVEL 1 *
* BLOCK 2, J = 3/2, LEVEL 1, 2, 3 (1-3) *
* BLOCK 3, J = 5/2, LEVEL 1 *
*******************************************************************************
>>jj2lsj
jj2lsj: Transformation of ASFs from a jj-coupled CSF basis
into an LS-coupled CSF basis (Fortran 95 version)
(C) Copyright by G. Gaigalas and Ch. F. Fischer,
(2017).
Input files: name.c, name.(c)m
Ouput files: name.lsj.lbl,
(optional) name.lsj.c, name.lsj.j,
name.uni.lsj.lbl, name.uni.lsj.sum
Name of state
>>2s22p3_2p5_3
Loading Configuration Symmetry List File ...
There are 4 relativistic subshells;
There are 4 relativistic CSFs;
... load complete;
Mixing coefficients from a CI calc.?
>>y
Do you need a unique labeling? (y/n)
>>n
nelec = 4
ncftot = 4
nw = 4
nblock = 3
block ncf nev 2j+1 parity
1 1 1 1 -1
2 2 2 3 -1
3 1 1 5 -1
Default settings? (y/n)
>>n
All levels (Y/N)
>>n
Maximum number of ASFs is: 7
Enter the level numbers of the ASF which are to be transformed,
Enter the block number
>>1
The block number is: 1
e.g., 1 3 4 7--20 48 69--85 :
>>1
Do you need to include more levels? (y/n)
>>y
Enter the block number
>>2
The block number is: 2
e.g., 1 3 4 7--20 48 69--85 :
>>1-3
Do you need to include more levels? (y/n)
>>y
Enter the block number
>>3
The block number is: 3
e.g., 1 3 4 7--20 48 69--85 :
>>1
Do you need to include more levels? (y/n)
>>n
Maximum % of omitted composition
>>0
What is the value below which an eigenvector composition
is to be neglected for printing?
>>0.01
jj2lsj: Execution complete.
*******************************************************************************
* RUN COUPLING TO GET THE IDENTIFICATION STATES IN DIFFERENT *
* COUPLING SCHEMES FOR THOSE LEVELS WHICH HAVE 1s, 2s, AND 2p *
* SHELLS IN IDENTIFICATION *
* INPUT FILES: 2s22p3_2p5_3.lsj.c, 2s22p3_2p5_3.lsj.j *
* OUPUT FILES: 2s22p3_2p5_3.coup3.LK3.lbl, 2s22p3_2p5_3.coup3.JK3.lbl *
* 2s22p3_2p5_3.LS.lbl, 2s22p3_2p5_3.coup3.LS3.lbl *
* 2s22p3_2p5_3.coup3.LSJ3.lbl, 2s22p3_2p5_3.coup3.jj.lbl *
* 2s22p3_2p5_3.coup3.cLSJ3.lbl, 2s22p3_2p5_3.coup3.sum *
*******************************************************************************
>>Coupling
Coupling: Transformation of ASFs from a LS-coupled CSF basis
into differete coupled CSF bases (Fortran 95)
(C) (2022) G. Gaigalas, A. Kramida.
Input files: *.lsj.c, *.lsj.j (ATSP (CPC) or GRASP2K types)
Output files: *.coup*.*.lbl, *.coup*.sum
Name of state
>>2s22p3_2p5_3
Default settings ? (Y/N)
>>y
Specify the number of coupled shells for evaluation (1,2 or 3):
>>3
3
What is the value below which an eigenvector composition
is to be neglected for printing?
>>0
0.0000000000000000
Specify shells for recoupling (no more than 12)
>>1s,2s,2p,3s,3p,3d
All transformations completed
There is one-to-one classification for LS coupling
There is one-to-one classification for LS3 coupling
There is one-to-one classification for LSJ3 coupling
There is one-to-one classification for LK3 coupling
There is one-to-one classification for JK3 coupling
There is one-to-one classification for cLSJ3 coupling
There is one-to-one classification for jj3 coupling
end subroutine generate_classification_data
Coupling: Execution complete.
*******************************************************************************
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* IN LK3-COUPLING FOR THOSE LEVELS WHICH HAVE 1s, 2s, AND 2p *
* SHELLS IN IDENTIFICATION AND FOR THE REST LS-COUPLING *
* *
* OBSERVE! *
* ABOVE WE TRANSFORMED ONLY A SUBSET OF THE LEVELS *
* WE NEED, HOWEVER, TO HAVE LABELS FOR ALL LEVELS. WE WILL USE *
* THE LSJ LABELS IN THE FILE 2s22p3_2p5_3.lsj.lbl_SAVE FOR THE *
* UNTRANSFORMED LEVELS. WE CAN BY HAND EDIT THE *
* 2s22p3_2p5_3.coup3.LK3.lbl FILE AND PASTE THE INFORMATION FOR *
* THE UNTRANSFORMED LEVELS FROM THE 2s22p3_2p5_3.lsj.lbl_SAVE *
* FILE AT THE APPROPRIATE PLACE IN 2s22p3_2p5_3.coup3.LK3.lbl *
* ALTERNATIVELY, AND THIS IS WHAT WE WILL DO BELOW, WE CAN USE THE *
* sed COMMAND TO COPY THE APPROPRIATE INFORMATION FOR THE TWO *
* UNTRANSFORMED LEVELS FROM 2s22p3_2p5_3.lsj.lbl_SAVE AND PUT *
* THE INFORMATION IN PATCH1 and PATCH2. *
* WE NOW USE sed TO PASTE THE INFORMATION IN PATCH1 AND PATCH2 *
* AT THE APPROPRIATE PLACE IN 2s22p3_2p5_3.coup3.LK3.lbl *
* THE USER IS ADVICED TO OPEN AND INSPECT BOTH THE *
* 2s22p3_2p5_3.lsj.lbl_SAVE FILE AND THE 2s22p3_2p5_3.coup3.LK3.lbl *
* FILE TO UNDERSTAND WHAT IS GOING ON *
*******************************************************************************
>>sed -n 25,28p 2s22p3_2p5_3.lsj.lbl_SAVE >patch1
>>sed -i 327rpatch1 2s22p3_2p5_3.coup3.LK3.lbl
>>sed -n 6,10p 2s22p3_2p5_3.lsj.lbl_SAVE >patch2
>>sed -i 57rpatch2 2s22p3_2p5_3.coup3.LK3.lbl
>>cp 2s22p3_2p5_3.coup3.LK3.lbl 2s22p3_2p5_3.lsj.lbl
>>rlevels 2s22p3_2p5_3.cm
nblock = 3 ncftot = 1165 nw = 9 nelec = 7
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
---------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
---------------------------------------------------------------------------------------------
1 1 3/2 - -263.2797841 0.00 0.00 1s2_ 2s2_.2p3(4S)(4S) S_4[0]<3/2>
2 2 3/2 - -262.9550555 71269.67 71269.67 1s2_ 2s2_.2p3(2D)(2D) D_2[2]<3/2>
3 1 5/2 - -262.9538206 71540.71 271.04 1s2_ 2s2_.2p3(2D)(2D) D_2[2]<5/2>
4 1 1/2 - -262.7906339 107356.06 35815.34 1s2_ 2s2_.2p3(2P)(2P) P_2[1]<1/2>
5 3 3/2 - -262.7882742 107873.94 517.88 1s2_ 2s2_.2p3(2P)(2P) P_2[1]<3/2>
6 4 3/2 - -259.5241179 824273.45 716399.51 1s(2).2p(5)_2P
7 2 1/2 - -259.4979399 830018.86 5745.41 1s(2).2p(5)_2P
---------------------------------------------------------------------------------------------
Please note that the labels for No 1–5 are in LK3 coupling and the rest (No 6 and 7) in LSJ coupling.
*******************************************************************************
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* IN JK3-COUPLING FOR THOSE LEVELS WHICH HAVE 1s, 2s, AND 2p *
* SHELLS IN IDENTIFICATION AND FOR THE REST LSJ-COUPLING *
* INFORMATION FROM 2s22p3_2p5_3.lsj.lbl_SAV ADDED *
*******************************************************************************
>>sed -i 327rpatch1 2s22p3_2p5_3.coup3.JK3.lbl
>>sed -i 57rpatch2 2s22p3_2p5_3.coup3.JK3.lbl
>>cp 2s22p3_2p5_3.coup3.JK3.lbl 2s22p3_2p5_3.lsj.lbl
>>rlevels 2s22p3_2p5_3.cm
nblock = 3 ncftot = 1165 nw = 9 nelec = 7
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 3/2 - -263.2797841 0.00 0.00 1s2_<0>2s2_.2p3(4S)(4S) 4[0]<3/2>
2 2 3/2 - -262.9550555 71269.67 71269.67 1s2_<0>2s2_.2p3(2D)(2D) 2[2]<3/2>
3 1 5/2 - -262.9538206 71540.71 271.04 1s2_<0>2s2_.2p3(2D)(2D) 2[2]<5/2>
4 1 1/2 - -262.7906339 107356.06 35815.34 1s2_<0>2s2_.2p3(2P)(2P) 2[1]<1/2>
5 3 3/2 - -262.7882742 107873.94 517.88 1s2_<0>2s2_.2p3(2P)(2P) 2[1]<3/2>
6 4 3/2 - -259.5241179 824273.45 716399.51 1s(2).2p(5)_2P
7 2 1/2 - -259.4979399 830018.86 5745.41 1s(2).2p(5)_2P
------------------------------------------------------------------------------------------
*******************************************************************************
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* IN LS3-COUPLING FOR THOSE LEVELS WHICH HAVE 1s, 2s, AND 2p *
* SHELLS IN IDENTIFICATION AND FOR THE REST LSJ-COUPLING *
* INFORMATION FROM 2s22p3_2p5_3.lsj.lbl_SAV ADDED *
*******************************************************************************
>>sed -i 320rpatch1 2s22p3_2p5_3.coup3.LS3.lbl
>>sed -i 56rpatch2 2s22p3_2p5_3.coup3.LS3.lbl
>>cp 2s22p3_2p5_3.coup3.LS3.lbl 2s22p3_2p5_3.lsj.lbl
>>rlevels 2s22p3_2p5_3.cm
nblock = 3 ncftot = 1165 nw = 9 nelec = 7
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 3/2 - -263.2797858 0.00 0.00 1s2_ 2s2_.2p3(4S)(4S) 4S<3/2>
2 2 3/2 - -262.9550573 71269.67 71269.67 1s2_ 2s2_.2p3(2D)(2D) 2D<3/2>
3 1 5/2 - -262.9538223 71540.71 271.04 1s2_ 2s2_.2p3(2D)(2D) 2D<5/2>
4 1 1/2 - -262.7906356 107356.06 35815.35 1s2_ 2s2_.2p3(2P)(2P) 2P<1/2>
5 3 3/2 - -262.7882760 107873.94 517.88 1s2_ 2s2_.2p3(2P)(2P) 2P<3/2>
6 4 3/2 - -259.5241195 824273.48 716399.54 1s(2).2p(5)_2P
7 2 1/2 - -259.4979415 830018.89 5745.41 1s(2).2p(5)_2P
------------------------------------------------------------------------------------------
*******************************************************************************
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* IN LSJ3-COUPLING FOR THOSE LEVELS WHICH HAVE 1s, 2s, AND 2p *
* SHELLS IN IDENTIFICATION AND FOR THE REST LSJ-COUPLING *
* INFORMATION FROM 2s22p3_2p5_3.lsj.lbl_SAV ADDED *
*******************************************************************************
>>sed -i 327rpatch1 2s22p3_2p5_3.coup3.LSJ3.lbl
>>sed -i 57rpatch2 2s22p3_2p5_3.coup3.LSJ3.lbl
>>cp 2s22p3_2p5_3.coup3.LSJ3.lbl 2s22p3_2p5_3.lsj.lbl
>>rlevels 2s22p3_2p5_3.cm
nblock = 3 ncftot = 1165 nw = 9 nelec = 7
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 3/2 - -263.2797841 0.00 0.00 1s2_ 2s2_.2p3(4S)(4S) (0,3/2)<3/2>
2 2 3/2 - -262.9550555 71269.67 71269.67 1s2_ 2s2_.2p3(2D)(2D) (0,3/2)<3/2>
3 1 5/2 - -262.9538206 71540.71 271.04 1s2_ 2s2_.2p3(2D)(2D) (0,5/2)<5/2>
4 1 1/2 - -262.7906339 107356.06 35815.34 1s2_ 2s2_.2p3(2P)(2P) (0,1/2)<1/2>
5 3 3/2 - -262.7882742 107873.94 517.88 1s2_ 2s2_.2p3(2P)(2P) (0,3/2)<3/2>
6 4 3/2 - -259.5241179 824273.45 716399.51 1s(2).2p(5)_2P
7 2 1/2 - -259.4979399 830018.86 5745.41 1s(2).2p(5)_2P
------------------------------------------------------------------------------------------
*******************************************************************************
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* IN jj-COUPLING FOR THOSE LEVELS WHICH HAVE 1s, 2s, AND 2p *
* SHELLS IN IDENTIFICATION AND FOR THE REST LSJ-COUPLING *
* INFORMATION FROM 2s22p3_2p5_3.lsj.lbl_SAV ADDED *
*******************************************************************************
>>sed -i 329rpatch1 2s22p3_2p5_3.coup3.jj.lbl
>>sed -i 59rpatch2 2s22p3_2p5_3.coup3.jj.lbl
>>cp 2s22p3_2p5_3.coup3.jj.lbl 2s22p3_2p5_3.lsj.lbl
>>rlevels 2s22p3_2p5_3.cm
nblock = 3 ncftot = 1165 nw = 9 nelec = 7
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 3/2 - -263.2797841 0.00 0.00 1s+2_2s+2_<0>.2p-_<1/2>.2p+2(2) <3/2>
2 2 3/2 - -262.9550555 71269.67 71269.67 1s+2_2s+2_<0>.2p-_<1/2>.2p+2(2) <3/2>
3 1 5/2 - -262.9538206 71540.71 271.04 1s+2_2s+2_<0>.2p-_<1/2>.2p+2(2) <5/2>
4 1 1/2 - -262.7906339 107356.06 35815.34 1s+2_2s+2_<0>.2p-_<1/2>.2p+2(0) <1/2>
5 3 3/2 - -262.7882742 107873.94 517.88 1s+2_2s+2_<0>.2p+3(3/2) <3/2>
6 4 3/2 - -259.5241179 824273.45 716399.51 1s(2).2p(5)_2P
7 2 1/2 - -259.4979399 830018.86 5745.41 1s(2).2p(5)_2P
------------------------------------------------------------------------------------------
*******************************************************************************
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* IN cLSJ3-COUPLING FOR THOSE LEVELS WHICH HAVE 1s, 2s, AND 2p *
* SHELLS IN IDENTIFICATION AND FOR THE REST LSJ-COUPLING *
* INFORMATION FROM 2s22p3_2p5_3.lsj.lbl_SAV ADDED *
*******************************************************************************
>>sed -i 327rpatch1 2s22p3_2p5_3.coup3.cLSJ3.lbl
>>sed -i 57rpatch2 2s22p3_2p5_3.coup3.cLSJ3.lbl
>>cp 2s22p3_2p5_3.coup3.cLSJ3.lbl 2s22p3_2p5_3.lsj.lbl
>>rlevels 2s22p3_2p5_3.cm
nblock = 3 ncftot = 1165 nw = 9 nelec = 7
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 3/2 - -263.2797841 0.00 0.00 1s+2_ (0,0)<0> 2s2_.2p3(4S)(4S)<3/2> (0,3/2)<3/2>
2 2 3/2 - -262.9550555 71269.67 71269.67 1s+2_ (0,0)<0> 2s2_.2p3(2D)(2D)<3/2> (0,3/2)<3/2>
3 1 5/2 - -262.9538206 71540.71 271.04 1s+2_ (0,0)<0> 2s2_.2p3(2D)(2D)<5/2> (0,5/2)<5/2>
4 1 1/2 - -262.7906339 107356.06 35815.34 1s+2_ (0,0)<0> 2s2_.2p3(2P)(2P)<1/2> (0,1/2)<1/2>
5 3 3/2 - -262.7882742 107873.94 517.88 1s+2_ (0,0)<0> 2s2_.2p3(2P)(2P)<3/2> (0,3/2)<3/2>
6 4 3/2 - -259.5241179 824273.45 716399.51 1s(2).2p(5)_2P
7 2 1/2 - -259.4979399 830018.86 5745.41 1s(2).2p(5)_2P
------------------------------------------------------------------------------------------
For definition of different coupling schemes see in [
14] and for interpretation of different coupling schemes notation produced by
Coupling see
Section 8.2, where we discuss in detail the transformation in different coupling schemes for
in B II.
*******************************************************************************
* WE WILL NOW COMPUTE THE M1 TRANSITION RATES *
* IN THIS CASE THE INITIAL AND FINAL STATE FILES ARE THE SAME *
* AND WE DO NOT NEED TO PERFORM A biorthonormal TRANSFORMATION *
* USING RBIOTRANSFORM. JUST COPY FILES TO name.bw AND name.cbm *
* THE 2s22p3_2p5_3.lsj.lbl FILE GIVES THE LABELS THAT WILL BE *
* USED BY THE TRANSITION PROGRAM. WE START BY USING THE LSJ LABELS *
* AND COPY 2s22p3_2p5_3.lsj.lbl_SAVE TO 2s22p3_2p5_3.lsj.lbl *
*******************************************************************************
>>cp 2s22p3_2p5_3.w 2s22p3_2p5_3.bw
>>cp 2s22p3_2p5_3.cm 2s22p3_2p5_3.cbm
>>cp 2s22p3_2p5_3.lsj.lbl_SAVE 2s22p3_2p5_3.lsj.lbl
*******************************************************************************
* RUN RTRANSITION FOR 2s22p3_2p5_3 TO COMPUTE M1 TRANSITION PARAMETERS*
* INPUT FILES: isodata, 2s22p3_2p5_3.c, 2s22p3_2p5_3.bw, *
* 2s22p3_2p5_3.cbm, *
* OUTPUT FILE: 2s22p3_2p5_3.2s22p3_2p5_3.ct, *
* 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj *
* 2s22p3_2p5_3.2s22p3_2p5_3.+1T (angular file) *
* *
* NOTE THAT THE LATTER OUTPUT FILE HAS ALL THE LABELS IN LSJ- *
* COUPLING WHICH IS VERY CONVENIENT *
* *
* PLEASE OBSERVE!! IF WE ARE GOING TO RUN RTRANSITION FOR AN RCI WAVE *
* FUNCTIONS THEN THE LSJ-INFORMATION SHOULD BE AVAILABLE FOR THE SAME *
* WAVE FUNCTION. IF FOR EXAMPLE THE LSJ-INFORMATION FROM JJ2LSJ IS *
* IS AVAILABLE FROM AN RMCDHF RUN AND WE RUN RTRANSITION ON THE RCI *
* WAVE FUNCTION THEN RTRANSITION WILL STOP. IN THIS CASE JUST RERUN *
* JJ2LSJ FOR THE RCI WAVE FUNCTION AND START RTRANSITION AGAIN FOR *
* THE SAME WAVE FUNCTION. IN OUR EXAMPLE JJ2LJS AND RTRANSITION ARE *
* RUN FOR RCI WAVE FUNCTIONS AND EVERYTHING IS OK. *
*******************************************************************************
>>rtransition
RTRANSITION
This program computes transition parameters from
transformed wave functions
Input files: isodata, name1.c, name1.bw, name1.(c)bm
name2.c, name2.bw, name2.(c)bm
optional, name1.lsj.lbl, name2.lsj.lbl
name1.name2.KT (optional angular files)
Output files: name1.name2.(c)t
optional, name1.name2.(c)t.lsj
name1.name2.KT (angular files)
Here K is parity and rank of transition: -1,+1 etc
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>2s22p3_2p5_3
Name of the Final state
>>2s22p3_2p5_3
MRGCSL: Execution begins ...
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
1 s
2 s
2 p-
2 p
3 s
3 p-
3 p
3 d-
3 d
3
274 864 1164
3
274 864 1164
Loading Configuration Symmetry List File ...
there are 9 relativistic subshells;
1
2 2
there are 2328 relativistic CSFs;
... load complete;
Enter the list of transition specifications
e.g., E1,M2 or E1 M2 or E1;M2 :
>>M1
M1 transitions only between levels with different J?
>>n
.....
RTRANSITION: Execution complete.
*******************************************************************************
* VIEW THE TRANSITION FILE WHERE THE LABELS ARE IN LSJ COUPLING *
*******************************************************************************
>>more 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj
Transition between files:
2s22p3_2p5_3
2s22p3_2p5_3
1 -262.79063388 1s(2).2s(2).2p(3)2P1_2P
1 -259.49793990 1s(2).2p(5)_2P
722662.80 CM-1 138.38 ANGS(VAC) 138.38 ANGS(AIR)
M1 S = 1.18001D-11 GF = 3.44839D-16 AKI = 6.00621D-05
1 -262.79063388 1s(2).2s(2).2p(3)2P1_2P
3 -262.78827423 1s(2).2s(2).2p(3)2P1_2P
517.88 CM-1 193094.26 ANGS(VAC) 193074.30 ANGS(AIR)
M1 S = 1.31018D+00 GF = 2.74383D-08 AKI = 1.22716D-03
1 -262.79063388 1s(2).2s(2).2p(3)2P1_2P
3 -259.52411791 1s(2).2p(5)_2P
716917.39 CM-1 139.49 ANGS(VAC) 139.49 ANGS(AIR)
M1 S = 1.73052D-06 GF = 5.01695D-11 AKI = 4.29992D+00
3 -263.27978407 1s(2).2s(2).2p(3)4S3_4S
1 -262.79063388 1s(2).2s(2).2p(3)2P1_2P
107356.06 CM-1 931.48 ANGS(VAC) 931.48 ANGS(AIR)
M1 S = 1.86549D-03 GF = 8.09867D-09 AKI = 3.11300D+01
3 -263.27978407 1s(2).2s(2).2p(3)4S3_4S
1 -259.49793990 1s(2).2p(5)_2P
830018.86 CM-1 120.48 ANGS(VAC) 120.48 ANGS(AIR)
M1 S = 5.92158D-07 GF = 1.98756D-11 AKI = 4.56677D+00
..........
5 -262.95382060 1s(2).2s(2).2p(3)2D3_2D
3 -262.78827423 1s(2).2s(2).2p(3)2P1_2P
36333.23 CM-1 2752.30 ANGS(VAC) 2752.01 ANGS(AIR)
M1 S = 3.63575D-02 GF = 5.34186D-08 AKI = 1.17593D+01
5 -262.95382060 1s(2).2s(2).2p(3)2D3_2D
3 -259.52411791 1s(2).2p(5)_2P
752732.73 CM-1 132.85 ANGS(VAC) 132.85 ANGS(AIR)
M1 S = 1.88251D-06 GF = 5.73023D-11 AKI = 5.41422D+00
*******************************************************************************
* GIVE THE TRANSITION FILE A NEW APPROPRIATE NAME FOR LATER USE *
*******************************************************************************
>>mv 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj_SAVE
*******************************************************************************
* RUN RTRANSITION FOR 2s22p3_2p5_3 TO COMPUTE M1 TRANSITION PARAMETERS*
* IN LK3-COUPLING FOR THOSE LEVELS WHICH HAVE 1s, 2s, AND 2p *
* SHELLS IN IDENTIFICATION AND FOR THE REST LS-COUPLING *
* INPUT FILES: isodata, 2s22p3_2p5_3.c, 2s22p3_2p5_3.bw, *
* 2s22p3_2p5_3.cbm, *
* OUTPUT FILE: 2s22p3_2p5_3.2s22p3_2p5_3.ct, *
* 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj *
* 2s22p3_2p5_3.2s22p3_2p5_3.+1T (angular file) *
* *
* COPY THE 2s22p3_2p5_3.coup3.LK3.lbl TO 2s22p3_2p5_3.lsj.lbl *
* IT IS THE LATTER FILE THAT IS READ AND USED BY THE TRANSITION *
* PROGRAM *
*******************************************************************************
>>cp 2s22p3_2p5_3.coup3.LK3.lbl 2s22p3_2p5_3.lsj.lbl
>>rtransition
RTRANSITION
This program computes transition parameters from
transformed wave functions
Input files: isodata, name1.c, name1.bw, name1.(c)bm
name2.c, name2.bw, name2.(c)bm
optional, name1.lsj.lbl, name2.lsj.lbl
name1.name2.KT (optional angular files)
Output files: name1.name2.(c)t
optional, name1.name2.(c)t.lsj
name1.name2.KT (angular files)
Here K is parity and rank of transition: -1,+1 etc
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>2s22p3_2p5_3
Name of the Final state
>>2s22p3_2p5_3
MRGCSL: Execution begins ...
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
1 s
2 s
2 p-
2 p
3 s
3 p-
3 p
3 d-
3 d
3
274 864 1164
3
274 864 1164
Loading Configuration Symmetry List File ...
there are 9 relativistic subshells;
1
2 2
there are 2328 relativistic CSFs;
... load complete;
Enter the list of transition specifications
e.g., E1,M2 or E1 M2 or E1;M2 :
>>M1
M1 transitions only between levels with different J?
>>n
.....
RTRANSITION: Execution complete.
*******************************************************************************
* VIEW THE TRANSITION FILE WHERE THE LABELS ARE IN LK3 COUPLING *
* THE DATA IS THE SAME AS ABOVE, ONLY THE LABELS OF THE STATES *
* DIFFER *
*******************************************************************************
>>more 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj
Transition between files:
2s22p3_2p5_3
2s22p3_2p5_3
1 -262.79063388 1s2_ 2s2_.2p3(2P)(2P) P_2[1]<1/2>
1 -259.49793990 1s(2).2p(5)_2P
722662.80 CM-1 138.38 ANGS(VAC) 138.38 ANGS(AIR)
M1 S = 1.18001D-11 GF = 3.44839D-16 AKI = 6.00621D-05
1 -262.79063388 1s2_ 2s2_.2p3(2P)(2P) P_2[1]<1/2>
3 -262.78827423 1s2_ 2s2_.2p3(2P)(2P) P_2[1]<3/2>
517.88 CM-1 193094.26 ANGS(VAC) 193074.30 ANGS(AIR)
M1 S = 1.31018D+00 GF = 2.74383D-08 AKI = 1.22716D-03
1 -262.79063388 1s2_ 2s2_.2p3(2P)(2P) P_2[1]<1/2>
3 -259.52411791 1s(2).2p(5)_2P
716917.39 CM-1 139.49 ANGS(VAC) 139.49 ANGS(AIR)
M1 S = 1.73052D-06 GF = 5.01695D-11 AKI = 4.29992D+00
3 -263.27978407 1s2_ 2s2_.2p3(4S)(4S) S_4[0]<3/2>
1 -262.79063388 1s2_ 2s2_.2p3(2P)(2P) P_2[1]<1/2>
107356.06 CM-1 931.48 ANGS(VAC) 931.48 ANGS(AIR)
M1 S = 1.86549D-03 GF = 8.09867D-09 AKI = 3.11300D+01
3 -263.27978407 1s2_ 2s2_.2p3(4S)(4S) S_4[0]<3/2>
1 -259.49793990 1s(2).2p(5)_2P
830018.86 CM-1 120.48 ANGS(VAC) 120.48 ANGS(AIR)
M1 S = 5.92158D-07 GF = 1.98756D-11 AKI = 4.56677D+00
..........
5 -262.95382060 1s2_ 2s2_.2p3(2D)(2D) D_2[2]<5/2>
3 -262.78827423 1s2_ 2s2_.2p3(2P)(2P) P_2[1]<3/2>
36333.23 CM-1 2752.30 ANGS(VAC) 2752.01 ANGS(AIR)
M1 S = 3.63575D-02 GF = 5.34186D-08 AKI = 1.17593D+01
5 -262.95382060 1s2_ 2s2_.2p3(2D)(2D) D_2[2]<5/2>
3 -259.52411791 1s(2).2p(5)_2P
752732.73 CM-1 132.85 ANGS(VAC) 132.85 ANGS(AIR)
M1 S = 1.88251D-06 GF = 5.73023D-11 AKI = 5.41422D+00
*******************************************************************************
* GIVE THE TRANSITION FILE A NEW APPROPRIATE NAME FOR LATER USE *
*******************************************************************************
>>mv 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj_LK3
*******************************************************************************
* RUN RTRANSITION FOR 2s22p3_2p5_3 TO COMPUTE M1 TRANSITION PARAMETERS*
* IN JK3-COUPLING FOR THOSE LEVELS WHICH HAVE 1s, 2s, AND 2p *
* SHELLS IN IDENTIFICATION AND FOR THE REST LS-COUPLING *
* INPUT FILES: isodata, 2s22p3_2p5_3.c, 2s22p3_2p5_3.bw, *
* 2s22p3_2p5_3.cbm, *
* OUTPUT FILE: 2s22p3_2p5_3.2s22p3_2p5_3.ct, *
* 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj *
* 2s22p3_2p5_3.2s22p3_2p5_3.+1T (angular file) *
* *
* COPY THE 2s22p3_2p5_3.coup3.JK3.lbl TO 2s22p3_2p5_3.lsj.lbl *
* IT IS THE LATTER FILE THAT IS READ AND USED BY THE TRANSITION *
* PROGRAM *
*******************************************************************************
>>cp 2s22p3_2p5_3.coup3.JK3.lbl 2s22p3_2p5_3.lsj.lbl
>>rtransition
RTRANSITION
This program computes transition parameters from
transformed wave functions
Input files: isodata, name1.c, name1.bw, name1.(c)bm
name2.c, name2.bw, name2.(c)bm
optional, name1.lsj.lbl, name2.lsj.lbl
name1.name2.KT (optional angular files)
Output files: name1.name2.(c)t
optional, name1.name2.(c)t.lsj
name1.name2.KT (angular files)
Here K is parity and rank of transition: -1,+1 etc
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>2s22p3_2p5_3
Name of the Final state
>>2s22p3_2p5_3
MRGCSL: Execution begins ...
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
1 s
2 s
2 p-
2 p
3 s
3 p-
3 p
3 d-
3 d
3
274 864 1164
3
274 864 1164
Loading Configuration Symmetry List File ...
there are 9 relativistic subshells;
1
2 2
there are 2328 relativistic CSFs;
... load complete;
Enter the list of transition specifications
e.g., E1,M2 or E1 M2 or E1;M2 :
>>M1
M1 transitions only between levels with different J?
>>n
.....
RTRANSITION: Execution complete.
>>mv 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj_JK3
*******************************************************************************
* RUN RTRANSITION FOR 2s22p3_2p5_3 TO COMPUTE M1 TRANSITION PARAMETERS*
* IN LS3-COUPLING FOR THOSE LEVELS WHICH HAVE 1s, 2s, AND 2p *
* SHELLS IN IDENTIFICATION AND FOR THE REST LS-COUPLING *
* INPUT FILES: isodata, 2s22p3_2p5_3.c, 2s22p3_2p5_3.bw, *
* 2s22p3_2p5_3.cbm, *
* OUTPUT FILE: 2s22p3_2p5_3.2s22p3_2p5_3.ct, *
* 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj *
* 2s22p3_2p5_3.2s22p3_2p5_3.+1T (angular file) *
* *
* COPY THE 2s22p3_2p5_3.coup3.LS3.lbl TO 2s22p3_2p5_3.lsj.lbl *
* IT IS THE LATTER FILE THAT IS READ AND USED BY THE TRANSITION *
* PROGRAM *
*******************************************************************************
>>cp 2s22p3_2p5_3.coup3.LS3.lbl 2s22p3_2p5_3.lsj.lbl
>>rtransition
RTRANSITION
This program computes transition parameters from
transformed wave functions
Input files: isodata, name1.c, name1.bw, name1.(c)bm
name2.c, name2.bw, name2.(c)bm
optional, name1.lsj.lbl, name2.lsj.lbl
name1.name2.KT (optional angular files)
Output files: name1.name2.(c)t
optional, name1.name2.(c)t.lsj
name1.name2.KT (angular files)
Here K is parity and rank of transition: -1,+1 etc
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>2s22p3_2p5_3
Name of the Final state
>>2s22p3_2p5_3
MRGCSL: Execution begins ...
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
1 s
2 s
2 p-
2 p
3 s
3 p-
3 p
3 d-
3 d
3
274 864 1164
3
274 864 1164
Loading Configuration Symmetry List File ...
there are 9 relativistic subshells;
1
2 2
there are 2328 relativistic CSFs;
... load complete;
Enter the list of transition specifications
e.g., E1,M2 or E1 M2 or E1;M2 :
>>M1
M1 transitions only between levels with different J?
>>n
.....
RTRANSITION: Execution complete.
*******************************************************************************
* GIVE THE TRANSITION FILE A NEW APPROPRIATE NAME FOR LATER USE *
*******************************************************************************
>>mv 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj_LS3
*******************************************************************************
* RUN RTRANSITION FOR 2s22p3_2p5_3 TO COMPUTE M1 TRANSITION PARAMETERS*
* IN LSJ3-COUPLING FOR THOSE LEVELS WHICH HAVE 1s, 2s, AND 2p *
* SHELLS IN IDENTIFICATION AND FOR THE REST LS-COUPLING *
* INPUT FILES: isodata, 2s22p3_2p5_3.c, 2s22p3_2p5_3.bw, *
* 2s22p3_2p5_3.cbm, *
* OUTPUT FILE: 2s22p3_2p5_3.2s22p3_2p5_3.ct, *
* 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj *
* 2s22p3_2p5_3.2s22p3_2p5_3.+1T (angular file) *
* *
* COPY THE 2s22p3_2p5_3.coup3.LSJ3.lbl TO 2s22p3_2p5_3.lsj.lbl *
* IT IS THE LATTER FILE THAT IS READ AND USED BY THE TRANSITION *
* PROGRAM *
*******************************************************************************
>>cp 2s22p3_2p5_3.coup3.LSJ3.lbl 2s22p3_2p5_3.lsj.lbl
>>rtransition
RTRANSITION
This program computes transition parameters from
transformed wave functions
Input files: isodata, name1.c, name1.bw, name1.(c)bm
name2.c, name2.bw, name2.(c)bm
optional, name1.lsj.lbl, name2.lsj.lbl
name1.name2.KT (optional angular files)
Output files: name1.name2.(c)t
optional, name1.name2.(c)t.lsj
name1.name2.KT (angular files)
Here K is parity and rank of transition: -1,+1 etc
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>2s22p3_2p5_3
Name of the Final state
>>2s22p3_2p5_3
MRGCSL: Execution begins ...
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
1 s
2 s
2 p-
2 p
3 s
3 p-
3 p
3 d-
3 d
3
274 864 1164
3
274 864 1164
Loading Configuration Symmetry List File ...
there are 9 relativistic subshells;
1
2 2
there are 2328 relativistic CSFs;
... load complete;
Enter the list of transition specifications
e.g., E1,M2 or E1 M2 or E1;M2 :
>>M1
M1 transitions only between levels with different J?
>>n
.....
RTRANSITION: Execution complete.
>>mv 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj_LSJ3
*******************************************************************************
* RUN RTRANSITION FOR 2s22p3_2p5_3 TO COMPUTE M1 TRANSITION PARAMETERS*
* IN jj-COUPLING FOR THOSE LEVELS WHICH HAVE 1s, 2s, AND 2p *
* SHELLS IN IDENTIFICATION AND FOR THE REST LS-COUPLING *
* INPUT FILES: isodata, 2s22p3_2p5_3.c, 2s22p3_2p5_3.bw, *
* 2s22p3_2p5_3.cbm, *
* OUTPUT FILE: 2s22p3_2p5_3.2s22p3_2p5_3.ct, *
* 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj *
* 2s22p3_2p5_3.2s22p3_2p5_3.+1T (angular file) *
* *
* COPY THE 2s22p3_2p5_3.coup3.jj.lbl TO 2s22p3_2p5_3.lsj.lbl *
* IT IS THE LATTER FILE THAT IS READ AND USED BY THE TRANSITION *
* PROGRAM *
*******************************************************************************
>>cp 2s22p3_2p5_3.coup3.jj.lbl 2s22p3_2p5_3.lsj.lbl
>>rtransition
RTRANSITION
This program computes transition parameters from
transformed wave functions
Input files: isodata, name1.c, name1.bw, name1.(c)bm
name2.c, name2.bw, name2.(c)bm
optional, name1.lsj.lbl, name2.lsj.lbl
name1.name2.KT (optional angular files)
Output files: name1.name2.(c)t
optional, name1.name2.(c)t.lsj
name1.name2.KT (angular files)
Here K is parity and rank of transition: -1,+1 etc
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>2s22p3_2p5_3
Name of the Final state
>>2s22p3_2p5_3
MRGCSL: Execution begins ...
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
1 s
2 s
2 p-
2 p
3 s
3 p-
3 p
3 d-
3 d
3
274 864 1164
3
274 864 1164
Loading Configuration Symmetry List File ...
there are 9 relativistic subshells;
1
2 2
there are 2328 relativistic CSFs;
... load complete;
Enter the list of transition specifications
e.g., E1,M2 or E1 M2 or E1;M2 :
>>M1
M1 transitions only between levels with different J?
>>n
.....
RTRANSITION: Execution complete.
*******************************************************************************
* GIVE THE TRANSITION FILE A NEW APPROPRIATE NAME FOR LATER USE *
*******************************************************************************
>>mv 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj_jj
*******************************************************************************
* RUN RTRANSITION FOR 2s22p3_2p5_3 TO COMPUTE M1 TRANSITION PARAMETERS*
* IN cLSJ3-COUPLING FOR THOSE LEVELS WHICH HAVE 1s, 2s, AND 2p *
* SHELLS IN IDENTIFICATION AND FOR THE REST LS-COUPLING *
* INPUT FILES: isodata, 2s22p3_2p5_3.c, 2s22p3_2p5_3.bw, *
* 2s22p3_2p5_3.cbm, *
* OUTPUT FILE: 2s22p3_2p5_3.2s22p3_2p5_3.ct, *
* 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj *
* 2s22p3_2p5_3.2s22p3_2p5_3.+1T (angular file) *
* *
* COPY THE 2s22p3_2p5_3.coup3.cLSJ3.lbl TO 2s22p3_2p5_3.lsj.lbl *
* IT IS THE LATTER FILE THAT IS READ AND USED BY THE TRANSITION *
* PROGRAM *
*******************************************************************************
>>cp 2s22p3_2p5_3.coup3.cLSJ3.lbl 2s22p3_2p5_3.lsj.lbl
>>rtransition
RTRANSITION
This program computes transition parameters from
transformed wave functions
Input files: isodata, name1.c, name1.bw, name1.(c)bm
name2.c, name2.bw, name2.(c)bm
optional, name1.lsj.lbl, name2.lsj.lbl
name1.name2.KT (optional angular files)
Output files: name1.name2.(c)t
optional, name1.name2.(c)t.lsj
name1.name2.KT (angular files)
Here K is parity and rank of transition: -1,+1 etc
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>2s22p3_2p5_3
Name of the Final state
>>2s22p3_2p5_3
MRGCSL: Execution begins ...
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 1164 relativistic CSFs;
... load complete;
1 s
2 s
2 p-
2 p
3 s
3 p-
3 p
3 d-
3 d
3
274 864 1164
3
274 864 1164
Loading Configuration Symmetry List File ...
there are 9 relativistic subshells;
1
2 2
there are 2328 relativistic CSFs;
... load complete;
Enter the list of transition specifications
e.g., E1,M2 or E1 M2 or E1;M2 :
>>M1
M1 transitions only between levels with different J?
>>n
.....
RTRANSITION: Execution complete.
*******************************************************************************
* GIVE THE TRANSITION FILE A NEW APPROPRIATE NAME FOR LATER USE *
*******************************************************************************
>>mv 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj 2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj_cLSJ3
6.4. Fourth Example: States in Fe XV Using MPI
The fourth example is to determine the energies for the 10 states belonging to the three even configurations and the 16 states belonging to the two odd configurations . In addition, the E1, M2 and M1 transition data should be computed. The NIST table for these states is shown below.
------------------------------------------------------------
Configuration | Term | J | Level |
--------------------|--------|-----|-----------------------|
| | | |
2p6.3s2 | 1S | 0 | 0 |
| | | |
3s.3p | 3P* | 0 | 233842 |
| | 1 | 239660 |
| | 2 | 253820 |
| | | |
3s.3p | 1P* | 1 | 351911 |
| | | |
3p2 | 3P | 0 | 554524 |
| | | |
3p2 | 1D | 2 | 559600 |
| | | |
3p2 | 3P | 1 | 564602 |
| | 2 | 581803 |
| | | |
3p2 | 1S | 0 | 659627 |
| | | |
3s.3d | 3D | 1 | 678772 |
| | 2 | 679785 |
| | 3 | 681416 |
| | | |
3s.3d | 1D | 2 | 762093 |
| | | |
3p.3d | 3F* | 2 | 928241 |
| | 3 | 938126 |
| | | |
3p.3d | 1D* | 2 | 948513 |
| | | |
3p.3d | 3F* | 4 | 949658 |
| | | |
3p.3d | 3D* | 1 | 982868 |
| | | |
3p.3d | 3P* | 2 | 983514 |
| | | |
3p.3d | 3D* | 3 | 994852 |
| | | |
3p.3d | 3P* | 0 | 995889 |
| | 1 | 996243 |
| | | |
3p.3d | 3D* | 2 | 996623 |
| | | |
3p.3d | 1F* | 3 | 1062515 |
| | | |
3p.3d | 1P* | 1 | 1074887 |
------------------------------------------------------------
The starting point is two separate rmcdhf calculations for the even and odd reference states, respectively. Then one layer of correlation orbitals is included describing valence–valence and core–valence correlation.
-
Define nuclear data.
-
Obtain common spectroscopic orbitals for the even parity MR set
- (a)
-
Generate list of CSFs describing the even states belonging to
- (b)
-
Perform angular integration.
- (c)
-
Generate initial estimates of radial orbitals.
- (d)
-
Perform SCF calculation on the weighted average of the even states.
- (e)
-
Save output to evenmr.
-
Improve even states
- (a)
-
Generate valence–valence and core–valence CSF expansions
- (b)
-
Perform angular integration using MPI.
- (c)
-
Generate initial estimates of radial orbitals.
- (d)
-
Perform SCF MPI calculation on the weighted average of the even states.
- (e)
-
Save output to even4.
- (f)
-
Perform rci MPI calculation in which Breit and QED effects are added.
-
Run jj2lsj to transform to -coupling.
-
Obtain common spectroscopic orbitals for the odd parity MR set
- (a)
-
Generate list of CSFs describing the even states belonging to
- (b)
-
Perform angular integration.
- (c)
-
Generate initial estimates of radial orbitals.
- (d)
-
Perform SCF calculation on the weighted average of the odd states.
- (e)
-
Save output to oddmr.
-
Improve odd states.
- (a)
-
Generate valence–valence and core–valence CSF expansions.
- (b)
-
Perform angular integration using MPI.
- (c)
-
Generate initial estimates of radial orbitals.
- (d)
-
Perform SCF MPI calculation on the weighted average of the odd states.
- (e)
-
Save output to odd4.
- (f)
-
Perform rci MPI calculation in which Breit and QED effects are added.
-
Run jj2lsj to transform to -coupling.
-
Run rlevels, rlevelseV to view energy separations in -coupling scheme.
-
Compute properties.
- (a)
-
Compute transition rates from the rci wave functions. Computation in two steps: biorthonormal transformation and then evaluation of transition matrix elements using standard Racah algebra methods. Both steps use MPI code.
We intend to run rangular_mpi, rmcdhf_mpi, and rci_mpi using four processors and a disks file defining the location of the directory (on the disk) in which temporary data should be stored. On our computer, we will run the MPI jobs on four processors from a directory called
/home/tspejo/GRASP2018/grasptest/example4/script
and store the temporary data in
/home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
The disks file corresponding to this case is shown below.
’/home/tspejo/GRASP2018/grasptest/example4/script’
’/home/tspejo/GRASP2018/grasptest/example4/tmp_mpi’
’/home/tspejo/GRASP2018/grasptest/example4/tmp_mpi’
’/home/tspejo/GRASP2018/grasptest/example4/tmp_mpi’
’/home/tspejo/GRASP2018/grasptest/example4/tmp_mpi’
If we use four processors for the MPI run, the full path to the directory storing temporary data should be given four times in the disks file. If we use eight processors for the MPI run, the full path to the directory storing temporary data should be given eight times, etc. The directory storing temporary data can be anywhere in the file system, and need not be on the same level in the file system as the working directory.
Provided the disks file is set up correctly according to the file structure of the local computer, the cpath.f90 routine of the grasp mpi90 library automatically creates the directory in which temporary data are stored along with subdirectories 000, 001, 002 etc. named after the processors, starting with 0. On our system cpath.f90 creates
/home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
along with four subdirectories 000, 001, 002, 003 named after the four processors, starting with 0. If cpath.f90 fails to create the directories specified in the disks file then temporary data are stored in the directory specified by the MPI_TMP environment variable.
On some computer systems, the MPI libraries need to be loaded before the calculation starts. The commands for this depend on the system, but could look like
module add openmpi
Make sure you load MPI libraries for
gfortran. For additional runs using the MPI codes, see
Section 9.7.
In the test-runs, prompt marked by >> or >>3, for example, indicates that the user should input 3 and then strike the return key. When >> is followed by blanks, just strike the return key.
*******************************************************************************
* RUN RNUCLEUS TO GENERATE NUCLEAR DATA AND DEFINE RADIAL GRID *
* OUTPUT FILE: isodata *
*******************************************************************************
>>rnucleus
RNUCLEUS
This program defines nuclear data and the radial grid
Outputfile: isodata
Enter the atomic number:
>>26
Enter the mass number (0 if the nucleus is to be modelled as a point source:
>>56
The default root mean squared radius is 3.7376999855041504 fm; (Angeli)
the default nuclear skin thickness is 2.2999999999999998 fm;
Revise these values?
>>n
Enter the mass of the neutral atom (in amu) (0 if the nucleus is to be static):
>>55.845
Enter the nuclear spin quantum number (I) (in units of h / 2 pi):
>>1
Enter the nuclear dipole moment (in nuclear magnetons):
>>1
Enter the nuclear quadrupole moment (in barns):
>>1
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE LIST OF CSFs FOR *
* CONFIGURATIONS 3s(2), 3p(2), 3s3p *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,i)2p(6,i)3s(2,i)
Give configuration 2
>>2s(2,i)2p(6,i)3p(2,i)
Give configuration 3
>>2s(2,i)2p(6,i)3s(1,i)3d(1,i)
Give configuration 4
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>3s,3p,3d
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,6
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>n
.........
4 blocks were created
block J/P NCSF
1 0+ 3
2 1+ 2
3 2+ 4
4 3+ 1
*******************************************************************************
* COPY FILES *
* IT IS ADVISABLE TO SAVE THE rcsfgenerate.log FILE TO HAVE A *
* RECORD ON HOW THE LIST OF CSFs WAS CREATED *
*******************************************************************************
>>cp rcsfgenerate.log evenmr.exc
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.alog, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
.....
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp, rwfnestimate.log *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.3098D+03 0.2951D+03 0.1000D+01 0.5759D-01 328 T-F
2s 0.6428D+02 0.1015D+03 0.1000D+01 0.2385D+00 346 T-F
2p- 0.6284D+02 0.7744D+00 0.1000D+01 0.2003D+00 346 T-F
2p 0.6217D+02 0.6941D+03 0.2000D+01 0.2028D+00 346 T-F
3s 0.2358D+02 0.5152D+02 0.1000D+01 0.5708D+00 358 T-F
3p- 0.2295D+02 0.4217D+00 0.1000D+01 0.5370D+00 358 T-F
3p 0.2278D+02 0.3794D+03 0.2000D+01 0.5409D+00 358 T-F
3d- 0.2170D+02 0.5565D+00 0.2000D+01 0.4629D+00 358 T-F
3d 0.2165D+02 0.6259D+03 0.3000D+01 0.4642D+00 358 T-F
RWFNESTIMATE: Execution complete.
*******************************************************************************
* RUN RMCDHF TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: ORBITALS BUILDING REFERENCE STATES ARE REQUIRED TO HAVE *
* THE CORRECT NUMBER OF NODES. THEY ARE REFERRED TO AS SPECTROSCOPIC *
* ORBITALS. IN THIS RUN WE VARY 1s,2s,2p,3s,3p,3d AND THEY ARE ALL *
* SPECTROSCOPIC. WE CAN USE WILD CARDS FOR SPECIFYING ORBITALS *
* *
* NOTE: INSTEAD OF SAYING THAT WE SHOULD OPTIMIZE ON, FOR EXAMPLE, *
* THE STATES 1,2,3,4 WE CAN WRITE 1-4 MEANING THE SAME THING *
* *
*******************************************************************************
>>rmcdhf
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Loading CSF File for ALL blocks
There are 10 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 4 blocks (block J/Parity NCF):
1 0+ 3 2 1+ 2 3 2+ 4 4 3+ 1
Enter ASF serial numbers for each block
Block 1 ncf = 3 id = 0+
>>1-3
Block 2 ncf = 2 id = 1+
>>1-2
Block 3 ncf = 4 id = 2+
>>1-4
Block 4 ncf = 1 id = 3+
>>1
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Enter orbitals to be varied (Updating order)
>>*
Which of these are spectroscopic orbitals?
>>*
Enter the maximum number of SCF cycles:
>>100
..............
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog name.log *
*******************************************************************************
>>rsave evenmr
Created evenmr.w, evenmr.c, evenmr.m, evenmr.sum, evenmr.alog and evenmr.log
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE n = 4 VALENCE-VALENCE AND *
* CORE-VALENCE LIST *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,i)2p(6,5)3s(2,*)
Give configuration 2
>>2s(2,1)2p(6,i)3s(2,*)
Give configuration 3
>>2s(2,i)2p(6,5)3p(2,*)
Give configuration 4
>>2s(2,1)2p(6,i)3p(2,*)
Give configuration 5
>>2s(2,i)2p(6,5)3s(1,*)3d(1,*)
Give configuration 6
>>2s(2,1)2p(6,i)3s(1,*)3d(1,*)
Give configuration 7
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>4s,4p,4d,4f
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,6
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
.........
4 blocks were found
block J/P NCSF
1 0+ 556
2 1+ 1448
3 2+ 1898
4 3+ 1810
*******************************************************************************
* COPY FILES *
* IT IS ADVISABLE TO SAVE THE rcsfgenerate.log FILE TO HAVE A *
* RECORD ON HOW THE LIST OF CSFs WAS CREATED *
*******************************************************************************
>>cp rcsfgenerate.log even4.exc
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR_MPI USING 4 PROCESSES TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.alog, mcp.30, mcp.31,..IN 000, 001, 002, 003 *
*******************************************************************************
>>mpirun -np 4 rangular_mpi
====================================================
RANGULAR_MPI: Execution Begins ...
====================================================
Participating nodes:
Host: atom1 ID: 000
Host: atom1 ID: 001
Host: atom1 ID: 002
Host: atom1 ID: 003
Date and Time:
atom1: Date: 20140812 Time: 011040.566 Zone: +0200
atom1: Date: 20140812 Time: 011040.566 Zone: +0200
atom1: Date: 20140812 Time: 011040.566 Zone: +0200
atom1: Date: 20140812 Time: 011040.566 Zone: +0200
Start Dir:
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
Serial I/O Dir (node-0 only):
atom1: /home/tspejo/GRASP2018/grasptest/example4/script
Work Dir (Parallel I/O):
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
Full interaction? (y/n)
>>y
.........
mpi stopped by node- 0 from RANGULAR_MPI: Execution complete.
mpi stopped by node- 2 from RANGULAR_MPI: Execution complete.
mpi stopped by node- 1 from RANGULAR_MPI: Execution complete.
mpi stopped by node- 3 from RANGULAR_MPI: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d 4s 4p- 4p 4d- 4d 4f- 4f
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>1
Enter the file name (Null then "rwfn.out")
Enter the list of relativistic subshells:
>>*
The following subshell radial wavefunctions remain to be estimated:
4s 4p- 4p 4d- 4d 4f- 4f
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4---Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.2803D+03 0.2923D+03 0.1000D+01 0.5839D-01 354 rwf
2s 0.4802D+02 0.9081D+02 0.1000D+01 0.2623D+00 358 rwf
2p- 0.4353D+02 0.6335D+00 0.1000D+01 0.2298D+00 358 rwf
2p 0.4305D+02 0.5671D+03 0.2000D+01 0.2326D+00 358 rwf
3s 0.1667D+02 0.4072D+02 0.1000D+01 0.6859D+00 362 rwf
3p- 0.1543D+02 0.2995D+00 0.1000D+01 0.6765D+00 363 rwf
3p 0.1534D+02 0.2693D+03 0.2000D+01 0.6810D+00 363 rwf
3d- 0.1358D+02 0.2668D+00 0.2000D+01 0.6260D+00 364 rwf
3d 0.1356D+02 0.3005D+03 0.3000D+01 0.6270D+00 364 rwf
4s 0.1141D+02 0.3095D+02 0.1000D+01 0.1086D+01 366 T-F
4p- 0.1110D+02 0.2579D+00 0.1000D+01 0.1061D+01 367 T-F
4p 0.1104D+02 0.2325D+03 0.2000D+01 0.1066D+01 367 T-F
4d- 0.1053D+02 0.3857D+00 0.2000D+01 0.1002D+01 367 T-F
4d 0.1051D+02 0.4342D+03 0.3000D+01 0.1004D+01 367 T-F
4f- 0.9897D+01 0.2129D+00 0.3000D+01 0.8833D+00 367 T-F
4f 0.9890D+01 0.2598D+03 0.4000D+01 0.8842D+00 367 T-F
RWFNESTIMATE: Execution complete.
*******************************************************************************
* RUN RMCDHF_MEM_MPI USING 4 PROCESSES TO OBTAIN SELF CONSISTENT *
* SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: FOR CORRELATION ORBITALS THERE ARE NO RESTRICTIONS ON THE *
* NUMBER OF NODES, I.E. THEY ARE NOT SPECTROSCOPIC. IN THIS RUN WE *
* VARY THE CORRELATION ORBITALS 4s,4p,4d,4f. NONE OF THESE ARE *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* 4* MEANS 4s, 4p-, 4p, 4d-, 4d, 4f-, 4f *
*******************************************************************************
>>mpirun -np 4 rmcdhf_mem_mpi
====================================================
RMCDHF_MPI: Execution Begins ...
====================================================
Participating nodes:
Host: atom1 ID: 000
Host: atom1 ID: 001
Host: atom1 ID: 002
Host: atom1 ID: 003
Date and Time:
atom1: Date: 20140812 Time: 011653.965 Zone: +0200
atom1: Date: 20140812 Time: 011653.965 Zone: +0200
atom1: Date: 20140812 Time: 011653.965 Zone: +0200
atom1: Date: 20140812 Time: 011653.965 Zone: +0200
Start Dir:
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
Serial I/O Dir (node-0 only):
atom1: /home/tspejo/GRASP2018/grasptest/example4/script
Work Dir (Parallel I/O):
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
Default settings? (y/n)
>>y
-----------------------------------------------------
Spin-angular coefficient are putting into the memory:
-----------------------------------------------------
Total memory on computer: 755.00 Gb
Free memory on computer: 630.17 Gb
Allocation for mcp.30:
Free memory on computer 630.16 Gb
Allocation for mcp.31:
Free memory on computer 630.16 Gb
Allocation for mcp.32:
Free memory on computer 630.16 Gb
Allocation for mcp.33:
Free memory on computer 630.15 Gb
Allocation for mcp.34:
Free memory on computer 630.14 Gb
Allocation for mcp.35:
Free memory on computer 630.13 Gb
Allocation for mcp.36:
Free memory on computer 630.13 Gb
Allocation for mcp.37:
Free memory on computer 630.13 Gb
Allocation for mcp.38:
Free memory on computer 630.13 Gb
Allocation for mcp.39:
Free memory on computer 630.13 Gb
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
Loading CSF File for ALL blocks
There are 5712 relativistic CSFs... load complete;
There are 4 blocks (block J/Parity NCF):
1 0+ 556 2 1+ 1448 3 2+ 1898 4 3+ 1810
Enter ASF serial numbers for each block
Block 1 ncf = 556 id = 0+
>>1-3
Block 2 ncf = 1448 id = 1+
>>1-2
Block 3 ncf = 1898 id = 2+
>>1-4
Block 4 ncf = 1810 id = 3+
>>1
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d 4s 4p- 4p 4d- 4d 4f- 4f
Enter orbitals to be varied (Updating order)
>>4*
Which of these are spectroscopic orbitals?
>>
Enter the maximum number of SCF cycles:
>>100
.....
mpi stopped by node- 1 from RMCDHF_MPI: Execution complete.
mpi stopped by node- 0 from RMCDHF_MPI: Execution complete.
mpi stopped by node- 3 from RMCDHF_MPI: Execution complete.
mpi stopped by node- 2 from RMCDHF_MPI: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog, name.log *
*******************************************************************************
>>rsave even4
Created even4.w, even4.c, even4.m, even4.sum, even4.alog and even4.log
*******************************************************************************
* RUN RCI_MPI USING 4 PROCESSES TO INCLUDE BREIT AND QED EFFECTS *
* INPUT FILES : isodata, even4.c, even4.w *
* OUTPUT FILES: even4.cm, even4.csum *
* *
* THE TRANSVERSE PHOTON FREQUENCIES CAN BE SET TO THE LOW FREQUENCY *
* LIMIT. RECOMMENDED IN CASES WHERE YOU HAVE CORRELATION ORBITALS *
* THE SELF ENERGY CORRECTION MAY FAIL FOR CORRELATION ORBITALS WITH *
* HIGH N. *
*******************************************************************************
>>mpirun -np 4 rci_mpi
====================================================
RCI_MPI: Execution Begins ...
====================================================
Participating nodes:
Host: atom1 ID: 000
Host: atom1 ID: 001
Host: atom1 ID: 002
Host: atom1 ID: 003
Date and Time:
atom1: Date: 20140812 Time: 012250.312 Zone: +0200
atom1: Date: 20140812 Time: 012250.312 Zone: +0200
atom1: Date: 20140812 Time: 012250.312 Zone: +0200
atom1: Date: 20140812 Time: 012250.312 Zone: +0200
Start Dir:
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
Serial I/O Dir (node-0 only):
atom1: /home/tspejo/GRASP2018/grasptest/example4/script
Work Dir (Parallel I/O):
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
Default settings?
>>y
Name of state:
>>even4
Block 1 , ncf = 556
Block 2 , ncf = 1448
Block 3 , ncf = 1898
Block 4 , ncf = 1810
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
Include contribution of H (Transverse)?
>>y
Modify all transverse photon frequencies?
>>y
Enter the scale factor:
>>1.d-6
Include H (Vacuum Polarisation)?
>>y
Include H (Normal Mass Shift)?
>>n
Include H (Specific Mass Shift)?
>>n
Estimate self-energy?
>>y
Largest n quantum number for including self-energy for orbital
n should be less or equal 8
>>4
There are 4 blocks (block J/Parity NCF):
1 0+ 556 2 1+ 1448 3 2+ 1898 4 3+ 1810
Enter ASF serial numbers for each block
Block 1 ncf = 556 id = 0+
>>1-3
Block 2 ncf = 1448 id = 1+
>>1-2
Block 3 ncf = 1898 id = 2+
>>1-4
Block 4 ncf = 1810 id = 3+
>>1
.....
mpi stopped by node- 0 from RCI_MPI: Execution complete.
mpi stopped by node- 3 from RCI_MPI: Execution complete.
mpi stopped by node- 1 from RCI_MPI: Execution complete.
mpi stopped by node- 2 from RCI_MPI: Execution complete.
*******************************************************************************
* RUN JJ2LSJ TO GET THE LSJ-COMPOSITION *
* INPUT FILE: even4.c, even4.cm *
* OUTPUT FILE: even4.lsj.lbl, even4.uni.lsj.lbl *
*******************************************************************************
>>jj2lsj
jj2lsj: Transformation of ASFs from a jj-coupled CSF basis
into an LSJ-coupled CSF basis (Fortran 95 version)
(C) Copyright by G. Gaigalas and Ch. F. Fischer,
(2017).
Input files: name.c, name.(c)m
Output files: name.lsj.lbl
(optional) name.lsj.c, name.lsj.j,
name.uni.lsj.lbl, name.uni.lsj.sum
Name of state
>>even4
Loading Configuration Symmetry List File ...
There are 16 relativistic subshells;
There are 5712 relativistic CSFs;
... load complete;
Mixing coefficients from a CI calc.?
>>y
Do you need a unique labeling? (y/n)
>>y
nelec = 12
ncftot = 5712
nw = 16
nblock = 4
block ncf nev 2j+1 parity
1 556 3 1 1
2 1448 2 3 1
3 1898 4 5 1
4 1810 1 7 1
Default settings? (y/n)
>>y
jj2lsj: Execution complete.
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE LIST OF CSFs FOR *
* CONFIGURATIONS 3s3p, 3p3d *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,i)2p(6,i)3s(1,i)3p(1,i)
Give configuration 2
>>2s(2,i)2p(6,i)3p(1,i)3d(1,i)
Give configuration 3
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>3s,3p,3d
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,8
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>n
.........
5 blocks were created
block J/P NCSF
1 0- 2
2 1- 5
3 2- 5
4 3- 3
5 4- 1
*******************************************************************************
* COPY FILES *
* IT IS ADVISABLE TO SAVE THE rcsfgenerate.log FILE TO HAVE A *
* RECORD ON HOW THE LIST OF CSFs WAS CREATED *
*******************************************************************************
>>cp rcsfgenerate.log oddmr.exc
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.alog, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
.....
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp, rwfnestimate.log *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.3098D+03 0.2951D+03 0.1000D+01 0.5759D-01 328 T-F
2s 0.6428D+02 0.1015D+03 0.1000D+01 0.2385D+00 346 T-F
2p- 0.6284D+02 0.7744D+00 0.1000D+01 0.2003D+00 346 T-F
2p 0.6217D+02 0.6941D+03 0.2000D+01 0.2028D+00 346 T-F
3s 0.2358D+02 0.5152D+02 0.1000D+01 0.5708D+00 358 T-F
3p- 0.2295D+02 0.4217D+00 0.1000D+01 0.5370D+00 358 T-F
3p 0.2278D+02 0.3794D+03 0.2000D+01 0.5409D+00 358 T-F
3d- 0.2170D+02 0.5565D+00 0.2000D+01 0.4629D+00 358 T-F
3d 0.2165D+02 0.6259D+03 0.3000D+01 0.4642D+00 358 T-F
RWFNESTIMATE: Execution complete.
*******************************************************************************
* RUN RMCDHF_MEM TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: ORBITALS BUILDING REFERENCE STATES ARE REQUIRED TO HAVE *
* THE CORRECT NUMBER OF NODES. THEY ARE REFERRED TO AS SPECTROSCOPIC *
* ORBITALS. IN THIS RUN WE VARY 1s,2s,2p,3s,3p,3d AND THEY ARE ALL *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
*******************************************************************************
>>rmcdhf_mem
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Loading CSF File for ALL blocks
There are 16 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 5 blocks (block J/Parity NCF):
1 0- 2 2 1- 5 3 2- 5 4 3- 3
5 4- 1
Enter ASF serial numbers for each block
Block 1 ncf = 2 id = 0-
>>1-2
Block 2 ncf = 5 id = 1-
>>1-5
Block 3 ncf = 5 id = 2-
>>1-5
Block 4 ncf = 3 id = 3-
>>1-3
Block 5 ncf = 1 id = 4-
>>1
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Enter orbitals to be varied (Updating order)
>>*
Which of these are spectroscopic orbitals?
>>*
Enter the maximum number of SCF cycles:
>>100
..............
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog, name.log *
*******************************************************************************
>>rsave oddmr
Created oddmr.w, oddmr.c, oddmr.m, oddmr.sum, oddmr.alog and oddmr.log
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE n = 4 VALENCE-VALENCE AND *
* CORE-VALENCE LIST *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,i)2p(6,5)3s(1,*)3p(1,*)
Give configuration 2
>>2s(2,1)2p(6,i)3s(1,*)3p(1,*)
Give configuration 3
>>2s(2,i)2p(6,5)3p(1,*)3d(1,*)
Give configuration 4
>>2s(2,1)2p(6,i)3p(1,*)3d(1,*)
Give configuration 5
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>4s,4p,4d,4f
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,8
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
.........
5 blocks were created
block J/P NCSF
1 0- 546
2 1- 1456
3 2- 1891
4 3- 1814
5 4- 1393
*******************************************************************************
* COPY FILES *
* IT IS ADVISABLE TO SAVE THE rcsfgenerate.log FILE TO HAVE A *
* RECORD ON HOW THE LIST OF CSFs WAS CREATED *
*******************************************************************************
>>cp rcsfgenerate.log odd4.exc
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR_MPI USING 4 PROCESSES TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.log, mcp.30, mcp.31,...IN 000, 001, 002, 003 *
*******************************************************************************
>>mpirun -np 4 rangular_mpi
====================================================
RANGULAR_MPI: Execution Begins ...
====================================================
Participating nodes:
Host: atom1 ID: 000
Host: atom1 ID: 001
Host: atom1 ID: 002
Host: atom1 ID: 003
Date and Time:
atom1: Date: 20140812 Time: 011040.566 Zone: +0200
atom1: Date: 20140812 Time: 011040.566 Zone: +0200
atom1: Date: 20140812 Time: 011040.566 Zone: +0200
atom1: Date: 20140812 Time: 011040.566 Zone: +0200
Start Dir:
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
Serial I/O Dir (node-0 only):
atom1: /home/tspejo/GRASP2018/grasptest/example4/script
Work Dir (Parallel I/O):
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
Full interaction? (y/n)
>>y
.........
mpi stopped by node- 0 from RANGULAR_MPI: Execution complete.
mpi stopped by node- 2 from RANGULAR_MPI: Execution complete.
mpi stopped by node- 1 from RANGULAR_MPI: Execution complete.
mpi stopped by node- 3 from RANGULAR_MPI: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d 4s 4p- 4p 4d- 4d 4f- 4f
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>1
Enter the file name (Null then "rwfn.out")
>>
Enter the list of relativistic subshells:
>>*
The following subshell radial wavefunctions remain to be estimated:
4s 4p- 4p 4d- 4d 4f- 4f
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.2803D+03 0.2923D+03 0.1000D+01 0.5838D-01 351 rwf
2s 0.4797D+02 0.9077D+02 0.1000D+01 0.2624D+00 357 rwf
2p- 0.4349D+02 0.6336D+00 0.1000D+01 0.2298D+00 358 rwf
2p 0.4301D+02 0.5672D+03 0.2000D+01 0.2326D+00 358 rwf
3s 0.1680D+02 0.4086D+02 0.1000D+01 0.6840D+00 362 rwf
3p- 0.1542D+02 0.2993D+00 0.1000D+01 0.6773D+00 363 rwf
3p 0.1533D+02 0.2692D+03 0.2000D+01 0.6818D+00 363 rwf
3d- 0.1358D+02 0.2660D+00 0.2000D+01 0.6264D+00 364 rwf
3d 0.1357D+02 0.2997D+03 0.3000D+01 0.6274D+00 364 rwf
4s 0.1141D+02 0.3095D+02 0.1000D+01 0.1086D+01 366 T-F
4p- 0.1110D+02 0.2579D+00 0.1000D+01 0.1061D+01 367 T-F
4p 0.1104D+02 0.2325D+03 0.2000D+01 0.1066D+01 367 T-F
4d- 0.1053D+02 0.3857D+00 0.2000D+01 0.1002D+01 367 T-F
4d 0.1051D+02 0.4342D+03 0.3000D+01 0.1004D+01 367 T-F
4f- 0.9897D+01 0.2129D+00 0.3000D+01 0.8833D+00 367 T-F
4f 0.9890D+01 0.2598D+03 0.4000D+01 0.8842D+00 367 T-F
RWFNESTIMATE: Execution complete.
*******************************************************************************
* RUN RMCDHF_MEM_MPI USING 4 PROCESSES TO OBTAIN SELF CONSISTENT *
* SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: FOR CORRELATION ORBITALS THERE ARE NO RESTRICTIONS ON THE *
* NUMBER OF NODES, I.E. THEY ARE NOT SPECTROSCOPIC. IN THIS RUN WE *
* VARY THE CORRELATION ORBITALS 4s,4p,4d,4f. NONE OF THESE ARE *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* 4* MEANS 4s, 4p-, 4p, 4d-, 4d, 4f-, 4f *
*******************************************************************************
>>mpirun -np 4 rmcdhf_mem_mpi
====================================================
RMCDHF_MPI: Execution Begins ...
====================================================
Participating nodes:
Host: atom1 ID: 000
Host: atom1 ID: 001
Host: atom1 ID: 002
Host: atom1 ID: 003
Date and Time:
atom1: Date: 20140812 Time: 020654.423 Zone: +0200
atom1: Date: 20140812 Time: 020654.422 Zone: +0200
atom1: Date: 20140812 Time: 020654.422 Zone: +0200
atom1: Date: 20140812 Time: 020654.422 Zone: +0200
Start Dir:
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
Serial I/O Dir (node-0 only):
atom1: /home/tspejo/GRASP2018/grasptest/example4/script
Work Dir (Parallel I/O):
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
Default settings? (y/n)
>>y
-----------------------------------------------------
Spin-angular coefficient are putting into the memory:
-----------------------------------------------------
Total memory on computer: 755.00 Gb
Free memory on computer: 630.12 Gb
Allocation for mcp.30:
Free memory on computer 630.12 Gb
Allocation for mcp.31:
Free memory on computer 630.12 Gb
Allocation for mcp.32:
Free memory on computer 630.12 Gb
Allocation for mcp.33:
Free memory on computer 630.11 Gb
Allocation for mcp.34:
Free memory on computer 630.09 Gb
Allocation for mcp.35:
Free memory on computer 630.08 Gb
Allocation for mcp.36:
Free memory on computer 630.08 Gb
Allocation for mcp.37:
Free memory on computer 630.08 Gb
Allocation for mcp.38:
Free memory on computer 630.08 Gb
Allocation for mcp.39:
Free memory on computer 630.08 Gb
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
Loading CSF File for ALL blocks
There are 7100 relativistic CSFs... load complete;
There are 5 blocks (block J/Parity NCF):
1 0- 546 2 1- 1456 3 2- 1891 4 3- 1814
5 4- 1393
Enter ASF serial numbers for each block
Block 1 ncf = 546 id = 0-
>>1-2
Block 2 ncf = 1456 id = 1-
>>1-5
Block 3 ncf = 1891 id = 2-
>>1-5
Block 4 ncf = 1814 id = 3-
>>1-3
Block 5 ncf = 1393 id = 4-
>>1
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d 4s 4p- 4p 4d- 4d 4f- 4f
Enter orbitals to be varied (Updating order)
>>4*
Which of these are spectroscopic orbitals?
>>
Enter the maximum number of SCF cycles:
>>100
............
mpi stopped by node- 0 from RMCDHF_MPI: Execution complete.
mpi stopped by node- 2 from RMCDHF_MPI: Execution complete.
mpi stopped by node- 1 from RMCDHF_MPI: Execution complete.
mpi stopped by node- 3 from RMCDHF_MPI: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog, name.log *
*******************************************************************************
>>rsave odd4
Created odd4.w, odd4.c, odd4.m, odd4.sum, odd4.alog and odd4.log
*******************************************************************************
* RUN RCI_MPI USING 4 PROCESSES TO INCLUDE BREIT AND QED EFFECTS *
* INPUT FILES : isodata, odd4.c, odd4.w *
* OUTPUT FILES: odd4.cm, odd4.csum *
* *
* THE TRANSVERSE PHOTON FREQUENCIES CAN BE SET TO THE LOW FREQUENCY *
* LIMIT. RECOMMENDED IN CASES WHERE YOU HAVE CORRELATION ORBITALS *
* THE SELF ENERGY CORRECTION MAY FAIL FOR CORRELATION ORBITALS WITH *
* HIGH N. *
*******************************************************************************
>>mpirun -np 4 rci_mpi
====================================================
RCI_MPI: Execution Begins ...
====================================================
Participating nodes:
Host: atom1 ID: 000
Host: atom1 ID: 001
Host: atom1 ID: 002
Host: atom1 ID: 003
Date and Time:
atom1: Date: 20140812 Time: 021251.038 Zone: +0200
atom1: Date: 20140812 Time: 021251.038 Zone: +0200
atom1: Date: 20140812 Time: 021251.038 Zone: +0200
atom1: Date: 20140812 Time: 021251.038 Zone: +0200
Start Dir:
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
atom1: /GRASP2018/grasptest/example4/script
Serial I/O Dir (node-0 only):
atom1: /home/tspejo/GRASP2018/grasptest/example4/script
Work Dir (Parallel I/O):
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
atom1: /home/tspejo/GRASP2018/grasptest/example4/tmp_mpi
Default settings?
>>y
Name of state:
>>odd4
Block 1 , ncf = 546
Block 2 , ncf = 1456
Block 3 , ncf = 1891
Block 4 , ncf = 1814
Block 5 , ncf = 1393
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
Include contribution of H (Transverse)?
>>y
Modify all transverse photon frequencies?
>>y
Enter the scale factor:
>>1.d-6
Include H (Vacuum Polarisation)?
>>y
Include H (Normal Mass Shift)?
>>n
Include H (Specific Mass Shift)?
>>n
Estimate self-energy?
>>y
Largest n quantum number for including self-energy for orbital
n should be less or equal 8
>>4
There are 5 blocks (block J/Parity NCF):
1 0- 546 2 1- 1456 3 2- 1891 4 3- 1814
5 4- 1393
Enter ASF serial numbers for each block
Block 1 ncf = 546 id = 0-
>>1-2
Block 2 ncf = 1456 id = 1-
>>1-5
Block 3 ncf = 1891 id = 2-
>>1-5
Block 4 ncf = 1814 id = 3-
>>1-3
Block 5 ncf = 1393 id = 4-
>>1
....
mpi stopped by node- 0 from RCI_MPI: Execution complete.
mpi stopped by node- 2 from RCI_MPI: Execution complete.
mpi stopped by node- 3 from RCI_MPI: Execution complete.
mpi stopped by node- 1 from RCI_MPI: Execution complete.
*******************************************************************************
* RUN JJ2LSJ TO GET THE LSJ-COMPOSITION *
* INPUT FILE: odd4.c, odd4.cm *
* OUTPUT FILE: odd4.lsj.lbl, odd4.uni.lsj.lbl *
*******************************************************************************
>>jj2lsj
jj2lsj: Transformation of ASFs from a jj-coupled CSF basis
into an LSJ-coupled CSF basis (Fortran 95 version)
(C) Copyright by G. Gaigalas and Ch. F. Fischer,
(2017).
Input files: name.c, name.(c)m
Output files: name.lsj.lbl
(optional) name.lsj.c, name.lsj.j,
name.uni.lsj.lbl, name.uni.lsj.sum
Name of state
>>odd4
Loading Configuration Symmetry List File ...
There are 16 relativistic subshells;
There are 7100 relativistic CSFs;
... load complete;
Mixing coefficients from a CI calc.?
>>y
Do you need a unique labeling? (y/n)
>>y
nelec = 12
ncftot = 7100
nw = 16
nblock = 5
block ncf nev 2j+1 parity
1 546 2 1 -1
2 1456 5 3 -1
3 1891 5 5 -1
4 1814 3 7 -1
5 1393 1 9 -1
Default settings? (y/n)
>>y
jj2lsj: Execution complete.
*******************************************************************************
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS *
* NOTE: SINCE LSJ-INFORMATION NOW IS AVAILABLE OUTPUT LABELS *
* WILL BE IN LSJ-COUPLING *
*******************************************************************************
>>rlevels even4.cm odd4.cm
nblock = 4 ncftot = 5712 nw = 16 nelec = 12
nblock = 5 ncftot = 7100 nw = 16 nelec = 12
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 0 + -1182.4117992 0.00 0.00 2s(2).2p(6).3s(2)_1S
2 1 0 - -1181.3459632 233923.97 233923.97 2s(2).2p(6).3s_2S.3p_3P
3 1 1 - -1181.3193175 239772.02 5848.05 2s(2).2p(6).3s_2S.3p_3P
4 1 2 - -1181.2548318 253925.01 14152.99 2s(2).2p(6).3s_2S.3p_3P
5 2 1 - -1180.8025239 353195.12 99270.11 2s(2).2p(6).3s_2S.3p_1P
6 2 0 + -1179.8828042 555050.24 201855.12 2s(2).2p(6).3p(2)3P2_3P
7 1 2 + -1179.8592139 560227.72 5177.47 2s(2).2p(6).3p(2)1D2_1D
8 1 1 + -1179.8374539 565003.49 4775.77 2s(2).2p(6).3p(2)3P2_3P
9 2 2 + -1179.7587696 582272.71 17269.22 2s(2).2p(6).3p(2)3P2_3P
10 3 0 + -1179.3935747 662423.72 80151.02 2s(2).2p(6).3p(2)1S0_1S
11 2 1 + -1179.3158027 679492.71 17068.98 2s(2).2p(6).3s_2S.3d_3D
12 3 2 + -1179.3111395 680516.16 1023.45 2s(2).2p(6).3s_2S.3d_3D
13 1 3 + -1179.3038352 682119.26 1603.10 2s(2).2p(6).3s_2S.3d_3D
14 4 2 + -1178.9201602 766326.18 84206.92 2s(2).2p(6).3s_2S.3d_1D
15 2 2 - -1178.1773931 929344.72 163018.54 2s(2).2p(6).3p_2P.3d_3F
16 1 3 - -1178.1321370 939277.28 9932.56 2s(2).2p(6).3p_2P.3d_3F
17 3 2 - -1178.0860896 949383.53 10106.25 2s(2).2p(6).3p_2P.3d_1D
18 1 4 - -1178.0797665 950771.28 1387.75 2s(2).2p(6).3p_2P.3d_3F
19 3 1 - -1177.9273160 984230.30 33459.02 2s(2).2p(6).3p_2P.3d_3D
20 4 2 - -1177.9244263 984864.51 634.21 2s(2).2p(6).3p_2P.3d_3P
21 2 3 - -1177.8730161 996147.75 11283.24 2s(2).2p(6).3p_2P.3d_3D
22 2 0 - -1177.8671996 997424.33 1276.58 2s(2).2p(6).3p_2P.3d_3P
23 4 1 - -1177.8658739 997715.29 290.96 2s(2).2p(6).3p_2P.3d_3P
24 5 2 - -1177.8645522 998005.36 290.07 2s(2).2p(6).3p_2P.3d_3D
25 3 3 - -1177.5443213 1068287.93 70282.57 2s(2).2p(6).3p_2P.3d_1F
26 5 1 - -1177.4837515 1081581.45 13293.52 2s(2).2p(6).3p_2P.3d_1P
------------------------------------------------------------------------------------------
We compare with the energy levels given in the NIST database.
------------------------------------------------------------
Configuration | Term | J | Level |
--------------------|--------|-----|-----------------------|
| | | |
2p6.3s2 | 1S | 0 | 0 |
| | | |
3s.3p | 3P* | 0 | 233842 |
| | 1 | 239660 |
| | 2 | 253820 |
| | | |
3s.3p | 1P* | 1 | 351911 |
| | | |
3p2 | 3P | 0 | 554524 |
| | | |
3p2 | 1D | 2 | 559600 |
| | | |
3p2 | 3P | 1 | 564602 |
| | 2 | 581803 |
| | | |
3p2 | 1S | 0 | 659627 |
| | | |
3s.3d | 3D | 1 | 678772 |
| | 2 | 679785 |
| | 3 | 681416 |
| | | |
3s.3d | 1D | 2 | 762093 |
| | | |
3p.3d | 3F* | 2 | 928241 |
| | 3 | 938126 |
| | | |
3p.3d | 1D* | 2 | 948513 |
| | | |
3p.3d | 3F* | 4 | 949658 |
| | | |
3p.3d | 3D* | 1 | 982868 |
| | | |
3p.3d | 3P* | 2 | 983514 |
| | | |
3p.3d | 3D* | 3 | 994852 |
| | | |
3p.3d | 3P* | 0 | 995889 |
| | 1 | 996243 |
| | | |
3p.3d | 3D* | 2 | 996623 |
| | | |
3p.3d | 1F* | 3 | 1062515 |
| | | |
3p.3d | 1P* | 1 | 1074887 |
------------------------------------------------------------
Please note that this is just a very small example calculation. The agreement between theory and experiment is improved when the active set is extended. If desired, we may display the energy separations in eV by running rlevelseV.
*******************************************************************************
* RUN RLEVELSEV TO VIEW ENERGIES AND ENERGY SEPARATIONS IN EV *
* NOTE: SINCE LSJ-INFORMATION NOW IS AVAILABLE OUTPUT LABELS *
* WILL BE IN LSJ-COUPLING *
*******************************************************************************
>>rlevelseV even4.cm odd4.cm
nblock = 4 ncftot = 5712 nw = 16 nelec = 12
nblock = 5 ncftot = 7100 nw = 16 nelec = 12
Energy levels for ...
Rydberg constant is 109737.31569
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (eV) (eV)
------------------------------------------------------------------------------------------
1 1 0 + -1182.4117992 0.00000 0.00000 2s(2).2p(6).3s(2)_1S
2 1 0 - -1181.3459632 29.00288 29.00288 2s(2).2p(6).3s_2S.3p_3P
3 1 1 - -1181.3193175 29.72794 0.72507 2s(2).2p(6).3s_2S.3p_3P
4 1 2 - -1181.2548318 31.48269 1.75475 2s(2).2p(6).3s_2S.3p_3P
5 2 1 - -1180.8025239 43.79061 12.30792 2s(2).2p(6).3s_2S.3p_1P
6 2 0 + -1179.8828042 68.81746 25.02685 2s(2).2p(6).3p(2)3P2_3P
7 1 2 + -1179.8592139 69.45938 0.64192 2s(2).2p(6).3p(2)1D2_1D
8 1 1 + -1179.8374539 70.05150 0.59212 2s(2).2p(6).3p(2)3P2_3P
9 2 2 + -1179.7587696 72.19261 2.14111 2s(2).2p(6).3p(2)3P2_3P
10 3 0 + -1179.3935747 82.13007 9.93746 2s(2).2p(6).3p(2)1S0_1S
11 2 1 + -1179.3158027 84.24636 2.11628 2s(2).2p(6).3s_2S.3d_3D
12 3 2 + -1179.3111395 84.37325 0.12689 2s(2).2p(6).3s_2S.3d_3D
13 1 3 + -1179.3038352 84.57201 0.19876 2s(2).2p(6).3s_2S.3d_3D
14 4 2 + -1178.9201602 95.01234 10.44033 2s(2).2p(6).3s_2S.3d_1D
15 2 2 - -1178.1773931 115.22406 20.21172 2s(2).2p(6).3p_2P.3d_3F
16 1 3 - -1178.1321370 116.45554 1.23148 2s(2).2p(6).3p_2P.3d_3F
17 3 2 - -1178.0860896 117.70856 1.25302 2s(2).2p(6).3p_2P.3d_1D
18 1 4 - -1178.0797665 117.88061 0.17206 2s(2).2p(6).3p_2P.3d_3F
19 3 1 - -1177.9273160 122.02900 4.14839 2s(2).2p(6).3p_2P.3d_3D
20 4 2 - -1177.9244263 122.10764 0.07863 2s(2).2p(6).3p_2P.3d_3P
21 2 3 - -1177.8730161 123.50658 1.39894 2s(2).2p(6).3p_2P.3d_3D
22 2 0 - -1177.8671996 123.66486 0.15828 2s(2).2p(6).3p_2P.3d_3P
23 4 1 - -1177.8658739 123.70093 0.03607 2s(2).2p(6).3p_2P.3d_3P
24 5 2 - -1177.8645522 123.73689 0.03596 2s(2).2p(6).3p_2P.3d_3D
25 3 3 - -1177.5443213 132.45082 8.71393 2s(2).2p(6).3p_2P.3d_1F
26 5 1 - -1177.4837515 134.09901 1.64819 2s(2).2p(6).3p_2P.3d_1P
------------------------------------------------------------------------------------------
*******************************************************************************
* RUN RBIOTRANSFORM_MPI USING 4 PROCESSES *
* INPUT FILES: even4.c, even4.w, even4.cm *
* odd4.c, odd4.w, odd4.cm, isodata *
* OUPUT FILES: even4.bw, even4.cbm, odd4.bw, odd4.cbm *
*******************************************************************************
>>mpirun -np 4 rbiotransform_mpi
====================================================
RBIOTRANSFORM_MPI: Execution Begins ...
====================================================
Participating nodes:
Host: per-vaio ID: 000
Host: per-vaio ID: 001
Host: per-vaio ID: 002
Host: per-vaio ID: 003
Date and Time:
per-vaio: Date: 20141120 Time: 002539.729 Zone: +0100
per-vaio: Date: 20141120 Time: 002539.729 Zone: +0100
per-vaio: Date: 20141120 Time: 002539.729 Zone: +0100
per-vaio: Date: 20141120 Time: 002539.729 Zone: +0100
Start Dir:
per-vaio: /home/per/graspruns/FeXIII
per-vaio: /home/per/graspruns/FeXIII
per-vaio: /home/per/graspruns/FeXIII
per-vaio: /home/per/graspruns/FeXIII
Serial I/O Dir (node-0 only):
per-vaio: /home/per/graspruns/FeXIII
Work Dir (Parallel I/O):
per-vaio: /home/per/tmp_mpi
per-vaio: /home/per/tmp_mpi
per-vaio: /home/per/tmp_mpi
per-vaio: /home/per/tmp_mpi
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>even4
Name of the Final state
>>odd4
Transformation of all J symmetries?
>>y
.....
mpi stopped by node- 0 from RBIOTRANSFORM_MPI: Execution complete.
mpi stopped by node- 1 from RBIOTRANSFORM_MPI: Execution complete.
mpi stopped by node- 2 from RBIOTRANSFORM_MPI: Execution complete.
mpi stopped by node- 3 from RBIOTRANSFORM_MPI: Execution complete.
*******************************************************************************
* RUN RTRANSITION_MPI USING 4 PROCESSES *
* INPUT FILES: even4.c, even4.bw, even4.cbm *
* odd4.c, odd4.bw, odd4.cbm, isodata *
* OUPUT FILES: even4.odd4.ct *
*******************************************************************************
>>mpirun -np 4 rtransition_mpi
====================================================
RTRANSITION_MPI: Execution Begins ...
====================================================
Participating nodes:
Host: per-vaio ID: 000
Host: per-vaio ID: 001
Host: per-vaio ID: 002
Host: per-vaio ID: 003
Date and Time:
per-vaio: Date: 20141120 Time: 003050.621 Zone: +0100
per-vaio: Date: 20141120 Time: 003050.621 Zone: +0100
per-vaio: Date: 20141120 Time: 003050.621 Zone: +0100
per-vaio: Date: 20141120 Time: 003050.621 Zone: +0100
Start Dir:
per-vaio: /home/per/graspruns/FeXIII
per-vaio: /home/per/graspruns/FeXIII
per-vaio: /home/per/graspruns/FeXIII
per-vaio: /home/per/graspruns/FeXIII
Serial I/O Dir (node-0 only):
per-vaio: /home/per/graspruns/FeXIII
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>even4
Name of the Final state
>>odd4
Enter the list of transition specifications
e.g., E1,M2 or E1 M2 or E1;M2 :
>>E1,M2
.....
mpi stopped by node- 0 from RTRANSITION_MPI: Execution complete.
mpi stopped by node- 3 from RTRANSITION_MPI: Execution complete.
mpi stopped by node- 1 from RTRANSITION_MPI: Execution complete.
mpi stopped by node- 2 from RTRANSITION_MPI: Execution complete.
Comment: it does not matter in which order the files even4 and odd4 are specified.
6.5. Fifth Example: The Study of Energy Spectra for Ni XIV, Obtaining Unique Labels
A wave function or a corresponding energy level is often designated by the label of the CSF with the largest expansion coefficient. This example presents a study of energy spectra for Ni XIV in which a few levels have the same identification. To get the energy spectra with unique labels, we use the unique option in the
jj2lsj program. The program uses the algorithm described in [
26,
40,
41]: for a given set of wave functions for the same
J and parity, the CSF with the largest expansion coefficient is used as the label for the function containing this largest component. Once a label is assigned, the corresponding CSF is removed from consideration in the determination of the next label. The last remaining label for a wave function may be based on a contribution that is tiny.
Define nuclear data.
Obtain common spectroscopic orbitals for the MR set.
- (a)
Generate list of CSFs describing the even states belonging to the , configurations and the odd states belonging to the configuration.
- (b)
Perform angular integration.
- (c)
Generate initial estimates of radial orbitals.
- (d)
Perform SCF calculation on the weighted average of all states belonging to , , and .
- (e)
Save output to Ni_mr.
Improve even states
- (a)
Generate CSF list from SD-excitations from and to .
- (b)
Run rcsfinteract to extract CSFs that interact with CSFs belonging to and .
- (c)
Perform angular integration.
- (d)
Generate initial estimates of radial orbitals.
- (e)
Perform SCF calculation on the weighted average of all states belonging to and .
- (f)
Save output to Ni_even_n4.
- (g)
Perform rci calculation in which the transverse photon interaction (Breit) and vacuum polarization and self-energy (QED) corrections are added.
Transform from - to -coupling
Improve odd states
- (a)
Generate CSF list from SD-excitations from to .
- (b)
Run rcsfinteract to extract CSFs that interact with CSFs belonging to .
- (c)
Perform angular integration.
- (d)
Generate initial estimates of radial orbitals.
- (e)
Perform SCF calculation on the weighted average of all states belonging to .
- (f)
Save output to Ni_odd_n4.
- (g)
Perform rci calculation in which the transverse photon interaction (Breit) and vacuum polarization and self-energy (QED) corrections are added.
Transform from - to -coupling using the unique label option.
Run rlevels to view energy separations (several states have the same label).
Copy files so that rlevels will display unique labels.
Run rlevels to view energy separations for levels now with unique labels.
Compute transition rates from the rci wave functions. Computation in two steps: biorthonormal transformation and then evaluation of transition matrix elements using standard Racah algebra methods.
In the test-runs, prompt marked by >> or >>3, for example, indicates that the user should input 3 and then strike the return key. When >> is followed by blanks, just strike the return key.
*******************************************************************************
* RUN RNUCLEUS TO GENERATE NUCLEAR DATA AND DEFINE RADIAL GRID *
* OUTPUT FILE: isodata *
*******************************************************************************
>>rnucleus
RNUCLEUS
This program defines nuclear data and the radial grid
Outputfile: isodata
Enter the atomic number:
>>28
Enter the mass number (0 if the nucleus is to be modelled as a point source:
>>61
The default root mean squared radius is 3.8224999904632568 fm; (Angeli)
the default nuclear skin thickness is 2.2999999999999998 fm;
Revise these values?
>>n
Enter the mass of the neutral atom (in amu) (0 if the nucleus is to be static):
>>58.6934
Enter the nuclear spin quantum number (I) (in units of h / 2 pi):
>>1
Enter the nuclear dipole moment (in nuclear magnetons):
>>1
Enter the nuclear quadrupole moment (in barns):
>>1
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE LIST FOR ALL *
* STATES OF 3s3p(4), 3s(2)3p(2)3d and 3s(2)3p(3) *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program generates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
OUTPUT FILES: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,i)2p(6,i)3s(1,i)3p(4,i)
Give configuration 2
>>2s(2,i)2p(6,i)3s(2,i)3p(2,i)3d(1,i)
Give configuration 3
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>3s,3p,3d
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,9
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>y
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*).
Give configuration 1
>>2s(2,i)2p(6,i)3s(2,i)3p(3,i)
Give configuration 2
>>
Give set of active orbitals in a comma delimited list ordered by l-symmetry, e.g., 5s,4p,3d
>>3s,3p
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,5
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>n
.........
8 blocks were created
block J/P NCSF
1 1/2+ 8
2 1/2- 1
3 3/2+ 11
4 3/2- 3
5 5/2+ 10
6 5/2- 1
7 7/2+ 5
8 9/2+ 2
*******************************************************************************
* COPY FILES *
* IT IS ADVISABLE TO SAVE THE rcsfgenerate.log FILE TO HAVE A *
* RECORD ON HOW THE LIST OF CSFs WAS CREATED *
*******************************************************************************
>>cp rcsfgenerate.log Ni_mr.exc
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.log, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
.....
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp, rwfnestimate.log *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
Shell e p0 gamma <r> MTP SRC
1s 0.3531D+03 0.3348D+03 0.1000D+01 0.5348D-01 329 T-F
2s 0.7017D+02 0.1144D+03 0.1000D+01 0.2231D+00 346 T-F
2p- 0.6820D+02 0.1007D+01 0.1000D+01 0.1878D+00 346 T-F
2p 0.6732D+02 0.8291D+03 0.2000D+01 0.1905D+00 346 T-F
3s 0.2444D+02 0.5706D+02 0.1000D+01 0.5420D+00 358 T-F
3p- 0.2358D+02 0.5370D+00 0.1000D+01 0.5120D+00 359 T-F
3p 0.2336D+02 0.4440D+03 0.2000D+01 0.5164D+00 359 T-F
3d- 0.2191D+02 0.7313D+00 0.2000D+01 0.4446D+00 359 T-F
3d 0.2185D+02 0.7607D+03 0.3000D+01 0.4461D+00 359 T-F
RWFNESTIMATE: Execution complete.
*******************************************************************************
* RUN RMCDHF_MEM TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: ORBITALS BUILDING REFERENCE STATES ARE REQUIRED TO HAVE *
* THE CORRECT NUMBER OF NODES. THEY ARE REFERRED TO AS SPECTROSCOPIC *
* ORBITALS. IN THIS RUN WE VARY 1s,2s,2p,3s,3p,3d AND THEY ARE ALL *
* SPECTROSCOPIC. WE CAN USE WILD CARDS FOR SPECIFYING ORBITALS *
* *
* NOTE: INSTEAD OF SAYING THAT WE SHOULD OPTIMIZE ON, FOR EXAMPLE, *
* THE STATES 1,2,3,4 WE CAN WRITE 1-4 MEANING THE SAME THING *
* *
*******************************************************************************
>>rmcdhf_mem
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Loading CSF File for ALL blocks
There are 41 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 8 blocks (block J/Parity NCF):
1 1/2+ 8 2 1/2- 1 3 3/2+ 11 4 3/2- 3
5 5/2+ 10 6 5/2- 1 7 7/2+ 5 8 9/2+ 2
Enter ASF serial numbers for each block
Block 1 ncf = 8 id = 1/2+
>>1-8
Block 2 ncf = 1 id = 1/2-
>>1
Block 3 ncf = 11 id = 3/2+
>>1-11
Block 4 ncf = 3 id = 3/2-
>>1-3
Block 5 ncf = 10 id = 5/2+
>>1-10
Block 6 ncf = 1 id = 5/2-
>>1
Block 7 ncf = 5 id = 7/2+
>>1-5
Block 8 ncf = 2 id = 9/2+
>>1-2
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Enter orbitals to be varied (Updating order)
>>*
Which of these are spectroscopic orbitals?
>>*
Enter the maximum number of SCF cycles:
>>999
..............
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog, name.log *
*******************************************************************************
>>rsave Ni_mr
Created Ni_mr.w, Ni_mr.c, Ni_mr.m, Ni_mr.sum, Ni_mr.alog and Ni_mr.log
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE LIST FOR ALL *
* STATES OF 3s3p(4), 3s(2)3p(2)3d *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program generates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
OUTPUT FILES: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,i)2p(6,i)3s(1,i)3p(4,i)
Give configuration 2
>>2s(2,i)2p(6,i)3s(2,i)3p(2,i)3d(1,i)
Give configuration 3
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>3s,3p,3d
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,9
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>n
.........
5 blocks were created
block J/P NCSF
1 1/2+ 8
2 3/2+ 11
3 5/2+ 10
4 7/2+ 5
5 9/2+ 2
*******************************************************************************
* COPY FILES *
* NOTE THAT WE COPY THE FILE TO RCSFMR.INP FOR USE *
* TOGETHER WITH RCSFINTERACT *
*******************************************************************************
>>cp rcsf.out rcsfmr.inp
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE LIST OBTAINED BY *
* SD-EXCITATIONS FROM 3s3p(4) and 3s(2)3p(2)3d TO n = 4 *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program generates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
OUTPUT FILES: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,i)2p(6,i)3s(1,*)3p(4,*)
Give configuration 2
>>2s(2,i)2p(6,i)3s(2,*)3p(2,*)3d(1,*)
Give configuration 3
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>4s,4p,4d,4f
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,9
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
.........
5 blocks were created
block J/P NCSF
1 1/2+ 1664
2 3/2+ 2837
3 5/2+ 3271
4 7/2+ 2972
5 9/2+ 2264
*******************************************************************************
* COPY FILES *
*******************************************************************************
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RCSFINTERACT PROGRAM TO DETERMINE WHICH OF THE CSFs IN THE *
* rcsf.inp LIST INTERACTS WITH THE CSFs IN rcsfmr.inp *
* THE INTERACTING CSFs ARE WRITTEN TO rcsf.out *
* INPUT FILES: rcsfmr.inp, rcsf.inp *
* OUTPUT FILE: rcsf.out *
*******************************************************************************
>>rcsfinteract
RCSFinteract: Determines all the CSFs (rcsf.inp) that interact
with the CSFs in the multireference (rcsfmr.inp)
(C) Copyright by G. Gaigalas and Ch. F. Fischer
(Fortran 95 version) NIST (2017).
Input files: rcsfmr.inp, rcsf.inp
Output file: rcsf.out
Reduction based on Dirac-Coulomb (1) or
Dirac-Coulomb-Breit (2) Hamiltonian?
>>2
....
There are 16 relativistic subshells;
Block MR NCSF Befor NCSF After NCSF
1 8 1664 1047
2 11 2837 1862
3 10 3271 2112
4 5 2972 1537
5 2 2264 801
RCSFINTERACT: Execution complete
*******************************************************************************
* COPY FILES *
*******************************************************************************
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.log, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
.....
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp, rwfnestimate.log *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d 4s 4p- 4p 4d- 4d 4f- 4f
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>1
Enter the file name (Null then "rwfn.out")
>>Ni_mr.w
Enter the list of relativistic subshells:
>>*
The following subshell radial wavefunctions remain to be estimated:
4s 4p- 4p 4d- 4d 4f- 4f
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
Shell e p0 gamma <r> MTP SRC
1s 0.3241D+03 0.3333D+03 0.1000D+01 0.5375D-01 357 Ni_
2s 0.5342D+02 0.1015D+03 0.1000D+01 0.2420D+00 359 Ni_
2p- 0.4833D+02 0.8507D+00 0.1000D+01 0.2110D+00 360 Ni_
2p 0.4768D+02 0.6996D+03 0.2000D+01 0.2140D+00 360 Ni_
3s 0.1702D+02 0.4580D+02 0.1000D+01 0.6442D+00 364 Ni_
3p- 0.1557D+02 0.3893D+00 0.1000D+01 0.6379D+00 364 Ni_
3p 0.1545D+02 0.3217D+03 0.2000D+01 0.6430D+00 364 Ni_
3d- 0.1320D+02 0.3700D+00 0.2000D+01 0.5968D+00 366 Ni_
3d 0.1319D+02 0.3854D+03 0.3000D+01 0.5980D+00 366 Ni_
4s 0.1130D+02 0.3354D+02 0.1000D+01 0.1050D+01 368 T-F
4p- 0.1090D+02 0.3205D+00 0.1000D+01 0.1031D+01 368 T-F
4p 0.1083D+02 0.2655D+03 0.2000D+01 0.1038D+01 368 T-F
4d- 0.1015D+02 0.4907D+00 0.2000D+01 0.9852D+00 369 T-F
4d 0.1013D+02 0.5108D+03 0.3000D+01 0.9872D+00 369 T-F
4f- 0.9324D+01 0.2669D+00 0.3000D+01 0.8778D+00 369 T-F
4f 0.9316D+01 0.3018D+03 0.4000D+01 0.8787D+00 369 T-F
RWFNESTIMATE: Execution complete.
*******************************************************************************
* RUN RMCDHF_MEM TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: FOR CORRELATION ORBITALS THERE ARE NO RESTRICTIONS ON THE *
* NUMBER OF NODES, I.E. THEY ARE NOT SPECTROSCOPIC. IN THIS RUN WE *
* VARY THE CORRELATION ORBITALS 4s, 4p, 4d, 4f. NONE OF THESE ARE *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* *
* NOTE: INSTEAD OF SAYING THAT WE SHOULD OPTIMIZE ON, FOR EXAMPLE, *
* THE STATES 1,2,3,4 WE CAN WRITE 1-4 MEANING THE SAME THING *
* *
*******************************************************************************
>>rmcdhf_mem
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
Loading CSF File for ALL blocks
There are 7359 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 5 blocks (block J/Parity NCF):
1 1/2+ 1047 2 3/2+ 1862 3 5/2+ 2112 4 7/2+ 1537
5 9/2+ 801
Enter ASF serial numbers for each block
Block 1 ncf = 1047 id = 1/2+
>>1-8
Block 2 ncf = 1862 id = 3/2+
>>1-11
Block 3 ncf = 2112 id = 5/2+
>>1-10
Block 4 ncf = 1537 id = 7/2+
>>1-5
Block 5 ncf = 801 id = 9/2+
>>1-2
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d 4s 4p- 4p 4d- 4d 4f- 4f
Enter orbitals to be varied (Updating order)
>>4*
Which of these are spectroscopic orbitals?
>>
Enter the maximum number of SCF cycles:
>>100
..............
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog, name.log *
*******************************************************************************
>>rsave Ni_even_n4
Created Ni_even_n4.w, Ni_even_n4.c, Ni_even_n4.m, Ni_even_n4.sum, Ni_even_n4.alog
and Ni_even_n4.log
*******************************************************************************
* RUN RCI TO INCLUDE TRANSVERSE PHOTON INTERACTION AND QED EFFECTS *
* INPUT FILES : isodata, Ni_even_n4.c, Ni_even_n4.w *
* OUTPUT FILES: Ni_even_n4.cm, Ni_even_n4.csum, Ni_even_n4.clog, *
* rci.res *
* *
* THE TRANSVERSE PHOTON FREQUENCIES CAN BE SET TO THE LOW FREQUENCY *
* LIMIT. RECOMMENDED IN CASES WHERE YOU HAVE CORRELATION ORBITALS *
* THE SELF ENERGY CORRECTION MAY FAIL FOR CORRELATION ORBITALS WITH *
* HIGH N. *
*******************************************************************************
>>rci
RCI
This is the configuration interaction program
Input file: isodata, name.c, name.w
Outputfiles: name.cm, name.csum, name.clog, rci.res
Default settings?
>>y
Name of state:
>>Ni_even_n4
Block 1 , ncf = 1047
Block 2 , ncf = 1862
Block 3 , ncf = 2112
Block 4 , ncf = 1537
Block 5 , ncf = 801
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
Include contribution of H (Transverse)?
>>y
Modify all transverse photon frequencies?
>>n
Include H (Vacuum Polarisation)?
>>y
Include H (Normal Mass Shift)?
>>n
Include H (Specific Mass Shift)?
>>n
Estimate self-energy?
>>y
Largest n quantum number for including self-energy for orbital
n should be less or equal 8
>>4
Loading Radial WaveFunction File ...
There are 5 blocks (block J/Parity NCF):
1 1/2+ 1047 2 3/2+ 1862 3 5/2+ 2112 4 7/2+ 1537
5 9/2+ 801
Enter ASF serial numbers for each block
Block 1 ncf = 1047 id = 1/2+
>>1-8
Block 2 ncf = 1862 id = 3/2+
>>1-11
Block 3 ncf = 2112 id = 5/2+
>>1-10
Block 4 ncf = 1537 id = 7/2+
>>1-5
Block 5 ncf = 801 id = 9/2+
>>1-2
....
RCI: Execution complete.
*******************************************************************************
* RUN JJ2LSJ TO GET THE LSJ-COMPOSITION *
* INPUT FILE: Ni_even_n4.c, Ni_even_n4.cm *
* OUTPUT FILE: Ni_even_n4.lsj.lbl, Ni_even_n4.uni.lsj.lbl *
*******************************************************************************
>>jj2lsj
jj2lsj: Transformation of ASFs from a jj-coupled CSF basis
into an LSJ-coupled CSF basis (Fortran 95 version)
(C) Copyright by G. Gaigalas and Ch. F. Fischer,
(2017).
Input files: name.c, name.(c)m
Output files: name.lsj.lbl
(optional) name.lsj.c, name.lsj.j,
name.uni.lsj.lbl, name.uni.lsj.sum
Name of state
>>Ni_even_n4
Loading Configuration Symmetry List File ...
There are 16 relativistic subshells;
There are 7359 relativistic CSFs;
... load complete;
Mixing coefficients from a CI calc.?
>>y
Do you need a unique labeling? (y/n)
>>y
nelec = 15
ncftot = 7359
nw = 16
nblock = 5
block ncf nev 2j+1 parity
1 1047 8 2 1
2 1862 11 4 1
3 2112 10 6 1
4 1537 5 8 1
5 801 2 10 1
Default settings? (y/n)
>>y
...........
jj2lsj: Execution complete.
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE LIST FOR ALL *
* STATES OF 3s(2)3p(3) *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program generates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
OUTPUT FILES: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,i)2p(6,i)3s(2,i)3p(3,i)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>3s,3p
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,5
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>n
.........
3 blocks were created
block J/P NCSF
1 1/2- 1
2 3/2- 3
3 5/2- 1
*******************************************************************************
* COPY FILES *
* NOTE THAT WE COPY THE FILE TO RCSFMR.INP FOR USE *
* TOGETHER WITH RCSFINTERACT *
*******************************************************************************
>>cp rcsf.out rcsfmr.inp
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE LIST OBTAINED BY *
* SD-EXCITATIONS FROM 3s(2)3p(3) TO n = 4 *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program generates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
OUTPUT FILES: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,i)2p(6,i)3s(2,*)3p(3,*)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>4s,4p,4d,4f
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,5
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
.........
3 blocks were created
block J/P NCSF
1 1/2- 481
2 3/2- 802
3 5/2- 868
*******************************************************************************
* COPY FILES *
*******************************************************************************
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RCSFINTERACT PROGRAM TO DETERMINE WHICH OF THE CSFs IN THE *
* rcsf.inp LIST INTERACTS WITH THE CSFs IN rcsfmr.inp *
* THE INTERACTING CSFs ARE WRITTEN TO rcsf.out *
* INPUT FILES: rcsfmr.inp, rcsf.inp *
* OUTPUT FILE: rcsf.out *
*******************************************************************************
>>rcsfinteract
RCSFinteract: Determines all the CSFs (rcsf.inp) that interact
with the CSFs in the multireference (rcsfmr.inp)
(C) Copyright by G. Gaigalas and Ch. F. Fischer
(Fortran 95 version) NIST (2017).
Input files: rcsfmr.inp, rcsf.inp
Output file: rcsf.out
Reduction based on Dirac-Coulomb (1) or
Dirac-Coulomb-Breit (2) Hamiltonian?
>>2
....
There are 16 relativistic subshells;
Block MR NCSF Befor NCSF After NCSF
1 1 481 237
2 3 802 577
3 1 868 480
RCSFINTERACT: Execution complete
*******************************************************************************
* COPY FILES *
*******************************************************************************
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.log, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
.....
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp, rwfnestimate.log *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d 4s 4p- 4p 4d- 4d 4f- 4f
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>1
Enter the file name (Null then "rwfn.out")
>>Ni_mr.w
Enter the list of relativistic subshells:
>>*
The following subshell radial wavefunctions remain to be estimated:
4s 4p- 4p 4d- 4d 4f- 4f
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.3241D+03 0.3333D+03 0.1000D+01 0.5375D-01 357 Ni_
2s 0.5342D+02 0.1015D+03 0.1000D+01 0.2420D+00 359 Ni_
2p- 0.4833D+02 0.8507D+00 0.1000D+01 0.2110D+00 360 Ni_
2p 0.4768D+02 0.6996D+03 0.2000D+01 0.2140D+00 360 Ni_
3s 0.1702D+02 0.4580D+02 0.1000D+01 0.6442D+00 364 Ni_
3p- 0.1557D+02 0.3893D+00 0.1000D+01 0.6379D+00 364 Ni_
3p 0.1545D+02 0.3217D+03 0.2000D+01 0.6430D+00 364 Ni_
3d- 0.1320D+02 0.3700D+00 0.2000D+01 0.5968D+00 366 Ni_
3d 0.1319D+02 0.3854D+03 0.3000D+01 0.5980D+00 366 Ni_
4s 0.1130D+02 0.3354D+02 0.1000D+01 0.1050D+01 368 T-F
4p- 0.1090D+02 0.3205D+00 0.1000D+01 0.1031D+01 368 T-F
4p 0.1083D+02 0.2655D+03 0.2000D+01 0.1038D+01 368 T-F
4d- 0.1015D+02 0.4907D+00 0.2000D+01 0.9852D+00 369 T-F
4d 0.1013D+02 0.5108D+03 0.3000D+01 0.9872D+00 369 T-F
4f- 0.9324D+01 0.2669D+00 0.3000D+01 0.8778D+00 369 T-F
4f 0.9316D+01 0.3018D+03 0.4000D+01 0.8787D+00 369 T-F
RWFNESTIMATE: Execution complete.
*******************************************************************************
* RUN RMCDHF_MEM TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: FOR CORRELATION ORBITALS THERE ARE NO RESTRICTIONS ON THE *
* NUMBER OF NODES, I.E. THEY ARE NOT SPECTROSCOPIC. IN THIS RUN WE *
* VARY THE CORRELATION ORBITALS 4s, 4p, 4d, 4f. NONE OF THESE ARE *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* *
* NOTE: INSTEAD OF SAYING THAT WE SHOULD OPTIMIZE ON, FOR EXAMPLE, *
* THE STATES 1,2,3,4 WE CAN WRITE 1-4 MEANING THE SAME THING *
* *
*******************************************************************************
>>rmcdhf_mem
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
Loading CSF File for ALL blocks
There are 1294 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 3 blocks (block J/Parity NCF):
1 1/2- 237 2 3/2- 577 3 5/2- 480
Enter ASF serial numbers for each block
Block 1 ncf = 237 id = 1/2-
>>1
Block 2 ncf = 577 id = 3/2-
>>1-3
Block 3 ncf = 480 id = 5/2-
>>1
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d 4s 4p- 4p 4d- 4d 4f- 4f
Enter orbitals to be varied (Updating order)
>>4*
Which of these are spectroscopic orbitals?
>>
Enter the maximum number of SCF cycles:
>>100
..............
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.log *
*******************************************************************************
>>rsave Ni_odd_n4
Created Ni_odd_n4.w, Ni_odd_n4.c, Ni_odd_n4.m, Ni_odd_n4.sum and Ni_odd_n4.log
*******************************************************************************
* RUN RCI TO INCLUDE TRANSVERSE PHOTON INTERACTION AND QED EFFECTS *
* INPUT FILES : isodata, Ni_odd_n4.c, Ni_odd_n4.w *
* OUTPUT FILES: Ni_odd_n4.cm, Ni_odd_n4.csum, Ni_odd_n4.clog, rci.res *
* *
* THE TRANSVERSE PHOTON FREQUENCIES CAN BE SET TO THE LOW FREQUENCY *
* LIMIT. RECOMMENDED IN CASES WHERE YOU HAVE CORRELATION ORBITALS *
* THE SELF ENERGY CORRECTION MAY FAIL FOR CORRELATION ORBITALS WITH *
* HIGH N. *
*******************************************************************************
>>rci
RCI
This is the configuration interaction program
Input file: isodata, name.c, name.w
Outputfiles: name.cm, name.csum, name.clog, rci.res
Default settings?
>>y
Name of state:
>>Ni_odd_n
Block 1 , ncf = 237
Block 2 , ncf = 577
Block 3 , ncf = 480
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
Include contribution of H (Transverse)?
>>y
Modify all transverse photon frequencies?
>>n
Include H (Vacuum Polarisation)?
>>y
Include H (Normal Mass Shift)?
>>n
Include H (Specific Mass Shift)?
>>n
Estimate self-energy?
>>y
Largest n quantum number for including self-energy for orbital
n should be less or equal 8
>>4
Loading Radial WaveFunction File ...
There are 3 blocks (block J/Parity NCF):
1 1/2- 237 2 3/2- 577 3 5/2- 480
Enter ASF serial numbers for each block
Block 1 ncf = 237 id = 1/2-
>>1
Block 2 ncf = 577 id = 3/2-
>>1-3
Block 3 ncf = 480 id = 5/2-
>>1
....
RCI: Execution complete.
*******************************************************************************
* RUN JJ2LSJ TO GET THE LSJ-COMPOSITION *
* INPUT FILE: Ni_odd_n4.c, Ni_odd_n4.cm *
* OUTPUT FILE: Ni_odd_n4.lsj.lbl, Ni_odd_n4.uni.lsj.lbl *
*******************************************************************************
>>jj2lsj
jj2lsj: Transformation of ASFs from a jj-coupled CSF basis
into an LSJ-coupled CSF basis (Fortran 95 version)
(C) Copyright by G. Gaigalas and Ch. F. Fischer,
(2017).
Input files: name.c, name.(c)m
Output files: name.lsj.lbl
(optional) name.lsj.c, name.lsj.j,
name.uni.lsj.lbl, name.uni.lsj.sum
Name of state
>>Ni_odd_n4
Loading Configuration Symmetry List File ...
There are 16 relativistic subshells;
There are 1294 relativistic CSFs;
... load complete;
Mixing coefficients from a CI calc.?
>>y
Do you need a unique labeling? (y/n)
>>y
nelec = 15
ncftot = 1294
nw = 16
nblock = 3
block ncf nev 2j+1 parity
1 237 1 2 -1
2 577 3 4 -1
3 480 1 6 -1
Default settings? (y/n)
>>y
...........
jj2lsj: Execution complete.
*******************************************************************************
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS. *
* IF DESIRED WE CAN INSTEAD RUN RLEVELSEV TO GET THE SEPARATION IN EV *
*******************************************************************************
>> rlevels Ni_even_n4.cm Ni_odd_n4.cm
nblock = 5 ncftot = 7359 nw = 16 nelec = 15
nblock = 3 ncftot = 1294 nw = 16 nelec = 15
Energy levels for ...
Rydberg constant is 109737.31534
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 3/2 - -1443.2224318 0.00 0.00 2s(2).2p(6).3s(2).3p(3)4S3_4S
2 2 3/2 - -1443.0055953 47590.12 47590.12 2s(2).2p(6).3s(2).3p(3)2D3_2D
3 1 5/2 - -1442.9699841 55405.86 7815.74 2s(2).2p(6).3s(2).3p(3)2D3_2D
4 1 1/2 - -1442.8231291 87636.81 32230.95 2s(2).2p(6).3s(2).3p(3)2P1_2P
5 3 3/2 - -1442.7718374 98894.05 11257.25 2s(2).2p(6).3s(2).3p(3)2P1_2P
6 1 5/2 + -1441.7819193 316155.95 217261.89 2s(2).2p(6).3s_2S.3p(4)3P2_4P
7 1 3/2 + -1441.7163490 330546.97 14391.02 2s(2).2p(6).3s_2S.3p(4)3P2_4P
8 1 1/2 + -1441.6893375 336475.31 5928.35 2s(2).2p(6).3s_2S.3p(4)3P2_4P
9 2 3/2 + -1441.4303647 393313.26 56837.95 2s(2).2p(6).3s_2S.3p(4)1D2_2D
10 2 5/2 + -1441.4141226 396877.99 3564.73 2s(2).2p(6).3s_2S.3p(4)1D2_2D
11 3 3/2 + -1441.1719219 450034.90 53156.91 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2P
12 2 1/2 + -1441.1457552 455777.83 5742.94 2s(2).2p(6).3s_2S.3p(4)1S0_2S
13 3 1/2 + -1441.0407477 478824.32 23046.48 2s(2).2p(6).3s_2S.3p(4)1S0_2S
14 4 3/2 + -1441.0025311 487211.88 8387.57 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4F
15 3 5/2 + -1440.9762734 492974.79 5762.91 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4F
16 1 7/2 + -1440.9366782 501664.93 8690.14 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4F
17 4 5/2 + -1440.9108204 507340.07 5675.14 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2F
18 1 9/2 + -1440.8905994 511778.07 4437.99 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4F
19 2 7/2 + -1440.8808794 513911.35 2133.28 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
20 4 1/2 + -1440.8807431 513941.26 29.92 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
21 5 3/2 + -1440.8744144 515330.27 1389.01 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
22 5 5/2 + -1440.8441958 521962.48 6632.21 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
23 3 7/2 + -1440.7788694 536299.97 14337.49 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
24 4 7/2 + -1440.6229634 570517.38 34217.41 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2G
25 2 9/2 + -1440.5997042 575622.19 5104.81 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2G
26 6 3/2 + -1440.5811088 579703.40 4081.21 2s(2).2p(6).3s_2S.3p(4)3P2_2P
27 6 5/2 + -1440.5348164 589863.39 10159.99 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
28 7 3/2 + -1440.5083509 595671.90 5808.50 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
29 5 1/2 + -1440.5059857 596191.02 519.12 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
30 6 1/2 + -1440.4811030 601652.14 5461.12 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
31 8 3/2 + -1440.4611905 606022.41 4370.27 2s(2).2p(6).3s(2).3p(2)1S0_1S.3d_2D
32 7 5/2 + -1440.3895597 621743.56 15721.15 2s(2).2p(6).3s(2).3p(2)1S0_1S.3d_2D
33 9 3/2 + -1440.3040979 640500.26 18756.70 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2D
34 8 5/2 + -1440.2991882 641577.81 1077.56 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2D
35 7 1/2 + -1440.2296306 656843.93 15266.12 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2P
36 9 5/2 + -1440.1955930 664314.33 7470.39 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2F
37 10 3/2 + -1440.1738626 669083.62 4769.29 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2P
38 8 1/2 + -1440.1603320 672053.23 2969.61 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2S
39 5 7/2 + -1440.1590801 672328.00 274.77 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2F
40 10 5/2 + -1440.0334583 699898.79 27570.79 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2D
41 11 3/2 + -1440.0284354 701001.19 1102.41 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2D
------------------------------------------------------------------------------------------
The
rlevels program reads the
name.cm files along with the
name.lsj.lbl files, which display the
composition as generated by
jj2lsj. From the energy spectra we see that some even levels have the same identification. These pairs of levels are 12 and 13; 19 and 23; 29 and 30. If a unique label was required,
jj2lsj also outputs files
name.uni.lsj.lb in which unique labels are determined according to the prescription given in [
26,
40,
41]. To display the energies with unique labels, we should copy
name.cm to
name.uni.cm and rerun
rlevels with
name.uni.cm as the input file.
*******************************************************************************
* COPY FILES TO HAVE EVEN PARITY LEVELS WITH UNIQUE LABELS *
* THAT SHOULD BE USED IN FURTHER CALCULATIONS *
*******************************************************************************
>>cp Ni_even_n4.cm Ni_even_n4.uni.cm
*******************************************************************************
* RUN RLEVELS TO VIEW ENERGIES AND ENERGY SEPARATIONS. *
* ENERGY LEVELS HAVE UNIQUE LABELS *
*******************************************************************************
>> rlevels Ni_even_n4.uni.cm Ni_odd_n4.cm
nblock = 5 ncftot = 7359 nw = 16 nelec = 15
nblock = 3 ncftot = 1294 nw = 16 nelec = 15
Energy levels for ...
Rydberg constant is 109737.31534
Splitting is the energy difference with the lower neighbor
------------------------------------------------------------------------------------------
No Pos J Parity Energy Total Levels Splitting Configuration
(a.u.) (cm^-1) (cm^-1)
------------------------------------------------------------------------------------------
1 1 3/2 - -1443.2223116 0.00 0.00 2s(2).2p(6).3s(2).3p(3)4S3_4S
2 2 3/2 - -1443.0054751 47590.12 47590.12 2s(2).2p(6).3s(2).3p(3)2D3_2D
3 1 5/2 - -1442.9698639 55405.86 7815.74 2s(2).2p(6).3s(2).3p(3)2D3_2D
4 1 1/2 - -1442.8230089 87636.81 32230.95 2s(2).2p(6).3s(2).3p(3)2P1_2P
5 3 3/2 - -1442.7717172 98894.05 11257.24 2s(2).2p(6).3s(2).3p(3)2P1_2P
6 1 5/2 + -1441.7818000 316155.75 217261.70 2s(2).2p(6).3s_2S.3p(4)3P2_4P
7 1 3/2 + -1441.7162297 330546.77 14391.02 2s(2).2p(6).3s_2S.3p(4)3P2_4P
8 1 1/2 + -1441.6892182 336475.11 5928.35 2s(2).2p(6).3s_2S.3p(4)3P2_4P
9 2 3/2 + -1441.4302453 393313.09 56837.98 2s(2).2p(6).3s_2S.3p(4)1D2_2D
10 2 5/2 + -1441.4140033 396877.81 3564.72 2s(2).2p(6).3s_2S.3p(4)1D2_2D
11 3 3/2 + -1441.1718022 450034.79 53156.98 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2P
12 2 1/2 + -1441.1456357 455777.69 5742.90 2s(2).2p(6).3s_2S.3p(4)3P2_2P
13 3 1/2 + -1441.0406282 478824.17 23046.48 2s(2).2p(6).3s_2S.3p(4)1S0_2S
14 4 3/2 + -1441.0024109 487211.89 8387.72 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4F
15 3 5/2 + -1440.9761532 492974.79 5762.91 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4F
16 1 7/2 + -1440.9365580 501664.94 8690.14 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4F
17 4 5/2 + -1440.9107001 507340.08 5675.14 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2F
18 1 9/2 + -1440.8904792 511778.07 4437.99 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4F
19 2 7/2 + -1440.8807592 513911.35 2133.28 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2F
20 4 1/2 + -1440.8806230 513941.26 29.91 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
21 5 3/2 + -1440.8742942 515330.26 1389.01 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
22 5 5/2 + -1440.8440756 521962.48 6632.21 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
23 3 7/2 + -1440.7787492 536299.97 14337.49 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
24 4 7/2 + -1440.6228432 570517.38 34217.42 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2G
25 2 9/2 + -1440.5995839 575622.19 5104.81 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2G
26 6 3/2 + -1440.5809891 579703.29 4081.10 2s(2).2p(6).3s_2S.3p(4)3P2_2P
27 6 5/2 + -1440.5346963 589863.37 10160.08 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
28 7 3/2 + -1440.5082308 595671.88 5808.50 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
29 5 1/2 + -1440.5058658 596190.95 519.07 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2P
30 6 1/2 + -1440.4809830 601652.08 5461.13 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
31 8 3/2 + -1440.4610704 606022.40 4370.32 2s(2).2p(6).3s(2).3p(2)1S0_1S.3d_2D
32 7 5/2 + -1440.3894396 621743.54 15721.14 2s(2).2p(6).3s(2).3p(2)1S0_1S.3d_2D
33 9 3/2 + -1440.3039779 640500.22 18756.68 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2D
34 8 5/2 + -1440.2990681 641577.79 1077.57 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2D
35 7 1/2 + -1440.2295105 656843.92 15266.12 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2P
36 9 5/2 + -1440.1954728 664314.33 7470.41 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2F
37 10 3/2 + -1440.1737424 669083.60 4769.27 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2P
38 8 1/2 + -1440.1602120 672053.18 2969.58 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2S
39 5 7/2 + -1440.1589598 672328.00 274.82 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2F
40 10 5/2 + -1440.0333381 699898.78 27570.78 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2D
41 11 3/2 + -1440.0283152 701001.19 1102.41 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2D
------------------------------------------------------------------------------------------
Comment: we now see that all states have different labels that allow for unambiguous identifications.
*******************************************************************************
* COPY FILES TO HAVE LEVELS WITH UNIQUE LABELS *
* THAT SHOULD BE USED IN FURTHER CALCULATIONS *
*******************************************************************************
>>cp Ni_even_n4.c Ni_even_n4.uni.c
>>cp Ni_even_n4.w Ni_even_n4.uni.w
Now we can use these files with unique labels in further calculations, e.g., transition properties.
*******************************************************************************
* RUN RBIOTRANSFORM FOR Ni_odd_n4 AND Ni_even_n4.uni *
* TO TRANSFORM WAVE FUNCTIONS *
* INPUT FILES: isodata, Ni_odd_n4.c, Ni_odd_n4.w, Ni_odd_n4.cm, *
* Ni_even_n4.uni.c, Ni_even_n4.uni.w, Ni_even_n4.uni.cm *
* OUTPUT FILES: Ni_odd_n4.cbm, Ni_odd_n4.bw, *
* Ni_even_n4.uni.cbm, Ni_even_n4.uni.bw *
* Ni_odd_n4.TB, Ni_even_n4.uni.TB (angular files) *
*******************************************************************************
>>rbiotransform
RBIOTRANSFORM
This program transforms the initial and final wave
functions so that standard tensor albegra can be
used in evaluation of the transition parameters
Input files: isodata, name1.c, name1.w, name1.(c)m
name2.c, name2.w, name2.(c)m
name1.TB, name2.TB (optional angular files)
Output files: name1.bw, name1.(c)bm,
name2.bw, name2.(c)bm
name1.TB, name2.TB (angular files)
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>Ni_odd_n4
Name of the Final state
>>Ni_even_n4.uni
Transformation of all J symmetries?
>>y
....
BIOTRANSFORM: Execution complete.
*******************************************************************************
* RUN RTRANSITION FOR Ni_odd_n4 AND Ni_even_n4.uni *
* TO COMPUTE TRANSITION PARAMETERS *
* INPUT FILES: isodata, Ni_odd_n4.c, Ni_odd_n4.bw, Ni_odd_n4.cbm, *
* Ni_even_n4.uni.c, Ni_even_n4.uni.bw, Ni_even_n4.uni.cbm*
* OUTPUT FILES: Ni_odd_n4.Ni_even_n4.uni.ct *
* Ni_odd_n4.Ni_even_n4.uni.-1T (angular files) *
*******************************************************************************
>>rtransition
RTRANSITION
This program computes transition parameters from
transformed wave functions
Input files: isodata, name1.c, name1.bw, name1.(c)bm
name2.c, name2.bw, name2.(c)bm
optional, name1.lsj.lbl, name2.lsj.lbl
name1.name2.KT (optional angular files)
Output files: name1.name2.(c)t
optional, name1.name2.(c)t.lsj
name1.name2.KT (angular files)
Here K is parity and rank of transition: -1,+1 etc
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>Ni_odd_n4
Name of the Final state
>>Ni_even_n4.uni
MRGCSL: Execution begins ...
Loading Configuration Symmetry List File ...
There are 16 relativistic subshells;
There are 1294 relativistic CSFs;
... load complete;
Loading Configuration Symmetry List File ...
There are 16 relativistic subshells;
There are 7359 relativistic CSFs;
... load complete;
1 s
2 s
2 p-
2 p
3 s
3 p-
3 p
3 d-
3 d
4 s
4 p-
4 p
4 d-
4 d
4 f-
4 f
3
237 814 1294
5
1047 2909 5021 6558 7359
Loading Configuration Symmetry List File ...
there are 16 relativistic subshells;
there are 8653 relativistic CSFs;
... load complete;
Enter the list of transition specifications
e.g., E1,M2 or E1 M2 or E1;M2 :
>>E1
.....
RTRANSITION: Execution complete.
Transition data are in Ni_odd_n4.Ni_even_n4.uni.ct.lsj file in which all levels have the unique labels.
An alternative way to get unique labels than the one described above is to denote the states by the
composition. This can be done with the PERL script
lscomp.pl and it is described in more detail in
Section 7.10.
6.6. Sixth Example: The Study of Energy Spectra for Ni XIV, Extended MR Using rcsfmr
To obtain good transition energies, it is often necessary to extend the MR. This is facilitated by the program rcsfmr. The rcsfmr program reads the name.lsj.lbl file produced by jj2lsj and extracts the configurations that give rise to -coupled CSFs with weights exceeding a user defined cut-off. Below is part of the Ni_even_n4.lsj.lbl file from the fifth example.
Pos J Parity Energy Total Comp. of ASF
1 1/2 + -1441.689593921 99.941%
-0.92754342 0.86033679 2s(2).2p(6).3s_2S.3p(4)3P2_4P
-0.31644623 0.10013822 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
-0.13107223 0.01717993 2s(2).2p(6).3s_2S.3p(4)1S0_2S
-0.06808224 0.00463519 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)1S0_4P
-0.06306024 0.00397659 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)1D2_4P
-0.06139607 0.00376948 2s(2).2p(6).3p(4)3P2_3P.3d_4P
-0.04384478 0.00192236 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)3P2_4P
0.04315453 0.00186231 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2S
0.04160917 0.00173132 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)3P2_4P
2 1/2 + -1441.146026942 99.870%
0.55236001 0.30510158 2s(2).2p(6).3s_2S.3p(4)1S0_2S
0.54901778 0.30142053 2s(2).2p(6).3s_2S.3p(4)3P2_2P
-0.51850029 0.26884256 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2P
-0.25241177 0.06371170 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2S
0.14974129 0.02242245 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2P
0.08843416 0.00782060 2s(2).2p(6).3p(4)1D2_1D.3d_2P
-0.07913818 0.00626285 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
0.06957348 0.00484047 2s(2).2p(6).3s_2S.3p(2)1S0_2S.3d(2)1S0_2S
-0.06792804 0.00461422 2s(2).2p(6).3s_2S.3p(4)3P2_4P
-0.04635416 0.00214871 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)1D2_2P
-0.04439733 0.00197112 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)3P2_2P
0.03795472 0.00144056 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)3P2_2P
-0.03450153 0.00119036 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)3P2_2S
-0.03371402 0.00113663 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
-0.03274764 0.00107241 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)1D2_2S
0.03171981 0.00100615 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)3F2_2P
3 1/2 + -1441.041027919 99.883%
0.69856599 0.48799445 2s(2).2p(6).3s_2S.3p(4)1S0_2S
0.44943909 0.20199550 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2P
-0.37641525 0.14168844 2s(2).2p(6).3s_2S.3p(4)3P2_2P
-0.31029154 0.09628084 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2S
-0.14516094 0.02107170 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2P
0.11017096 0.01213764 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
-0.10592606 0.01122033 2s(2).2p(6).3s_2S.3p(4)3P2_4P
0.08894930 0.00791198 2s(2).2p(6).3s_2S.3p(2)1S0_2S.3d(2)1S0_2S
-0.06646514 0.00441762 2s(2).2p(6).3p(4)1D2_1D.3d_2P
-0.04537257 0.00205867 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
-0.04336944 0.00188091 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)3P2_2S
-0.04274245 0.00182692 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)1D2_2S
0.04115897 0.00169406 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)1D2_2P
0.03553871 0.00126300 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)3P2_2P
.....................
1 9/2 + -1440.890865311 99.130%
0.96823013 0.93746959 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4F
-0.18911784 0.03576556 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2G
0.09063449 0.00821461 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)3F2_4F
0.04435216 0.00196711 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)1G2_4F
-0.04085208 0.00166889 2s(2).2p(6).3s_2S.3p(2)3P2_2P.3d(2)3F2_4F
-0.03263660 0.00106515 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)1D2_4F
2 9/2 + -1440.599976411 99.058%
0.96757720 0.93620564 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2G
0.18856813 0.03555794 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4F
-0.08465547 0.00716655 2s(2).2p(6).3s_2S.3p(2)3P2_2P.3d(2)1G2_2G
-0.06715953 0.00451040 2s(2).2p(6).3s_2S.3p(2)3P2_2P.3d(2)3F2_2G
-0.04376060 0.00191499 2s(2).2p(6).3s(2).3d(3)2G3_2G
-0.04015861 0.00161271 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)1D2_2G
We see that the states are strongly mixed and that it is desirable to extend the MR. The size of the extended MR is a compromise between available computational resources and the desired accuracy of computed properties. Often an exploratory approach is needed. In this example, we will somewhat ad hoc determine an MR from the -coupled CSFs with weights larger than 0.03.
-
Run rcsfmr for Ni_even_n4.lsj.lbl with a cut-off 0.03.
-
Use the output from rcsfmr as an input to rcsfgenerate with no excitations. Copy to rcsfmr.inp
-
Use the output from rcsfmr as an input to rcsfgenerate and allow SD excitations from the extended MR. Copy to rcsf.inp
-
Run rcsfinteract
*******************************************************************************
* RUN RCSFMR FOR Ni_even_n4 *
*******************************************************************************
>>rcsfmr
RCSFMR
This program reads the name.lsj.lbl file and extracts a
set of MR configuartions that give rise to LSJ coupled
CSFs with absolute weights larger than a specified cut-off
Input file: namel.lsj.lbl
Ouput is written to screen
Name of state
>>Ni_even_n4
Give cut-off for weight
>>0.03
Configurations in the MR
2s(2,*)2p(6,*)3s(1,*)3p(4,*)
2s(2,*)2p(6,*)3s(2,*)3p(2,*)3d(1,*)
2s(2,*)2p(6,*)3s(1,*)3p(2,*)3d(2,*)
2s(2,*)2p(6,*)3p(4,*)3d(1,*)
2s(2,*)2p(6,*)3s(2,*)3d(3,*)
2s(2,*)2p(6,*)3s(2,*)3p(1,*)3d(1,*)4f(1,*)
2s(2,*)2p(6,*)3s(1,*)3p(3,*)4f(1,*)
*******************************************************************************
* RUN RCSFGENERATE USING THE OBTAINED CONFIGURATIONS FROM RCSFMR *
* BY REQUESTING ZERO EXCITATIONS WE WILL GET THE CSFs OF THE MR *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program generates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,i)2p(6,i)3s(1,*)3p(4,*)
Give configuration 2
>>2s(2,i)2p(6,i)3s(2,*)3p(2,*)3d(1,*)
Give configuration 3
>>2s(2,i)2p(6,i)3s(1,*)3p(2,*)3d(2,*)
Give configuration 4
>>2s(2,i)2p(6,i)3p(4,*)3d(1,*)
Give configuration 5
>>2s(2,i)2p(6,i)3s(2,*)3d(3,*)
Give configuration 6
>>2s(2,i)2p(6,i)3s(2,*)3p(1,*)3d(1,*)4f(1,*)
Give configuration 7
>>2s(2,i)2p(6,i)3s(1,*)3p(3,*)4f(1,*)
Give configuration 8
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>4s,4p,4d,4f
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,9
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>n
..................
Group CSFs into symmetry blocks
5 blocks were created
block J/P NCSF
1 1/2+ 61
2 3/2+ 104
3 5/2+ 116
4 7/2+ 96
5 9/2+ 67
*******************************************************************************
* COPY FILES *
*******************************************************************************
>>cp rcsf.out rcsfmr.inp
It is very important to realize that the orbital order in rcsfmr.inp needs to be the same as in the larger list rcsf.inp to be reduced. For this reason, we need a user defined orbital ordering that starts with the orbitals in the MR and then adds the correlation orbitals. The clist.ref file is, thus
1s
2s
2p
3s
3p
3d
4f
4s
4p
4d
We are now in the position to run rcsfgenerate.
*******************************************************************************
* RUN RCSFGENERATE USING THE OBTAINED CONFIGURATIONS FROM RCSFMR *
* REQUEST TWO EXCITATIONS. USER DEFINED ORBITAL ORDERING *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program generates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>u
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,i)2p(6,i)3s(1,*)3p(4,*)
Give configuration 2
>>2s(2,i)2p(6,i)3s(2,*)3p(2,*)3d(1,*)
Give configuration 3
>>2s(2,i)2p(6,i)3s(1,*)3p(2,*)3d(2,*)
Give configuration 4
>>2s(2,i)2p(6,i)3p(4,*)3d(1,*)
Give configuration 5
>>2s(2,i)2p(6,i)3s(2,*)3d(3,*)
Give configuration 6
>>2s(2,i)2p(6,i)3s(2,*)3p(1,*)3d(1,*)4f(1,*)
Give configuration 7
>>2s(2,i)2p(6,i)3s(1,*)3p(3,*)4f(1,*)
Give configuration 8
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>4s,4p,4d,4f
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>1,9
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
..................
Group CSFs into symmetry blocks
5 blocks were created
block J/P NCSF
1 1/2+ 5061
2 3/2+ 8907
3 5/2+ 10810
4 7/2+ 10604
5 9/2+ 8889
*******************************************************************************
* COPY FILES *
*******************************************************************************
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RCSFINTERACT *
*******************************************************************************
RCSFinteract: Determines all the CSFs (rcsf.inp) that interact
with the CSFs in the multireference (rcsfmr.inp)
(C) Copyright by G. Gaigalas and Ch. F. Fischer
(Fortran 95 version) NIST (2017).
Input files: rcsfmr.inp, rcsf.inp
Output file: rcsf.out
Reduction based on Dirac-Coulomb (1) or
Dirac-Coulomb-Breit (2) Hamiltonian?
>>2
Loading Configuration Symmetry List File ...
There are 16 relativistic subshells;
Block MR NCSF Before NCSF After NCSF
1 61 5061 3551
2 104 8907 6489
3 116 10810 7824
4 96 10604 7398
5 67 8889 5936
Wall time:
5 seconds
Finish Date and Time:
Date (Yr/Mon/Day): 2018/05/17
Time (Hr/Min/Sec): 00/54/36.490
Zone: +0200
RCSFinteract: Execution complete.
The same procedure can be applied to Ni_even_n4.lsj.lbl.
6.7. Seventh Example: Restarting rci
Follow the fifth example up to the rci calculation for Ni_even_n4. During the rci calculation the Hamiltonian matrix elements, in sparse representation, are successively written to the file rci.res. If the calculation stalls at some point, the rci program can be restarted. During a restart, all radial integrals are recomputed, and then the computation starts with computing the matrix elements following the last matrix element that was saved to rci.res. In this example, we assume that the rci calculation for Ni_even_n4 stalled in the middle of block 3, and we show how to make a restart.
*******************************************************************************
* RUN RCI TO INCLUDE TRANSVERSE PHOTON INTERACTION AND QED EFFECTS *
* INPUT FILES : isodata, Ni_even_n4.c, Ni_even_n4.w, rci.res *
* OUTPUT FILES: Ni_even_n4.cm, Ni_even_n4.csum, Ni_even_n4.clog, *
* rci.res *
* This is a restart that reads the rci.res file *
*******************************************************************************
>>rci
RCI
This is the configuration interaction program
Input file: isodata, name.c, name.w
Outputfiles: name.cm, name.csum, name.clog
rci.res (can be used for restart)
Default settings?
>>n
Name of state:
>>Ni_even_n4
Block 1 , ncf = 1047
Block 2 , ncf = 1862
Block 3 , ncf = 2112
Block 4 , ncf = 1537
Block 5 , ncf = 801
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
Restarting RCI90 ?
>>y
Calling lodres ...
Estimate contributions from the self-energy?
>>y
There are 5 blocks (block J/Parity NCF):
1 1/2+ 1047 2 3/2+ 1862 3 5/2+ 2112 4 7/2+ 1537
5 9/2+ 801
Enter ASF serial numbers for each block
Block 1 ncf = 1047 id = 1/2+
>>1-8
Block 2 ncf = 1862 id = 3/2+
>>1-11
Block 3 ncf = 2112 id = 5/2+
>>1-10
Block 4 ncf = 1537 id = 7/2+
>>1-5
Block 5 ncf = 801 id = 9/2+
>>1-2
Calling STRSUM...
Calling FACTT...
Calling GENINTRK...
Allocating space for 6071 Rk integrals
Calling GENINTBREIT1...
Computing 53106 Breit integrals of type 1
Calling GENINTBREIT2...
Computing 26494 Breit integrals of type 2
Calling MATRIX...
Loading CSF File for block 1
There are 1047 relativistic CSFs... load complete;
Entering QED ...
1047 (total 1047 ) rows read from .res
LAPACK routine DSPEVX selected for eigenvalue problem.
RCI92 MIXing coefficients File generated.
Loading CSF File for block 2
There are 1862 relativistic CSFs... load complete;
Entering QED ...
1862 (total 1862 ) rows read from .res
LAPACK routine DSPEVX selected for eigenvalue problem.
RCI92 MIXing coefficients File generated.
Loading CSF File for block 3
There are 2112 relativistic CSFs... load complete;
Entering QED ...
739 (total 2112 ) rows read from .res
Calling setham ...
Row 800 : 283 nonzero elements; block = 3
Row 900 : 258 nonzero elements; block = 3
Row 1000 : 393 nonzero elements; block = 3
Row 1100 : 224 nonzero elements; block = 3
Row 1200 : 250 nonzero elements; block = 3
Row 1300 : 104 nonzero elements; block = 3
Row 1400 : 297 nonzero elements; block = 3
Row 1500 : 375 nonzero elements; block = 3
Row 1600 : 224 nonzero elements; block = 3
Row 1700 : 106 nonzero elements; block = 3
Row 1800 : 141 nonzero elements; block = 3
Row 1900 : 128 nonzero elements; block = 3
Row 2000 : 208 nonzero elements; block = 3
Row 2100 : 111 nonzero elements; block = 3
Row 2111 : 74 nonzero elements; block = 3
Row 2112 : 113 nonzero elements; block = 3
nelmnt = 436242
Sparse - Memory, iniest2
RCI92 MIXing coefficients File generated.
Loading CSF File for block 4
There are 1537 relativistic CSFs... load complete;
Entering QED ...
0 (total 1537 ) rows read from .res
Calling setham ...
Row 1 : 1 nonzero elements; block = 4
Row 100 : 67 nonzero elements; block = 4
Row 200 : 62 nonzero elements; block = 4
Row 300 : 100 nonzero elements; block = 4
Row 400 : 83 nonzero elements; block = 4
Row 500 : 139 nonzero elements; block = 4
Row 600 : 223 nonzero elements; block = 4
Row 700 : 234 nonzero elements; block = 4
Row 800 : 321 nonzero elements; block = 4
Row 900 : 341 nonzero elements; block = 4
Row 1000 : 289 nonzero elements; block = 4
Row 1100 : 337 nonzero elements; block = 4
Row 1200 : 311 nonzero elements; block = 4
Row 1300 : 192 nonzero elements; block = 4
Row 1400 : 191 nonzero elements; block = 4
Row 1500 : 282 nonzero elements; block = 4
Row 1536 : 150 nonzero elements; block = 4
Row 1537 : 176 nonzero elements; block = 4
LAPACK routine DSPEVX selected for eigenvalue problem.
RCI92 MIXing coefficients File generated.
Loading CSF File for block 5
There are 801 relativistic CSFs... load complete;
Entering QED ...
0 (total 801 ) rows read from .res
Calling setham ...
Row 1 : 1 nonzero elements; block = 5
Row 100 : 13 nonzero elements; block = 5
Row 200 : 81 nonzero elements; block = 5
Row 300 : 48 nonzero elements; block = 5
Row 400 : 183 nonzero elements; block = 5
Row 500 : 207 nonzero elements; block = 5
Row 600 : 118 nonzero elements; block = 5
Row 700 : 162 nonzero elements; block = 5
Row 800 : 163 nonzero elements; block = 5
Row 801 : 184 nonzero elements; block = 5
LAPACK routine DSPEVX selected for eigenvalue problem.
RCI92 MIXing coefficients File generated.
Finish time, Statistics
Wall time:
54 seconds
Finish Date and Time:
Date (Yr/Mon/Day): 2018/07/20
Time (Hr/Min/Sec): 23/40/40.589
Zone: +0200
RCI: Execution complete.
During the restart, all matrix elements for blocks 1 and 2 were read from rci.res. For block 3 matrix elements up to row 739 were read and the restarted computation carries on from this point. Transforming from to coupling and displaying energies with rlevels shows that all the energies from the restarted rci calculation are identical to the ones in the fifth example. The restart option works the same for rci_mpi with the difference that the rci.res are read from the files defined in the disks file.
6.8. Eighth Example: in Be I, Transforming to Natural Orbitals, Using Option 4 in
rwfnestimate
The eighth example is for in Be I. The example shows the computation of rmcdhf and rci wave functions, and the subsequent transformation to natural orbitals TP Section 3.4. The rci calculation is redone in the natural orbitals basis, and we see how the expansion coefficients are concentrated to relatively fewer CSFs, potentially leading to smaller MR sets. In addition, we plot the radial density function , see TP Section 3.4.
Define nuclear data.
Obtain spectroscopic orbitals for the MR set.
- (a)
Generate configuration state list containing three CSFs generated from the configurations.
- (b)
Perform angular integration.
- (c)
Generate initial estimates of radial orbitals.
- (d)
Perform SCF calculation
- (e)
Save output to DF.
Improve the wave function
- (a)
Generate valence correlation expansion.
- (b)
Perform angular integration.
- (c)
Generate initial estimates of radial orbitals.
- (d)
Perform SCF calculation.
- (e)
Save output to n3.
- (f)
Generate valence correlation expansion.
- (g)
Perform angular integration.
- (h)
Generate initial estimates of radial orbitals, use option 4.
- (i)
Perform SCF calculation.
- (j)
Save output to n4.
- (k)
Perform rci calculation in which the transverse photon interaction (Breit) and vacuum polarization and self-energy (QED) corrections are added.
- (l)
Display the expansion coefficients for the rci wave function
Transform to natural orbitals
Perform rci calculation in the natural orbital basis in which the transverse photon interaction (Breit) and vacuum polarization and self-energy (QED) corrections are added.
Display the expansion coefficients for the rci wave functions in the natural orbital basis.
Plot the radial density distribution.
In the test-runs, prompt marked by >> or >>3, for example, indicates that the user should input 3 and then strike the return key. When >> is followed by blanks, just strike the return key.
*******************************************************************************
* RUN RNUCLEUS TO GENERATE NUCLEAR DATA AND DEFINE RADIAL GRID *
* OUTPUT FILE: isodata *
*******************************************************************************
>>rnucleus
Enter the atomic number:
>>4
Enter the mass number (0 if the nucleus is to be modelled as a point source:
>>9
The default root mean squared radius is 2.5190000534057617 fm; (Angeli)
the default nuclear skin thickness is 2.2999999999999998 fm;
Revise these values?
>>n
Enter the mass of the neutral atom (in amu) (0 if the nucleus is to be static):
>>9
Enter the nuclear spin quantum number (I) (in units of h / 2 pi):
>>1
Enter the nuclear dipole moment (in nuclear magnetons):
>>1
Enter the nuclear quadrupole moment (in barns):
>>1
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE LIST OF CSFs FOR 1s(2)2s(2) 1S J = 0. *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program generates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
OUTPUT FILES: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,i)
Give configuration 2
>>2p(2,i)
Give configuration 3
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>2s,2p
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,0
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>n
.........
1 blocks were created
block J/P NCSF
1 0+ 3
*******************************************************************************
* COPY FILES *
* IT IS ADVISABLE TO SAVE THE rcsfgenerate.log FILE TO HAVE A *
* RECORD ON HOW THE LIST OF CSFs WAS CREATED *
*******************************************************************************
>>cp rcsfgenerate.log DF.exc
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.log, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
.....
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* * MEANS ALL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp, rwfnestimate.log *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 4 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.4307D+01 0.1434D+02 0.1000D+01 0.4246D+00 332 T-F
2s 0.4126D+00 0.3424D+01 0.1000D+01 0.2336D+01 357 T-F
2p- 0.2827D+00 0.3699D-03 0.1000D+01 0.2430D+01 361 T-F
2p 0.2827D+00 0.2310D+01 0.2000D+01 0.2431D+01 361 T-F
RWFNESTIMATE: Execution complete.
Comment: <r> is the mean orbital radius in a.u. MTP is the extension of the orbitals on the grid, for which the upper limit in the default installation is 590 points. SRC is the source of the estimate, in this case T-F (Thomas-Fermi).
*******************************************************************************
* RUN RMCDHF_MEM TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: ORBITALS BUILDING REFERENCE STATES ARE REQUIRED TO HAVE *
* THE CORRECT NUMBER OF NODES. THEY ARE REFERRED TO AS SPECTROSCOPIC *
* ORBITALS. IN THIS RUN WE VARY 1s, 2s, 2p AND THEY ARE ALL *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* * MEANS ALL ORBITALS *
*******************************************************************************
>>rmcdhf_mem
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 4 relativistic subshells;
Loading CSF File for ALL blocks
There are 3 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 1 blocks (block J/Parity NCF):
1 0+ 3
Enter ASF serial numbers for each block
Block 1 ncf = 3 id = 0+
>>1
Radial functions
1s 2s 2p- 2p
Enter orbitals to be varied (Updating order)
>>*
Which of these are spectroscopic orbitals?
>>*
Enter the maximum number of SCF cycles:
>>100
..........
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog, name.log *
*******************************************************************************
>>rsave DF
Created DF.w, DF.c, DF.m, DF.sum DF.alog and DF.log
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE n = 3 VV CORRELATION LIST *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfile: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,*)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>3s,3p,3d
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,0
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
.........
1 blocks were created
block J/P NCSF
1 1/2+ 11
*******************************************************************************
* COPY FILES *
* IT IS ADVISABLE TO SAVE THE rcsfgenerate.log FILE TO HAVE A *
* RECORD ON HOW THE LIST OF CSFs WAS CREATED *
*******************************************************************************
>>cp rcsfgenerate.log n3.exc
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.log, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
...........
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>1
Enter the file name (Null then "rwfn.out")
>>
Enter the list of relativistic subshells:
>>*
The following subshell radial wavefunctions remain to be estimated:
3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.4712D+01 0.1475D+02 0.1000D+01 0.4128D+00 357 rwf
2s 0.3498D+00 0.2557D+01 0.1000D+01 0.2582D+01 360 rwf
2p- 0.4927D+00 0.2396D-03 0.1000D+01 0.2498D+01 358 rwf
2p 0.4927D+00 0.1497D+01 0.2000D+01 0.2498D+01 358 rwf
3s 0.1104D+00 0.1312D+01 0.1000D+01 0.7134D+01 372 T-F
3p- 0.8539D-01 0.1497D-03 0.1000D+01 0.8182D+01 375 T-F
3p 0.8538D-01 0.9348D+00 0.2000D+01 0.8183D+01 375 T-F
3d- 0.6409D-01 0.6710D-05 0.2000D+01 0.8778D+01 378 T-F
3d 0.6409D-01 0.5036D-01 0.3000D+01 0.8778D+01 378 T-F
RWFNESTIMATE: Execution complete.
Comment: please note how we used the wild card * twice. We start by reading the orbitals from a grasp file (previous run rwfn.out). Using the wild card * the program reads as many orbitals as possible, i.e., , , -, . The orbitals , -, , -, then remain to be estimated, and we use Thomas-Fermi estimates. By again using the wild card * all the remaining orbitals will be Thomas-Fermi estimates.
*******************************************************************************
* RUN RMCDHF_MEM TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: FOR CORRELATION ORBITALS THERE ARE NO RESTRICTIONS ON THE *
* NUMBER OF NODES, I.E. THEY ARE NOT SPECTROSCOPIC. IN THIS RUN WE *
* VARY THE CORRELATION ORBITALS 3s,3p, 3d. NONE OF THESE ARE *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* 3* MEANS 3s, 3p-, 3p, 3d-, 3d *
*******************************************************************************
>>rmcdhf_mem
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-consistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Loading CSF File for ALL blocks
There are 11 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 1 blocks (block J/Parity NCF):
1 1/2+ 11
Enter ASF serial numbers for each block
Block 1 ncf = 11 id = 1/2+
>>1
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Enter orbitals to be varied (Updating order)
>>3*
Which of these are spectroscopic orbitals?
>>
Enter the maximum number of SCF cycles:
>>100
..........
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog, name.log *
*******************************************************************************
>>rsave n3
Created n3.w, n3.c, n3.m, n3.sum n3.alog and n3.log
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE n = 4 VV CORRELATION LIST *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfile: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>1
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>2s(2,*)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>4s,4p,4d,4f
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,0
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
.........
1 blocks were created
block J/P NCSF
1 1/2+ 26
*******************************************************************************
* COPY FILES *
* IT IS ADVISABLE TO SAVE THE rcsfgenerate.log FILE TO HAVE A *
* RECORD ON HOW THE LIST OF CSFs WAS CREATED *
*******************************************************************************
>>cp rcsfgenerate.log n4.exc
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.log, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
...........
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d 4s 4p- 4p 4d- 4d 4f- 4f
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>1
Enter the file name (Null then "rwfn.out")
>>
Enter the list of relativistic subshells:
>>*
The following subshell radial wavefunctions remain to be estimated:
4s 4p- 4p 4d- 4d 4f- 4f
Read subshell radial wavefunctions. Choose one below
1--GRASP92 File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>4
Enter the list of relativistic subshells:
>>*
Enter increase in Z for correlation orbitals
>>5
Orbital Z_eff for hydrogenic orbitals
4s 7.00
4p- 7.00
4p 7.00
4d- 7.00
4d 7.00
4f- 7.00
4f 7.00
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.4712D+01 0.1475D+02 0.1000D+01 0.4128D+00 357 n3.
2s 0.3498D+00 0.2557D+01 0.1000D+01 0.2582D+01 360 n3.
2p- 0.4927D+00 0.2396D-03 0.1000D+01 0.2498D+01 358 n3.
2p 0.4927D+00 0.1497D+01 0.2000D+01 0.2498D+01 358 n3.
3s 0.8407D+00 0.4206D+01 0.1000D+01 0.3214D+01 361 n3.
3p- 0.1383D+01 0.3979D-03 0.1000D+01 0.3091D+01 358 n3.
3p 0.1382D+01 0.2507D+01 0.2000D+01 0.3093D+01 358 n3.
3d- 0.1001D+01 0.1148D-04 0.2000D+01 0.2655D+01 357 n3.
3d 0.1001D+01 0.8634D-01 0.3000D+01 0.2655D+01 357 n3.
4s 0.1532D+01 0.4624D+01 0.1000D+01 0.3427D+01 350 Hyd
4p- 0.1532D+01 0.2924D-02 0.1000D+01 0.3284D+01 350 Hyd
4p 0.1532D+01 0.1046D+02 0.2000D+01 0.3285D+01 350 Hyd
4d- 0.1532D+01 0.1478D-02 0.2000D+01 0.2999D+01 350 Hyd
4d 0.1531D+01 0.6343D+01 0.3000D+01 0.3000D+01 350 Hyd
4f- 0.1531D+01 0.3041D-03 0.3000D+01 0.2571D+01 349 Hyd
4f 0.1531D+01 0.1399D+01 0.4000D+01 0.2571D+01 349 Hyd
RWFNESTIMATE: Execution complete.
Please note how we use option 4. We have tested an increase in Z, in this case 5, so that the mean radii <r> of the new orbitals overlap approximately the region in space where we expect them. Since they describe valence correlation, they should have about the same radii as the orbitals.
*******************************************************************************
* RUN RMCDHF_MEM TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: FOR CORRELATION ORBITALS THERE ARE NO RESTRICTIONS ON THE *
* NUMBER OF NODES, I.E. THEY ARE NOT SPECTROSCOPIC. IN THIS RUN WE *
* VARY THE CORRELATION ORBITALS 4s,4p,4d,4f. NONE OF THESE ARE *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* 4* MEANS 4s, 4p-, 4p, 4d-, 4d, 4f-, 4f *
*******************************************************************************
>>rmcdhf_mem
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-consistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
Loading CSF File for ALL blocks
There are 26 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 1 blocks (block J/Parity NCF):
1 0+ 26
Enter ASF serial numbers for each block
Block 1 ncf = 26 id = 0+
>>1
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d 4s 4p- 4p 4d- 4d 4f- 4f
Enter orbitals to be varied (Updating order)
>>4*
Which of these are spectroscopic orbitals?
Enter the maximum number of SCF cycles:
>>100
..........
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog, name.log *
*******************************************************************************
>>rsave n4
Created n4.w, n4.c, n4.m, n4.sum n4.alog and n4.log
*******************************************************************************
* RUN RCI TO INCLUDE TRANSVERSE PHOTON INTERACTION AND QED EFFECTS *
* INPUT FILES : isodata, n4.c, n4.w *
* OUTPUT FILES: n4.cm, n4.csum, n4.clog, rci.res *
* *
* THE TRANSVERSE PHOTON FREQUENCIES CAN BE SET TO THE LOW FREQUENCY *
* LIMIT. RECOMMENDED IN CASES WHERE YOU HAVE CORRELATION ORBITALS *
* THE SELF ENERGY CORRECTION MAY FAIL FOR CORRELATION ORBITALS WITH *
* HIGH N. *
*******************************************************************************
>>rci
RCI
This is the configuration interaction program
Input file: isodata, name.c, name.w
Outputfiles: name.cm, name.csum, name.clog, rci.res
Default settings?
>>y
Name of state:
>>n4
Block 1 , ncf = 26
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
Include contribution of H (Transverse)?
>>y
Modify all transverse photon frequencies?
>>n
Include H (Vacuum Polarisation)?
>>y
Include H (Normal Mass Shift)?
>>n
Include H (Specific Mass Shift)?
>>n
Estimate self-energy?
>>y
Largest n quantum number for including self-energy for orbital
n should be less or equal 8
>>3
Loading Radial WaveFunction File ...
There are 1 blocks (block J/Parity NCF):
1 0+ 26
Enter ASF serial numbers for each block
Block 1 ncf = 26 id = 0+
>>1
......
RCI: Execution complete.
*******************************************************************************
* RUN RMIXEXTRACT TO DISPLAY THE MIXING COEFFICIENTS *
*******************************************************************************
>>rmixextract
RMIXEXTRACT
Extract and prints mixing coefficient above a
cut-off. Corresponding CSFs written to screen and
to rcsf.out
Input files: name.c, name.(c)m
Output file: rcsf.out
Name of state
>>n4
Mixing coefficients from CI calc. ?
>>y
Enter the cut-off value for the coefficients [0--1]
>>0
Sort extracted CSFs according to mixingcoeffcients? (y/n)
>>n
nblock = 1 ncftot = 26 nw = 16 nelec = 4
===========================================================================
nb = 1 ncfblk = 26 nevblk = 1 2J+1 = 1 parity = 1
nb = 1 ncfblk = 26 nevblk = 1 2J+1 = 1 parity = 1
===========================================================================
Average Energy = -12.602608908837407 ncf_reduced = 26
Energy = -14.620897436670143 Coefficients and CSF :
1 0.953738
2s ( 2)
0+
2 -0.001117
2s ( 1) 3s ( 1)
1/2 1/2
0+
3 -0.001846
2s ( 1) 4s ( 1)
1/2 1/2
0+
4 0.242750
2p ( 2)
0
0+
5 0.171674
2p-( 2)
0+
6 0.000254
2p ( 1) 3p ( 1)
3/2 3/2
0+
7 0.000302
2p ( 1) 4p ( 1)
3/2 3/2
0+
8 0.000178
2p-( 1) 3p-( 1)
1/2 1/2
0+
9 0.000214
2p-( 1) 4p-( 1)
1/2 1/2
0+
10 -0.039770
3s ( 2)
0+
11 -0.001052
3s ( 1) 4s ( 1)
1/2 1/2
0+
12 0.004905
3p ( 2)
0
0+
13 0.003467
3p-( 2)
0+
14 -0.000333
3p ( 1) 4p ( 1)
3/2 3/2
0+
15 -0.000237
3p-( 1) 4p-( 1)
1/2 1/2
0+
16 -0.013120
3d ( 2)
0
0+
17 -0.010712
3d-( 2)
0
0+
18 0.000530
3d ( 1) 4d ( 1)
5/2 5/2
0+
19 0.000432
3d-( 1) 4d-( 1)
3/2 3/2
0+
20 -0.004103
4s ( 2)
0+
21 0.001628
4p ( 2)
0
0+
22 0.001150
4p-( 2)
0+
23 -0.002808
4d ( 2)
0
0+
24 -0.002291
4d-( 2)
0
0+
25 0.004766
4f ( 2)
0
0+
26 0.004127
4f-( 2)
0
0+
RMIXEXTRACT: Execution complete.
*******************************************************************************
* RUN RDENSITY TO COMPUTE THE RADIAL DENSITY FUNCTION AND TRANSFORM *
* TO NATURAL ORBITALS. *
* INPUT FILES: isodata, n4.c, n4.w, n4.cm *
* OUTPUT FILE: n4.cd (density), n4.nw (natural orbitals) *
*******************************************************************************
>>redensity
RDENSITY: Execution begins ...
Default settings?
>>y
Name of state
>>n4
Mixing coefficients from a CI calc.?
>>y
Loading Configuration Symmetry List File ...
There are 16 relativistic subshells;
There are 26 relativistic CSFs;
... load complete;
Loading Radial WaveFunction File ...
nelec = 4
ncftot = 26
nw = 16
nblock = 1
block ncf nev 2j+1 parity
1 26 1 1 1
How do you want to order your egvc ?
1) By looking at the dominant component
2) Following the decreasing order of the egvl
>>2
......
RDENSITY: Execution complete.
*******************************************************************************
* COPY FILES TO PERFORM RCI CALCULATION IN THE NATURAL ORBITAL BASIS. *
*******************************************************************************
>>cp n4.c n4NO.c
>>cp n4.nw n4NO.w
*******************************************************************************
* RUN RCI TO INCLUDE TRANSVERSE PHOTON INTERACTION AND QED EFFECTS *
* INPUT FILES : isodata, n4NO.c, n4NO.w *
* OUTPUT FILES: n4NO.cm, n4NO.csum, n4NO.clog, rci.res *
* *
* THE TRANSVERSE PHOTON FREQUENCIES CAN BE SET TO THE LOW FREQUENCY *
* LIMIT. RECOMMENDED IN CASES WHERE YOU HAVE CORRELATION ORBITALS *
* THE SELF ENERGY CORRECTION MAY FAIL FOR CORRELATION ORBITALS WITH *
* HIGH N. *
*******************************************************************************
>>rci
RCI
This is the configuration interaction program
Input file: isodata, name.c, name.w
Outputfiles: name.cm, name.csum, name.clog, rci.res
Default settings?
>>y
Name of state:
>>n4NO
Block 1 , ncf = 26
Loading CSF file ... Header only
There are/is 16 relativistic subshells;
Include contribution of H (Transverse)?
>>y
Modify all transverse photon frequencies?
>>n
Include H (Vacuum Polarisation)?
>>y
Include H (Normal Mass Shift)?
>>n
Include H (Specific Mass Shift)?
>>n
Estimate self-energy?
>>y
Largest n quantum number for including self-energy for orbital
n should be less or equal 8
>>3
Loading Radial WaveFunction File ...
There are 1 blocks (block J/Parity NCF):
1 0+ 26
Enter ASF serial numbers for each block
Block 1 ncf = 26 id = 0+
>>1
......
RCI: Execution complete.
*******************************************************************************
* RUN RMIXEXTRACT TO DISPLAY THE MIXING COEFFICIENTS *
*******************************************************************************
>>rmixextract
RMIXEXTRACT
Extract and prints mixing coefficient above a
cut-off. Corresponding CSFs written to screen and
to rcsf.out
Input files: name.c, name.(c)m
Output file: rcsf.out
Name of state
>>n4NO
Mixing coefficients from CI calc. ?
>>y
Enter the cut-off value for the coefficients [0--1]
>>0
Sort extracted CSFs according to mixingcoeffcients? (y/n)
>>n
nblock = 1 ncftot = 26 nw = 16 nelec = 4
===========================================================================
nb = 1 ncfblk = 26 nevblk = 1 2J+1 = 1 parity = 1
nb = 1 ncfblk = 26 nevblk = 1 2J+1 = 1 parity = 1
===========================================================================
Average Energy = -12.602609084238223 ncf_reduced = 26
Energy = -14.620897382271725 Coefficients and CSF :
1 0.953740
2s ( 2)
0+
2 -0.000000
2s ( 1) 3s ( 1)
1/2 1/2
0+
3 -0.000000
2s ( 1) 4s ( 1)
1/2 1/2
0+
4 0.242750
2p ( 2)
0
0+
5 0.171674
2p-( 2)
0+
6 0.000000
2p ( 1) 3p ( 1)
3/2 3/2
0+
7 0.000000
2p ( 1) 4p ( 1)
3/2 3/2
0+
8 0.000000
2p-( 1) 3p-( 1)
1/2 1/2
0+
9 0.000000
2p-( 1) 4p-( 1)
1/2 1/2
0+
10 -0.039787
3s ( 2)
0+
11 -0.000000
3s ( 1) 4s ( 1)
1/2 1/2
0+
12 0.004922
3p ( 2)
0
0+
13 0.003479
3p-( 2)
0+
14 -0.000000
3p ( 1) 4p ( 1)
3/2 3/2
0+
15 -0.000000
3p-( 1) 4p-( 1)
1/2 1/2
0+
16 -0.013134
3d ( 2)
0
0+
17 -0.010723
3d-( 2)
0
0+
18 -0.000000
3d ( 1) 4d ( 1)
5/2 5/2
0+
19 -0.000000
3d-( 1) 4d-( 1)
3/2 3/2
0+
20 -0.004089
4s ( 2)
0+
21 0.001611
4p ( 2)
0
0+
22 0.001138
4p-( 2)
0+
23 -0.002794
4d ( 2)
0
0+
24 -0.002280
4d-( 2)
0
0+
25 0.004766
4f ( 2)
0
0+
26 0.004127
4f-( 2)
0
0+
RMIXEXTRACT: Execution complete.
We see that the energy is invariant, but the weights have been concentrated to relatively fewer CSFs. The weights for many CSFs are now zero.
The
rdensity program also outputs the file
n4.cd file that contains the radial density distribution
for each grid point, see
Section 8.4 for a discussion of the file structure. In
Figure 3 we have plotted
as a function of
r in a.u.
6.9. Ninth Example: Magnetic-Field- and Hyperfine-Induced Transitions in Ni XXV
The ninth example is for the unexpected transition
in Ni XXV, see [
42]. The example shows the computation of Zeeman and hyperfine interaction matrix using the
hfszeeman95 program, with given
rci wave functions, and the use of Matlab program
mithit to compute the transition rates between magnetic fine-structure substates in the presence of an external magnetic field and the rates of hyperfine induced transitions in the field-free limit.
Define nuclear data.
Obtain common spectroscopic orbitals for the MR set.
- (a)
Generate configuration state list for MR set {, , and }.
- (b)
Perform angular integration.
- (c)
Generate initial estimates of radial orbitals.
- (d)
Perform SCF calculation on the weighted average of all states belonging to and .
- (e)
Save output to mr.
Improve even states
- (a)
Generate n = 3 valence–valence CSF expansions.
- (b)
Perform angular integration.
- (c)
Generate initial estimates of radial orbitals.
- (d)
Perform SCF calculation on the weighted average of the even state.
- (e)
Save output to even_n3.
- (f)
Perform rci calculation in which the transverse photon interaction (Breit) and vacuum polarization and self-energy (QED) corrections are added.
Transform from - to -coupling
Improve odd states
- (a)
Generate n = 3 valence–valence CSF expansions.
- (b)
Perform angular integration.
- (c)
Generate initial estimates of radial orbitals.
- (d)
Perform SCF calculation on the weighted average of the odd states.
- (e)
Save output to odd_n3.
- (f)
Perform rci calculation in which the transverse photon interaction (Breit) and vacuum polarization and self-energy (QED) corrections are added.
Transform from - to -coupling
Calculate properties
- (a)
Calculate Zeeman and hyperfine interaction matrix using the rci wave functions.
- (b)
Compute the transition rates from the rci wave functions. Calculation in two steps: biorthonormal transformation and evaluation of transition matrix elements using standard Racah algebra methods. The latter procedure is done using rtransition_phase.
- (c)
Compute the magnetic-field-induced transition rate at B = 3 tesla.
- (d)
Compute the hyperfine-induced transition rate in the field-free limit.
In the test-runs, prompt marked by >> or >>3, for example, indicates that the user should input 3 and then strike the return key. When >> is followed by blanks, just strike the return key.
*******************************************************************************
* RUN RNUCLEUS TO GENERATE NUCLEAR DATA AND DEFINE RADIAL GRID *
* OUTPUT FILE: isodata *
*******************************************************************************
>>rnucleus
Enter the atomic number:
>>28
Enter the mass number (0 if the nucleus is to be modelled as a point source:
>>61
The default root mean squared radius is 3.8224999904632568 fm; (Angeli)
the default nuclear skin thickness is 2.2999999999999998 fm;
Revise these values?
>>n
Enter the mass of the neutral atom (in amu) (0 if the nucleus is to be static):
>>58.6934
Enter the nuclear spin quantum number (I) (in units of h / 2 pi):
>>1.5
Enter the nuclear dipole moment (in nuclear magnetons):
>>-0.75002
Enter the nuclear quadrupole moment (in barns):
>>0.162
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE LIST OF CSFs FOR *
* CONFIGURATIONS 2s(2), 2p(2), 2s2p *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program creates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,i)2s(2,i)
Give configuration 2
>>1s(2,i)2p(2,i)
Give configuration 3
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>2s,2p
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,0
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>y
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,i)2s(1,i)2p(1,i)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>2s,2p
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,4
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>0
Generate more lists ? (y/n)
>>n
.........
4 blocks were created
block J/P NCSF
1 0+ 3
2 0- 1
3 1- 2
4 2- 1
*******************************************************************************
* COPY FILES *
*******************************************************************************
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.log, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
.....
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* * MEANS ALL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp, rwfnestimate.log *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 4 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.3902D+03 0.3381D+03 0.1000D+01 0.5289D-01 328 T-F
2s 0.9484D+02 0.1211D+03 0.1000D+01 0.2123D+00 344 T-F
2p- 0.9460D+02 0.1104D+01 0.1000D+01 0.1766D+00 344 T-F
2p 0.9357D+02 0.9101D+03 0.2000D+01 0.1791D+00 344 T-F
RWFNESTIMATE: Execution complete.
*******************************************************************************
* RUN RMCDHF TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: ORBITALS BUILDING REFERENCE STATES ARE REQUIRED TO HAVE *
* THE CORRECT NUMBER OF NODES. THEY ARE REFERRED TO AS SPECTROSCOPIC *
* ORBITALS. IN THIS RUN WE VARY 1s, 2s, 2p AND THEY ARE ALL *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* * MEANS ALL ORBITALS *
*******************************************************************************
>>rmcdhf
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 4 relativistic subshells;
Loading CSF File for ALL blocks
There are 7 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 4 blocks (block J/Parity NCF):
1 0+ 3 2 0- 1 3 1- 2 4 2- 1
Enter ASF serial numbers for each block
Block 1 ncf = 3 id = 0+
>>1
Block 2 ncf = 1 id = 0-
>>1
Block 3 ncf = 2 id = 1-
>>1,2
Block 4 ncf = 1 id = 2-
>>1
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p
Enter orbitals to be varied (Updating order)
>>*
Which of these are spectroscopic orbitals?
>>*
Enter the maximum number of SCF cycles:
>>100
..........
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog, name.log *
*******************************************************************************
>>rsave mr
Created mr.w, mr.c, mr.m, mr.sum mr.alog and mr.log
*******************************************************************************
* RUN RCSFGENERATE TO GENERATE n = 3 VALENCE-VALENCE *
* CORRELATION LIST FOR EVEN STATE *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
*******************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program generates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,i)2s(2,*)
Give configuration 2
>>1s(2,i)2p(2,*)
Give configuration 3
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>3s,3p,3d
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,0
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
.........
1 blocks were created
block J/P NCSF
1 0+ 11
*******************************************************************************
* COPY FILES *
*******************************************************************************
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.log, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
...........
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>1
Enter the file name (Null then "rwfn.out")
>>mr.w
Enter the list of relativistic subshells:
>>*
The following subshell radial wavefunctions remain to be estimated:
3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
2
Enter the list of relativistic subshells:
>>*
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.3671D+03 0.3339D+03 0.1000D+01 0.5367D-01 341 mr.
2s 0.8431D+02 0.1134D+03 0.1000D+01 0.2235D+00 347 mr.
2p- 0.8236D+02 0.9734D+00 0.1000D+01 0.1894D+00 347 mr.
2p 0.8154D+02 0.8034D+03 0.2000D+01 0.1919D+00 347 mr.
3s 0.4070D+02 0.6530D+02 0.1000D+01 0.4835D+00 355 T-F
3p- 0.4058D+02 0.6470D+00 0.1000D+01 0.4481D+00 354 T-F
3p 0.4028D+02 0.5359D+03 0.2000D+01 0.4517D+00 355 T-F
3d- 0.4007D+02 0.1028D+01 0.2000D+01 0.3798D+00 354 T-F
3d 0.3997D+02 0.1070D+04 0.3000D+01 0.3812D+00 354 T-F
RWFNESTIMATE: Execution complete.
*******************************************************************************
* RUN RMCDHF TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: FOR CORRELATION ORBITALS THERE ARE NO RESTRICTIONS ON THE *
* NUMBER OF NODES, I.E. THEY ARE NOT SPECTROSCOPIC. IN THIS RUN WE *
* VARY THE CORRELATION ORBITALS 3s,3p, 3d. NONE OF THESE ARE *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* 3* MEANS 3s, 3p-, 3p, 3d-, 3d *
*******************************************************************************
>>rmcdhf
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Loading CSF File for ALL blocks
There are 11 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 1 blocks (block J/Parity NCF):
1 0+ 11
Enter ASF serial numbers for each block
Block 1 ncf = 11 id = 0+
>>1
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Enter orbitals to be varied (Updating order)
>>3*
Which of these are spectroscopic orbitals?
>>
Enter the maximum number of SCF cycles:
>>100
..........
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog, name.log *
*******************************************************************************
>>rsave even_n3
Created even_n3.w, even_n3.c, even_n3.m, even_n3.sum even_n3.alog and even_n3.log
*******************************************************************************
* RUN RCI TO INCLUDE TRANSVERSE PHOTON INTERACTION AND QED EFFECTS *
* INPUT FILES : isodata, even_n3.c, even_n3.w *
* OUTPUT FILES: even_n3.cm, even_n3.csum, even_n3.clog, rci.res *
* *
* THE TRANSVERSE PHOTON FREQUENCIES CAN BE SET TO THE LOW FREQUENCY *
* LIMIT. RECOMMENDED IN CASES WHERE YOU HAVE CORRELATION ORBITALS *
* THE SELF ENERGY CORRECTION MAY FAIL FOR CORRELATION ORBITALS WITH *
* HIGH N. *
*******************************************************************************
>>rci
RCI
This is the configuration interaction program
Input file: isodata, name.c, name.w
Outputfiles: name.cm, name.csum, name.clog
rci.res (can be used for restart)
Default settings?
>>y
Name of state:
>>even_n3
Block 1 , ncf = 11
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Include contribution of H (Transverse)?
>>y
Modify all transverse photon frequencies?
>>y
Enter the scale factor:
>>1.d-6
Include H (Vacuum Polarisation)?
>>y
Include H (Normal Mass Shift)?
>>n
Include H (Specific Mass Shift)?
>>n
Estimate self-energy?
>>y
Largest n quantum number for including self-energy for orbital
n should be less or equal 8
>>3
Loading Radial WaveFunction File ...
There are 1 blocks (block J/Parity NCF):
1 0+ 11
Enter ASF serial numbers for each block
Block 1 ncf = 11 id = 0+
>>1
......
RCI: Execution complete.
*******************************************************************************
* RUN JJ2LSJ TO TRANSFORM FROM JJ- TO LSJ-COUPLING *
* INPUT FILES: even_n3.c, even_n3.cm *
* OUTPUT FILE: even_n3.lsj.lbl, even_n3.uni.lsj.lbl *
*******************************************************************************
>>jj2lsj
jj2lsj: Transformation of ASFs from a jj-coupled CSF basis
into an LSJ-coupled CSF basis (Fortran 95 version)
(C) Copyright by G. Gaigalas and Ch. F. Fischer,
(2021).
Input files: name.c, name.(c)m
Output files: name.lsj.lbl
(optional) name.lsj.c, name.lsj.j,
name.uni.lsj.lbl, name.uni.lsj.sum
Name of state
>>even_n3
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 11 relativistic CSFs;
... load complete;
Mixing coefficients from a CI calc.?
>>y
Do you need a unique labeling? (y/n)
>>y
nelec = 4
ncftot = 11
nw = 9
nblock = 1
block ncf nev 2j+1 parity
1 11 1 1 1
Default settings? (y/n)
>>y
....
jj2lsj: Execution Complete
********************************************************************************
* RUN RCSFGENERATE TO GENERATE n = 3 VALENCE-VALENCE *
* CORRELATION LIST FOR ODD STATE *
* OUTPUT FILES: rcsfgenerate.log, rcsf.out *
********************************************************************************
>>rcsfgenerate
RCSFGENERATE
This program generates a list of CSFs
Configurations should be entered in spectroscopic notation
with occupation numbers and indications if orbitals are
closed (c), inactive (i), active (*) or has a minimal
occupation e.g., 1s(2,1)2s(2,*)
Outputfiles: rcsf.out, rcsfgenerate.log
Default, reverse, symmetry or user specified ordering? (*/r/s/u)
>>*
Select core
0: No core
1: He ( 1s(2) = 2 electrons)
2: Ne ([He] + 2s(2)2p(6) = 10 electrons)
3: Ar ([Ne] + 3s(2)3p(6) = 18 electrons)
4: Kr ([Ar] + 3d(10)4s(2)4p(6) = 36 electrons)
5: Xe ([Kr] + 4d(10)5s(2)5p(6) = 54 electrons)
6: Rn ([Xe] + 4f(14)5d(10)6s(2)6p(6) = 86 electrons)
>>0
Enter list of (maximum 100) configurations. End list with a blank line or an asterisk (*)
Give configuration 1
>>1s(2,i)2s(1,*)2p(1,*)
Give configuration 2
>>
Give set of active orbitals, as defined by the highest principal quantum number
per l-symmetry, in a comma delimited list in s,p,d etc order, e.g., 5s,4p,3d
>>3s,3p,3d
Resulting 2*J-number? lower, higher (J=1 -> 2*J=2 etc.)
>>0,4
Number of excitations (if negative number e.g., -2, correlation
orbitals will always be doubly occupied)
>>2
Generate more lists ? (y/n)
>>n
.........
3 blocks were created
block J/P NCSF
1 0- 6
2 1- 14
3 2- 12
*******************************************************************************
* COPY FILES *
*******************************************************************************
>>cp rcsf.out rcsf.inp
*******************************************************************************
* RUN RANGULAR TO GENERATE ENERGY EXPRESSION *
* INPUT FILE : rcsf.inp *
* OUTPUT FILES: rangular.log, mcp.30, mcp.31,.... *
*******************************************************************************
>>rangular
RANGULAR
This program performs angular integration
Input file: rcsf.inp
Outputfiles: mcp.30, mcp.31, ....
rangular.log
Full interaction? (y/n)
>>y
...........
RANGULAR: Execution complete.
*******************************************************************************
* RUN RWFNESTIMATE TO GENERATE INITIAL ESTIMATES FOR RADIAL ORBITALS *
* INPUT FILES: isodata, rcsf.inp, previous rwfn files *
* OUTPUT FILE: rwfn.inp *
*******************************************************************************
>>rwfnestimate
RWFNESTIMATE
This program estimates radial wave functions
for orbitals
Input files: isodata, rcsf.inp, optional rwfn file
Output file: rwfn.inp
Default settings ?
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
The following subshell radial wavefunctions remain to be estimated:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>1
Enter the file name (Null then "rwfn.out")
>>mr.w
Enter the list of relativistic subshells:
>>*
The following subshell radial wavefunctions remain to be estimated:
3s 3p- 3p 3d- 3d
Read subshell radial wavefunctions. Choose one below
1--GRASP2K File
2--Thomas-Fermi
3--Screened Hydrogenic
4--Screened Hydrogenic [custom Z]
>>2
Enter the list of relativistic subshells:
>>*
All required subshell radial wavefunctions have been estimated:
Shell e p0 gamma <r> MTP SRC
1s 0.3671D+03 0.3339D+03 0.1000D+01 0.5367D-01 341 mr.
2s 0.8431D+02 0.1134D+03 0.1000D+01 0.2235D+00 347 mr.
2p- 0.8236D+02 0.9734D+00 0.1000D+01 0.1894D+00 347 mr.
2p 0.8154D+02 0.8034D+03 0.2000D+01 0.1919D+00 347 mr.
3s 0.4070D+02 0.6530D+02 0.1000D+01 0.4835D+00 355 T-F
3p- 0.4058D+02 0.6470D+00 0.1000D+01 0.4481D+00 354 T-F
3p 0.4028D+02 0.5359D+03 0.2000D+01 0.4517D+00 355 T-F
3d- 0.4007D+02 0.1028D+01 0.2000D+01 0.3798D+00 354 T-F
3d 0.3997D+02 0.1070D+04 0.3000D+01 0.3812D+00 354 T-F
RWFNESTIMATE: Execution complete.
*******************************************************************************
* RUN RMCDHF TO OBTAIN SELF CONSISTENT SOLUTIONS *
* INPUT FILES: isodata, rcsf.inp, rwfn.inp, mcp.30, mcp.31,... *
* OUTPUT FILES: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log *
* *
* NOTE: FOR CORRELATION ORBITALS THERE ARE NO RESTRICTIONS ON THE *
* NUMBER OF NODES, I.E. THEY ARE NOT SPECTROSCOPIC. IN THIS RUN WE *
* VARY THE CORRELATION ORBITALS 3s,3p, 3d. NONE OF THESE ARE *
* SPECTROSCOPIC. WE CAN USE WILD CARDS * FOR SPECIFYING ORBITALS *
* 3* MEANS 3s, 3p-, 3p, 3d-, 3d *
*******************************************************************************
>>rmcdhf
RMCDHF
This program determines the radial orbitals
and the expansion coefficients of the CSFs
in a self-onsistent field proceedure
Input file: isodata, rcsf.inp, rwfn.inp, mcp.30, ...
Outputfiles: rwfn.out, rmix.out, rmcdhf.sum, rmcdhf.log
Default settings? (y/n)
>>y
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Loading CSF File for ALL blocks
There are 32 relativistic CSFs... load complete;
Loading Radial WaveFunction File ...
There are 3 blocks (block J/Parity NCF):
1 0- 6 2 1- 14 3 2- 12
Enter ASF serial numbers for each block
Block 1 ncf = 6 id = 0-
>>1
Block 2 ncf = 14 id = 1-
>>1,2
Block 3 ncf = 12 id = 2-
>>1
level weights (1 equal; 5 standard; 9 user)
>>5
Radial functions
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
Enter orbitals to be varied (Updating order)
>>3*
Which of these are spectroscopic orbitals?
>>
Enter the maximum number of SCF cycles:
>>100
..........
RMCDHF: Execution complete.
*******************************************************************************
* RUN RSAVE TO SAVE OUTPUT FILES: name.c, name.w, name.m, name.sum *
* name.alog, name.log *
*******************************************************************************
>>rsave odd_n3
Created odd_n3.w, odd_n3.c, odd_n3.m, odd_n3.sum odd_n3.alog and odd_n3.log
*******************************************************************************
* RUN RCI TO INCLUDE TRANSVERSE PHOTON INTERACTION AND QED EFFECTS *
* INPUT FILES : isodata, odd_n3.c, odd_n3.w *
* OUTPUT FILES: odd_n3.cm, odd_n3.csum, odd_n3.clog, rci.res *
* *
* THE TRANSVERSE PHOTON FREQUENCIES CAN BE SET TO THE LOW FREQUENCY *
* LIMIT. RECOMMENDED IN CASES WHERE YOU HAVE CORRELATION ORBITALS *
* THE SELF ENERGY CORRECTION MAY FAIL FOR CORRELATION ORBITALS WITH *
* HIGH N. *
*******************************************************************************
>>rci
RCI
This is the configuration interaction program
Input file: isodata, name.c, name.w
Outputfiles: name.cm, name.csum, name.clog
rci.res (can be used for restart)
Default settings?
>>y
Name of state:
>>odd_n3
Block 1 , ncf = 6
Block 2 , ncf = 14
Block 3 , ncf = 12
Loading CSF file ... Header only
There are/is 9 relativistic subshells;
Include contribution of H (Transverse)?
>>y
Modify all transverse photon frequencies?
>>y
Enter the scale factor:
>>1.d-6
Include H (Vacuum Polarisation)?
>>y
Include H (Normal Mass Shift)?
>>n
Include H (Specific Mass Shift)?
>>n
Estimate self-energy?
>>y
Largest n quantum number for including self-energy for orbital
n should be less or equal 8
>>3
Loading Radial WaveFunction File ...
There are 3 blocks (block J/Parity NCF):
1 0- 6 2 1- 14 3 2- 12
Enter ASF serial numbers for each block
Block 1 ncf = 6 id = 0-
>>1
Block 2 ncf = 14 id = 1-
>>1,2
Block 3 ncf = 12 id = 2-
>>1
......
RCI: Execution complete.
*******************************************************************************
* RUN JJ2LSJ TO TRANSFORM FROM JJ- TO LSJ-COUPLING *
* INPUT FILES: odd_n3.c, odd_n3.cm *
* OUTPUT FILE: odd_n3.lsj.lbl, odd_n3.uni.lsj.lbl *
*******************************************************************************
>>jj2lsj
jj2lsj: Transformation of ASFs from a jj-coupled CSF basis
into an LSJ-coupled CSF basis (Fortran 95 version)
(C) Copyright by G. Gaigalas and Ch. F. Fischer,
(2021).
Input files: name.c, name.(c)m
Output files: name.lsj.lbl
(optional) name.lsj.c, name.lsj.j,
name.uni.lsj.lbl, name.uni.lsj.sum
Name of state
>>odd_n3
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 11 relativistic CSFs;
... load complete;
Mixing coefficients from a CI calc.?
>>y
Do you need a unique labeling? (y/n)
>>y
nelec = 4
ncftot = 32
nw = 9
nblock = 3
block ncf nev 2j+1 parity
1 6 1 1 -1
2 14 2 3 -1
3 12 1 5 -1
Default settings? (y/n)
>>y
....
jj2lsj: Execution Complete
********************************************************************************
* RUN HFSZEEMAN95 FOR even_n3 *
* INPUT FILES: isodata, even_n3 .c, even_n3 .w, even_n3 .cm *
* OUTPUT FILE: even_n3 .ch, even_n3 .cgjhfs *
********************************************************************************
>>hfszeeman95
HFSZEEMAN95
This is the magnetic interaction program
Input files: isodata, name.c, name.(c)m, name.w
Output files: name.(c)h, name.(c)gjhfs
HFSZEEMAN95: Execution begins ...
Default settings?
>>y
Name of state
>>even_n3
Mixing coefficients from a CI calc.?
>>y
Calculate off-diagonal matrix elements?
>>y
....
HFSZEEMAN95: Execution complete.
********************************************************************************
* RUN HFSZEEMAN95 FOR odd_n3 *
* INPUT FILES: isodata, odd_n3 .c, odd_n3 .w, odd_n3 .cm *
* OUTPUT FILE: odd_n3 .ch, odd_n3 .cgjhfs *
********************************************************************************
>>hfszeeman95
HFSZEEMAN95
This is the magnetic interaction program
Input files: isodata, name.c, name.(c)m, name.w
Output files: name.(c)h, name.(c)gjhfs
HFSZEEMAN95: Execution begins ...
Default settings?
>>y
Name of state
>>odd_n3
Mixing coefficients from a CI calc.?
>>y
Calculate off-diagonal matrix elements?
>>y
....
HFSZEEMAN95: Execution complete.
********************************************************************************
* VIEW ZEEMAN AND HYPERFINE INTERACTION MATRX FOR ODD STATES *
********************************************************************************
>>more odd_n3.cgjhfs
Number of relativistic eigenvalues
4
Lev J Parity E
1 2.0 - -944.099340681
1 1.0 - -944.694737339
2 1.0 - -942.723168239
1 0.0 - -944.876941705
Zeeman interaction matrix
0.18322E+01 -0.34691E+00 0.68227E-01 0.00000E+00
0.44786E+00 0.10439E+01 -0.67174E-01 0.40125E+00
-0.88081E-01 -0.67174E-01 0.71718E+00 -0.78350E-01
0.00000E+00 -0.69499E+00 0.13571E+00 0.00000E+00
HFI-matrix for the magnetic dipole operator
0.36367E+02 -0.10508E+02 0.27000E+02 0.00000E+00
0.13566E+02 0.36112E+02 0.22640E+02 0.15292E+02
-0.34857E+02 0.22640E+02 -0.18141E+01 0.81431E+01
0.00000E+00 -0.26486E+02 -0.14104E+02 0.00000E+00
HFI-matrix for the electric quadrupole operator
0.28620E+03 0.32475E+03 -0.59600E+02 -0.22196E+03
-0.41925E+03 -0.22145E+03 0.14396E+03 0.00000E+00
0.76944E+02 0.14396E+03 0.46833E+03 0.00000E+00
-0.49632E+03 -0.00000E+00 -0.00000E+00 0.00000E+00
********************************************************************************
* RUN RBIOTRANSFORM FOR even_n3 AND odd_n3 TO TRANSFORM WAVE FUNCTIONS *
* INPUT FILES: isodata, even_n3.c, even_n3.w, even_n3.cm, *
* odd_n3.c, odd_n3.w, odd_n3.cm *
* OUTPUT FILES: even_n3 .cbm, even_n3 .bw, odd_n3.cbm, odd_n3.bw *
* even_n3.TB, odd_n3.TB (angular files) *
* NOTE THAT THE ORDER OF INITIAL AND FINAL STATE DOES NOT MATTER *
********************************************************************************
>>rbiotransform
RBIOTRANSFORM
This program transforms the initial and final wave
functions so that standard tensor albegra can be
used in evaluation of the transition parameters
Input files: isodata, name1.c, name1.w, name1.(c)m
name2.c, name2.w, name2.(c)m
name1.TB, name2.TB (optional angular files)
Output files: name1.bw, name1.(c)bm,
name2.bw, name2.(c)bm
name1.TB, name2.TB (angular files)
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>even_n3
Name of the Final state
>>odd_n3
Transformation of all J symmetries?
>>y
....
BIOTRANSFORM: Execution complete.
*****************************************************************************************
* RUN RTRANSITION_PHASE FOR even_n3 and odd_n3 TO COMPUTE TRANSITION PARAMETERS *
* INPUT FILES: isodata, even_n3.c, even_n3.bw, even_n3.cbm *
* odd_n3.c, odd_n3.bw, odd_n3.cbm *
* OUTPUT FILES: even_n3.odd_n3.ct *
* odd_n3.odd_n3.-1T (angular file) *
* NOTE THAT THE ORDER OF INITIAL AND FINAL STATE DOES NOT MATTER *
*****************************************************************************************
>>rtransition_phase
RTRANSITION
This program computes transition parameters from
transformed wave functions
Input files: isodata, name1.c, name1.bw, name1.(c)bm
name2.c, name2.bw, name2.(c)bm
optional, name1.lsj.lbl, name2.lsj.lbl
name1.name2.KT (optional angular files)
Output files: name1.name2.(c)t
optional, name1.name2.(c)t.lsj
name1.name2.KT (angular files)
Here K is parity and rank of transition: -1,+1 etc
Default settings?
>>y
Input from a CI calculation?
>>y
Name of the Initial state
>>even_n3
Name of the Final state
>>odd_n3
MRGCSL: Execution begins ...
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 11 relativistic CSFs;
... load complete;
Loading Configuration Symmetry List File ...
There are 9 relativistic subshells;
There are 32 relativistic CSFs;
... load complete;
1 s
2 s
2 p-
2 p
3 s
3 p-
3 p
3 d-
3 d
1
11
3
6 20 32
Loading Configuration Symmetry List File ...
there are 9 relativistic subshells;
there are 43 relativistic CSFs;
... load complete;
Enter the list of transition specifications
e.g., E1,M2 or E1 M2 or E1;M2 :
>>E1,M2
.....
RTRANSITION: Execution complete.
*******************************************************************************
* VIEW COMPUTED TRANSITION PARAMETERS *
*******************************************************************************
>>more even_n3.odd_n3.ct
Transition between files:
f1 = even_n3
f2 = odd_n3
Electric 2**( 1)-pole transitions
=================================
Upper Lower
Lev J P Lev J P E (Kays) A (s-1) gf S M
f2 1 1 - f1 1 0 + 419612.86 C 1.04269D+08 2.66340D-03 2.08960D-03 -4.57121D-02
B 8.48871D+07 2.16832D-03 1.70118D-03 -4.12454D-02
f2 2 1 - f1 1 0 + 852322.26 C 2.56686D+10 1.58918D-01 6.13827D-02 -2.47755D-01
B 2.38815D+10 1.47854D-01 5.71090D-02 -2.38975D-01
Magnetic 2**( 2)-pole transitions
=================================
Upper Lower
Lev J P Lev J P E (Kays) A (s-1) gf S M
f2 1 2 - f1 1 0 + 550287.32 M 1.40086D+01 3.46773D-10 9.30999D-01 -9.64883D-01
Comment: The reduced transition matrix elements M given in the even_n3.odd_n3.ct are need by the Matlab program mithit for the computation of magnetic-field- and hyperfine-induced transitions.
*******************************************************************************
* RUN MATLAB PROGRAM MITHIT FOR MAGNETIC-FIELD-INDUCED TRANSITION AT *
* MAGNETIC FIELD STRENGTH B = 3 TESLA *
* INPUT FILES: even_n3.cgjhfs, odd_n3.cgjhfs, even_n3.odd_n3.ct *
* OUTPUT FILES: even_n3.czm, odd_n3.czm, even_n3.odd_n3.fs.mit.mtrans *
*******************************************************************************
>>mithit
Name of the Initial state:
>>even_n3
Name of the Final state:
>>odd_n3
Are the calculations based on a relativistic CI calculation? (Y/N)
>>y
MIT-fs(0), HIT(1) or MIT-hfs(2):
>>0
B-field in Tesla (0) or Gauss (1):
>>0
Give the upper limit for the B-field:
>>3
Energies in a.u. (0), cm-1 (1) or MHz (2) ?
>>1
Start Computation of Energies and Mixing Coefficients
of the Magnetic Sublevels of Initial States
level E_fs (a.u.) J
-----------------------------------------------
1 -946.606634183 0
Would you like a plot of Zeeman splitting with B field? (Y/N)
>>n
Finished even_n3
Start Computation of Energies and Mixing Coefficients
of the Magnetic Sublevels of Final States
level E_fs (a.u.) J
-----------------------------------------------
1 -944.099340681 2
2 -944.694737339 1
3 -942.723168239 1
4 -944.876941705 0
Would you like a plot of Zeeman splitting with B field? (Y/N)
>> n
Finished odd_n3
Would you like to compute the transition rates? (Y/N)
>>y
level E_BP (a.u.) J
-------------------------------
Initial levels:
1 -946.606634183 0
Final levels:
1 -944.099340681 2
2 -944.694737339 1
3 -942.723168239 1
4 -944.876941705 0
Give an index vector of the initial levels(lower level):
>>1
Give an index vector of the final levels(upper level):
>>4
Would you like a plot of synthetic spectra? (Y/N)
>>n
MITHIT finished
********************************************************************************
* VIEW COMPUTED MAGNETIC-FIELD-INDUCED TRANSITION RATE *
********************************************************************************
>>more even_n3.odd_n3.fs.mit.mtrans
Magnetic field
B = 3.0000000 Tesla
Fine structure energies in a.u.
even_n3
level J E_BP (a.u.)
1 0.0 -946.606634
odd_n3
level J E_BP (a.u.)
1 2.0 -944.099341
2 1.0 -944.694737
3 1.0 -942.723168
4 0.0 -944.876942
Transition rates and wavenumbers in Kays
Upper Lower
level J M_J E_hfs (a.u.) FS-LEV level J M_J E_hfs (a.u.) FS-LEV A (s-1) E (Kays)
4 0.0 0.0 -944.876941705 4 1 0.0 0.0 -946.606634183 1 4.0607E-02 379623.6178
********************************************************************************
* RUN MATLAB PROGRAM MITHIT FOR HYPERFINE-INDUCED TRANSITION *
* INPUT FILES: even_n3.cgjhfs, odd_n3.cgjhfs, even_n3.odd_n3.ct *
* OUTPUT FILES: even_n3.czm, odd_n3.czm, even_n3.odd_n3.hfs.hit.trans *
********************************************************************************
>>mithit
Name of the Initial state:
>>even_n3
Name of the Final state:
>>odd_n3
Are the calculations based on a relativistic CI calculation? (Y/N)
>>y
MIT-fs(0), HIT(1) or MIT-hfs(2):
>>1
Nuclear spin I:
>>1.5
Nuclear magnetic dipole moment mu:
>> -0.75002
Nuclear electric quadrupole moment Q:
>>0.162
Start Computation of Energies and Mixing Coefficients
of the Magnetic Sublevels of Initial States
level E_hfs (a.u.) FS-LEV J F
-----------------------------------------------
1 -946.606634183 1 0 3/2
Finished even_n3
Start Computation of Energies and Mixing Coefficients
of the Magnetic Sublevels of Final States
level E_hfs (a.u.) FS-LEV J F
-----------------------------------------------
1 -944.099384680 1 2 7/2
2 -944.694775514 2 1 5/2
3 -944.099333945 1 2 5/2
4 -942.723166078 3 1 5/2
5 -944.876941710 4 0 3/2
6 -944.694711495 2 1 3/2
7 -944.099296427 1 2 3/2
8 -942.723170502 3 1 3/2
9 -944.694674496 2 1 1/2
10 -944.099273405 1 2 1/2
11 -942.723170188 3 1 1/2
Finished odd_n3
Would you like to compute the transition rates? (Y/N)
>>y
level E_hfs (a.u.) FS-LEV J F
------------------------------------------------------------
Initial levels:
1 -946.606634183 1 0 3/2
Final levels:
1 -944.099384680 1 2 7/2
2 -944.099333945 1 2 5/2
3 -944.099296427 1 2 3/2
4 -944.099273405 1 2 1/2
5 -944.694775514 2 1 5/2
6 -944.694711495 2 1 3/2
7 -944.694674496 2 1 1/2
8 -942.723166078 3 1 5/2
9 -942.723170502 3 1 3/2
10 -942.723170188 3 1 1/2
11 -944.876941710 4 0 3/2
Give an index vector of the initial levels(lower level):
>>1
Give an index vector of the final levels(upper level):
>>11
Would you like a plot of synthetic spectra? (Y/N)
>>n
MITHIT finished
********************************************************************************
* VIEW COMPUTED HYPERFINE-INDUCED TRANSITION RATE *
********************************************************************************
>>more even_n3.odd_n3.hfs.hit.trans
Nuclear data
Nuclear spin 1.500000 au
Nuclear magnetic dipole moment -0.750020 n.m.
Nuclear electric quadrupole moment 0.162000 barns
Hyperfine structure energies in a.u.
even_n3
level J F E_hfs (a.u.) FS-LEV
1 0.0 1.5 -946.606634 1
odd_n3
level J F E_hfs (a.u.) FS-LEV
1 2.0 3.5 -944.099385 1
2 2.0 2.5 -944.099334 1
3 2.0 1.5 -944.099296 1
4 2.0 0.5 -944.099273 1
5 1.0 2.5 -944.694776 2
6 1.0 1.5 -944.694711 2
7 1.0 0.5 -944.694674 2
8 1.0 2.5 -942.723166 3
9 1.0 1.5 -942.723171 3
10 1.0 0.5 -942.723170 3
11 0.0 1.5 -944.876942 4
Transition rates and wavenumbers in Kays
Upper Lower
level J F E_hfs (a.u.) FS-LEV level J F E_hfs (a.u.) FS-LEV A (s-1) E (Kays)
11 0.0 1.5 -944.876941710 4 1 0.0 1.5 -946.606634183 1 2.6057E+00 379623.6167
8. Interpreting the Output Files
In this section, we describe in detail what information can be found in the different output files and how this information should be interpreted
8.1. Output Files from the First Example
Below is the
isodata file for the Li example,
Section 6.1.
Atomic number:
3.0000000000000000
Mass number (integer) :
7.0000000000000000
Fermi distribution parameter a:
0.52338755531043146
Fermi distribution parameter c:
1.2520789669753825
Mass of nucleus (in amu):
6.9393542602910001
Nuclear spin (I) (in units of h / 2 pi):
1.5000000000000000
Nuclear dipole moment (in nuclear magnetons):
3.2564267999999998
Nuclear quadrupole moment (in barns):
-4.0000000000000001E-002
The calculation was for Li with and . The nuclear charge distribution was modelled as an extended Fermi distribution with
The parameters a and c are computed from the root mean squared radius and the skin thickness. The root mean squared radius is taken from the tables of Angeli and Marinova [Atomic Data and Nuclear Data Tables Volume 99, Issue 1, 69-95, (2013)]. This gives fm and fm. On the lines following these quantities, the nuclear mass and nuclear spin I are given, along with the nuclear magnetic dipole moment in nuclear magnetons and the nuclear quadrupole moment Q in barns.
After each rcsfgenerate run, there is a log-file displaying the response to the different questions. Below is the rcsfgenerate.log file from the complete active space expansion for .
* ! Orbital order
0 ! Selected core
1s(2,*)2p(1,*)
*
3s,3p,3d
1 3 ! Lower and higher 2*J
3 ! Number of excitations
n
The log-file is a copy of the input data. By executing the command
rcsfgenerate < rcsfgenerate.log
the rcsf.out file will be reproduced. The rcsfgenerate.log file can easily be edited to give a new list of CSFs. For example
* ! Orbital order
0 ! Selected core
1s(2,*)2p(1,*)
*
4s,4p,4d,4f
1 3 ! Lower and higher 2*J
3 ! Number of excitations
n
will give a file rcsf.out with CSFs corresponding to an active set .
The rcsfgenerate program produces an rcsf.out file. The file has a header with information about the radial orbitals and the closed shells (core shells). After this information, there is a list of CSFs. The CSFs are ordered in blocks with specified value of J. Each block is separated by a line with an asterisk. Below is the file 2p_3.c.
Core subshells:
Peel subshells:
1s 2s 2p- 2p 3s 3p- 3p 3d- 3d
CSF(s):
1s ( 2) 2p-( 1)
1/2
1/2-
1s ( 2) 3p-( 1)
1/2
1/2-
1s ( 1) 2s ( 1) 2p ( 1)
1/2 1/2 3/2
1 1/2-
..............
3p-( 1) 3d-( 1) 3d ( 1)
1/2 3/2 5/2
2 1/2-
3p-( 1) 3d-( 2)
1/2 0
1/2-
*
1s ( 2) 2p ( 1)
3/2
3/2-
1s ( 2) 3p ( 1)
3/2
3/2-
1s ( 1) 2s ( 1) 2p ( 1)
1/2 1/2 3/2
0 3/2-
............
3p-( 1) 3d-( 1) 3d ( 1)
1/2 3/2 5/2
2 3/2-
3p-( 1) 3d-( 2)
1/2 2
3/2-
The line with core subshells is empty, and in this case we have no closed core. The radial orbitals are , , -, , , -, , -, . After the radial orbitals, there are lists of CSFs arranged in blocks. The first block of CSFs has . The second block has . An asterisk is separating the blocks. In the file, each CSF occupies three lines. On the first line the subshells and their occupations are listed in a linear form where, for example, becomes 1s ( 2). The second line shows the coupling of each subshell to a J quantum number, and the third line shows how the J quantum numbers of each subshell are coupled from left to right to a final J, see TP Section 2.4.
The program rangular produces a log-file displaying the response to the different questions. After running rsave this file is saved in name.alog. Below is the log-file 2p_3.alog
y ! Full interaction
The log-file is a copy of the input data. We see that angular data were computed for all interactions. The log-file is more useful in cases where we do not have full interaction, see
Section 14.1. In these cases, the file contains information about the zero-order space.
The SCF program rmcdhf produces a log-file displaying the response to the different questions. After running rsave this file is saved in name.log. Below is the log-file 2p_3.log from the run on weighted average of the states.
y ! Default settings
1
1
5 ! level weights
3*
100 ! Number of SCF cycles
The log-file is a copy of the input data. We see that the run was with default settings (there will be no log-file for non-default settings). It was a calculation targeting the first levels of the two blocks (ASF serial numbers 1 for each of the two blocks). The level weight is 5 (default option), meaning that the levels, in the energy functional, are weighted according to the statistical weight , see TP Section 2.7, e.g., (44). On the line that follows, 3* means that orbitals with principal quantum numbers 3 are optimized. The blank line indicates that none of the optimized orbitals are spectroscopic. 100 SCF cycles were requested. By executing the command
rmcdhf < rmcdhf.log (or rmcdhf < name.log)
the rmcdhf run will be executed again with the settings in rmcdhf.log. The log-file can easily be edited and used as an input also to other runs.
The SCF program rmcdhf produces a summary file. After running rsave this file is saved in name.sum. Below is the summary file 2p_3.sum from the run on weighted average of the states.
There are 3 electrons in the cloud
in 186 relativistic CSFs
based on 9 relativistic subshells.
The atomic number is 3.0000000000;
the mass of the nucleus is 1.264966898269D+04 electron masses;
Fermi nucleus:
c = 3.612753059646D-05 Bohr radii,
a = 9.890591370096D-06 Bohr radii;
there are 82 tabulation points in the nucleus.
Speed of light = 137.0359991390D+00 atomic units.
Radial grid: R(I) = RNT*(exp((I-1)*H)-1), I = 1, ..., N;
RNT = 6.666666666667D-07 Bohr radii;
H = 5.000000000000D-02 Bohr radii;
N = 590;
R(1) = 0.000000000000D+00 Bohr radii;
R(2) = 3.418073091735D-08 Bohr radii;
R(N) = 4.110372988964D+06 Bohr radii.
EOL calculation.
2 levels will be optimised;
their indices are: 1, 1.
Each is assigned its statistical weight;
Radial wavefunction summary:
Self
Subshell e p0 gamma P(2) Q(2) Consistency MTP
1s 2.5177395314D+00 9.280D+00 1.00 3.172D-07 -3.513D-12 0.000D+00 355
2s 1.9634308400D-01 1.452D+00 1.00 4.965D-08 -5.499D-13 0.000D+00 361
2p- 1.2867248397D-01 5.116D-05 1.00 2.518D-15 1.598D-10 0.000D+00 366
2p 1.2866992757D-01 4.265D-01 2.00 4.983D-16 -5.519D-21 0.000D+00 366
3s 8.0600844816D+00 1.181D+01 1.00 5.027D-07 -7.981D-13 8.965D-08 360
3p- 8.7786093395D+00 2.853D-03 1.00 1.994D-13 8.921D-09 8.940D-07 364
3p 8.7823537644D+00 2.381D+01 2.00 2.786D-14 -1.173D-18 1.303D-06 364
3d- 1.6298092328D+01 8.146D-03 2.00 2.740D-20 1.739D-15 6.785D-06 358
3d 1.6306599649D+01 8.169D+01 3.00 3.262D-21 -3.617D-26 8.697D-06 358
-3 -1 2 4 Generalised
Subshell < r > < r > < r > < r > < r > occupation
1s 0.00000D+00 2.68556D+00 5.73199D-01 4.47081D-01 5.33751D-01 1.99386D+00
2s 0.00000D+00 3.45596D-01 3.87317D+00 1.77347D+01 5.65669D+02 2.01584D-04
2p- 0.00000D+00 2.65023D-01 4.79564D+00 2.78265D+01 1.47509D+03 3.33335D-01
2p 5.85643D-02 2.65011D-01 4.79578D+00 2.78280D+01 1.47522D+03 6.66669D-01
3s 0.00000D+00 3.09463D+00 8.79428D-01 1.76019D+00 3.83411D+01 2.52035D-03
3p- 0.00000D+00 1.94750D+00 6.74894D-01 8.40900D-01 2.20221D+01 1.08534D-03
3p 1.72790D+01 1.94712D+00 6.74750D-01 8.40046D-01 2.19775D+01 2.17087D-03
3d- 1.01792D+01 1.82956D+00 6.31485D-01 4.57970D-01 4.14736D-01 6.48046D-05
3d 1.01575D+01 1.82935D+00 6.31441D-01 4.57852D-01 4.14179D-01 9.72851D-05
Eigenenergies:
Level J Parity Hartrees Kaysers eV
1 1/2 - -7.404576963163D+00 -1.625116799442D+06 -2.014888020446D+02
1 3/2 - -7.404574103806D+00 -1.625116171886D+06 -2.014887242376D+02
Weights of major contributors to ASF:
Block Level J Parity CSF contributions
1 1 1/2 - 0.9985 -0.0343 0.0326 0.0230 0.0107
1 56 58 60 30
2 1 3/2 - 0.9985 -0.0343 0.0326 0.0230 0.0099
1 62 67 71 32
The first lines of the file tell us that the calculation was for a three electron system and that there were in total 186 CSFs built on 9 relativistic radial orbitals. After this, there is information about the nucleus. In this case, the nucleus has
and a mass of
electron masses. The nuclear charge distribution is modelled by a Fermi distribution with
Bohr radii, and
Bohr radii. There is information about the radial grid used in the calculation. The grid is given by
with
Bohr radii and
Bohr radii. In the current implementation of
grasp,
with
, see TP Section 2.2, Equation (8). There are
grid points.
We see that it is an EOL calculation and that the calculation was on the lowest state (first eigenvalue) of each block (
and
). In the optimization, each state is weighted according to the statistical weight
, see TP Section 2.7, Equation (44). The information on optimization is followed by a radial orbital summary. Important characteristics of a radial orbital are the orbital energy eigenvalue and parameters that determine the behavior near
. The radial amplitudes
can be expanded in power series
near the origin where the index
,
, and
are constants that depend on the nuclear potential model. In the radial orbital summary
e is orbital energy eigenvalue,
p0 is a parameter related to the leading expansion coefficients of the radial amplitudes and
gamma is the exponent
, for details see I.P. Grant, Relativistic Quantum Theory of Atoms and Molecules, Springer 2007, p 272–273 and also the subroutine
start in
lib92.
P(2) and
Q(2) are the values of the radial amplitudes at the first grid point
R(2) away from zero. Then the self-consistency (weighted change of an orbital during an iteration) is given for each orbital. In this case, the orbitals
,
,
-,
were kept frozen, and they thus have a self-consistency of zero. The orbitals
,
-,
,
-,
were optimized and the self-consistency is between
for
and
for
. Finally, the value
MTP gives the number of the outermost grid point used for representing the radial amplitudes of the orbital. At the remaining grid points, the radial amplitudes of the orbital are set to zero. Around 360 of the available 590 grid points are utilized.
Different radial expectation values
of the orbitals are given along with the generalized occupation numbers. The generalized occupation number
of an orbital
is defined as
where
is the number of electrons in subshell
in CSF
r and
is the generalized weight
In the expression for the generalized weight the sum is over all levels in the EOL calculation. are the mixing coefficients of level i in the basis of the CSFs. An orbital with a small generalized occupation number is associated with CSFs that have small expansion coefficients.
At the end of the summary file, the eigenenergies for the states are displayed in different energy units, where Kayser is the synonym of cm. The weights of the major CSF contributors are also given. Please note that the CSFs in this case are counted block wise.
The relativistic configuration interaction program rci produces a log-file displaying the response to the different questions. This file is saved in name.clog. Below is the log-file 2p_3.clog from the run of the states.
y ! Default settings
2p_3
y ! Contribution of H Transverse?
y ! Modify photon frequencies?
9.9999999999999995E-007 ! Scale factor
y ! Vacuum polarization?
n ! Normal mass shift?
n ! Specific mass shift?
y ! Self energy?
3 ! Max n for including self energy
1
1
The log-file is a copy of the input data. The name of the state is 2p_3 and full interaction was included. Contributions from the transverse interaction (Breit) were added, where the photon frequencies were multiplied with a factor , see TP Section 2.3, Equation (11). Vacuum polarization as well as self-energy corrections were added. The self-energy corrections are based on estimations for the orbitals. For correlation orbitals with high principal quantum numbers, these estimations may fail. In this case, self-energy corrections were based on orbitals with principal quantum numbers smaller than 3. Finally, we see that the calculation determined the first levels of the two blocks (ASF serial numbers 1 for each of the two blocks). By executing the command
rci < 2p_3.clog
the rci run will be executed again with the settings in 2p_3.clog. The log-file can easily be edited and used as an input also to other runs.
The rci program produces a summary file name.csum. Below is the summary file 2p_3.csum from the run on weighted average of the states
There are 3 electrons in the cloud
in 186 relativistic CSFs
based on 9 relativistic subshells.
The atomic number is 3.0000000000;
the mass of the nucleus is 1.264966898269D+04 electron masses;
Fermi nucleus:
c = 3.612753059646D-05 Bohr radii,
a = 9.890591370096D-06 Bohr radii;
there are 82 tabulation points in the nucleus.
Speed of light = 1.370359991390D+02 atomic units.
To H (Dirac Coulomb) is added
H (Transverse) --- factor multiplying the photon frequency: 1.00000000D-06;
H (Vacuum Polarisation);
the total will be diagonalised.
Diagonal contributions from H (Self Energy) will be estimated
from a screened hydrogenic approximation.
Radial grid: R(I) = RNT*(exp((I-1)*H)-1), I = 1, ..., N;
RNT = 6.666666666667D-07 Bohr radii;
H = 5.000000000000D-02 Bohr radii;
N = 590;
R(1) = 0.000000000000D+00 Bohr radii;
R(2) = 3.418073091735D-08 Bohr radii;
R(N) = 4.110372988964D+06 Bohr radii.
Subshell radial wavefunction summary:
Subshell e p0 gamma P(2) Q(2) MTP
1s 2.5177395314D+00 9.280D+00 1.00 3.172D-07 -3.513D-12 355
2s 1.9634308400D-01 1.452D+00 1.00 4.965D-08 -5.499D-13 361
2p- 1.2867248397D-01 5.116D-05 1.00 2.518D-15 1.598D-10 366
2p 1.2866992757D-01 4.265D-01 2.00 4.983D-16 -5.519D-21 366
3s 8.0600844816D+00 1.181D+01 1.00 5.027D-07 -7.981D-13 360
3p- 8.7786093395D+00 2.853D-03 1.00 1.994D-13 8.921D-09 364
3p 8.7823537644D+00 2.381D+01 2.00 2.786D-14 -1.173D-18 364
3d- 1.6298092328D+01 8.146D-03 2.00 2.740D-20 1.739D-15 358
3d 1.6306599649D+01 8.169D+01 3.00 3.262D-21 -3.617D-26 358
...........
Information about number of radial integrals, density of the
Hamiltonian matrix etc, the energies and the leading CSFs for each
level etc.
From the summary file, we again see what operators were included in the Hamiltonian. Information about the grid and the orbitals, same as in the name.sum file is also available.
The rhfs program computes hyperfine structure data. In addition, the Landé -factor is computed. Below is the output file 2p_3.ch, edited to fit the page, from the rhfs run for the rci wave function, given in the 2p_3.c, 2p_3.w and 2p_3.cm files, of the states.
Nuclear spin 1.500000000000000D+00 au
Nuclear magnetic dipole moment 3.256426800000000D+00 n.m.
Nuclear electric quadrupole moment -4.000000000000000D-02 barns
Interaction constants:
Level1 J Parity A (MHz) B (MHz) total g_J
1 1/2 - 4.4821853986D+01 -0.0000000000D+00 6.6588395646D-01
1 3/2 - -3.5378452915D+00 -1.7729096327D-01 1.3340987050D+00
At the top, the nuclear spin and moments are displayed. Then, for each level, the A and B hyperfine interaction constants, see TP Section 3.1 Equations (59)–(60) are given in MHz. In addition, the Landé -factors, TP Section 3.2 Equation (66), are given.
The rhfs program gives another file 2p_3.choffd, which contains off-diagonal hyperfine data
Nuclear spin 1.500000000000000D+00 au
Nuclear magnetic dipole moment 3.256426800000000D+00 n.m.
Nuclear electric quadrupole moment -4.000000000000000D-02 barns
Interaction constants:
Level1 J Parity Level2 J Parity A (MHz) B (MHz)
1 1/2 - 1 1/2 - 4.4822178831D+01 -0.0000000000D+00
1 3/2 - 1 1/2 - 1.1768857887D+01 -3.8388220012D-02
1 3/2 - 1 3/2 - -3.5381700218D+00 -1.7729096288D-01
Matrix elements:
Level1 J Parity Level2 J Parity F Matrix element (a.u.)
1 1/2 - 1 1/2 - 1 -8.5152606438D-09
1 1/2 - 1 1/2 - 2 5.1091563863D-09
Matrix elements:
Level1 J Parity Level2 J Parity F Matrix element (a.u.)
1 3/2 - 1 1/2 - 1 4.0297075799D-09
1 3/2 - 1 1/2 - 2 5.3525245724D-09
Matrix elements:
Level1 J Parity Level2 J Parity F Matrix element (a.u.)
1 3/2 - 1 3/2 - 0 1.9828496377D-09
1 3/2 - 1 3/2 - 1 1.4720532074D-09
1 3/2 - 1 3/2 - 2 4.2351513722D-10
1 3/2 - 1 3/2 - 3 -1.2166549923D-09
Given are diagonal and off-diagonal hyperfine interaction constants
A and
B in MHz and the
F dependent hyperfine matrix elements in atomic units. The above quantities are defined in [
8], Equations (13)–(17) and Equations (7)–(8).
The ris4 program computes mass- and field shift isotope data. Below is the output file 2p_3.ci, edited to fit the page, from the ris4 run for the rci wave function, given in the 2p_3.c, 2p_3.w and 2p_3.cm files, of the states.
Number of eigenvalues: 2
Level J Parity Energy
1 1/2 - -0.7404260995D+01 (a.u.)
1 3/2 - -0.7404259683D+01 (a.u.)
Level J Parity Normal mass shift parameter
<K^1 > <K^2+K^3> <K^1+K^2+K^3>
1 1/2 - 0.7409611828D+01 -0.6671237484D-02 0.7402940590D+01 (a.u.)
0.2674486353D+05 -0.2407971433D+02 0.2672078382D+05 (GHz u)
<K^1 > <K^2+K^3> <K^1+K^2+K^3>
1 3/2 - 0.7409602908D+01 -0.6657064450D-02 0.7402945843D+01 (a.u.)
0.2674483134D+05 -0.2402855701D+02 0.2672080278D+05 (GHz u)
Level J Parity Specific mass shift parameter
<K^1 > <K^2+K^3> <K^1+K^2+K^3>
1 1/2 - 0.2425644688D+00 -0.1746264308D-03 0.2423898424D+00 (a.u.)
0.8755321826D+03 -0.6303110296D+00 0.8749018716D+03 (GHz u)
<K^1 > <K^2+K^3> <K^1+K^2+K^3>
1 3/2 - 0.2425741100D+00 -0.1915018511D-03 0.2423826081D+00 (a.u.)
0.8755669823D+03 -0.6912225626D+00 0.8748757597D+03 (GHz u)
Level J Parity Electron density in atomic units
Dens. (a.u.)
1 1/2 - 0.1372240739D+02
1 3/2 - 0.1372240990D+02
Level J Parity Field shift electronic factors and average point discrepancy in fit
F0 (GHz/fm^2) F2 (GHz/fm^4) F4 (GHz/fm^6)
1 1/2 - 0.2025876387D+00 -0.3303847114D-05 0.5227748000D-07
1 3/2 - 0.2025876757D+00 -0.3303847831D-05 0.5227749057D-07
F6 (GHz/fm^8) Disc. (per mille)
1 1/2 - -0.6985943239D-09 0.0000
1 3/2 - -0.6985944586D-09 0.0000
Level J Parity Field shift electronic factors (corrected for varying density inside nucleus)
F0VED0 (GHz/fm^2) F0VED1 (GHz/fm^4)
1 1/2 - 0.2025433326D+00 -0.2805899138D-05
1 3/2 - 0.2025433696D+00 -0.2805899756D-05
We see that there are two eigenvalues for which the energies are printed. After that, for each level, the normal mass shift parameters, decomposed in three parts, see [
12] Section 3.2, Equation (41) and TP Section 3.3, Equation (73), are given in (a.u.) and (GHz u). After the normal mass shift parameters, the specific mass shift parameters, decomposed in three parts, see [
12] Section 3.2, Equation (41) and TP Section 3.3, Equation (74), are given in (a.u.) and (GHz u). Next, the electron density at the origin,
, is given in a.u. After that follow the field shift electronic factors,
, as defined in [
12], Section 3.3, Equation (18), see also TP Section 3.3, Equation (79). To estimate the effect on the field shift from the varying electronic density (ved) inside the nuclear volume, the quantity
is introduced, see [
12] Section 4, Equation (39). The latter can be expressed in terms of
and
, see [
12] Section 4, Equations (47) and (48). These parameters are displayed at the end of the output file.
The rtransition program computes transition data. Below is the output file 2s_3.2p_3.ct from the rtransition electric dipole E1 run for rci wave functions given in the 2s_3.c, 2s_3.w, 2s_3.cm and 2p_3.c, 2p_3.w and 2p_3.cm files.
Transition between files:
f1 = 2s_3
f2 = 2p_3
Electric 2**( 1)-pole transitions
=================================
Upper Lower
File Lev J P File Lev J P E (Kays) A (s-1) gf S
f2 1 1/2 - f1 1 1/2 + 14861.28 C 3.81311D+07 5.17671D-01 1.14676D+01
B 3.74756D+07 5.08773D-01 1.12705D+01
f2 1 3/2 - f1 1 1/2 + 14861.57 C 3.81334D+07 1.03537D+00 2.29353D+01
B 3.74782D+07 1.01758D+00 2.25413D+01
The first lines of the file give the name of the files defining the wave functions. Then data are given for the electric dipole transition E1. The first transition is from the upper level 1 with and negative parity in file f2, i.e., to the lower level 1 with and positive parity in file f1, i.e., . The second transition is from the upper level 1 with and negative parity in file f2, i.e., to the lower level 1 with and positive parity in file f1, i.e., . For each transition the transition energy E is given in Kaysers (cm). Additionally, the transition rate A in emission (Einstein A-coefficient), the weighted oscillator strength and the line strength S are given in Coulomb (velocity) and Babushkin (length) gauge.
If the 2s_3.lsj.lbl and 2p_3.lsj.lbl files produced by jj2lsj are available at the run of rtransition an additional output file 2s_3.2p_3.ct.lsj is produced. This file is shown below
Transition between files:
2s_3
2p_3
1 -7.47197402 1s(2).2s_2S
1 -7.40426103 1s(2).2p_2P
14861.28 CM-1 6728.89 ANGS(VAC) 6728.20 ANGS(AIR)
E1 S = 1.12705D+01 GF = 5.08773D-01 AKI = 3.74756D+07 dT = 0.01719
1.14676D+01 5.17671D-01 3.81311D+07
1 -7.47197402 1s(2).2s_2S
3 -7.40425972 1s(2).2p_2P
14861.57 CM-1 6728.76 ANGS(VAC) 6728.06 ANGS(AIR)
E1 S = 2.25413D+01 GF = 1.01758D+00 AKI = 3.74782D+07 dT = 0.01718
2.29353D+01 1.03537D+00 3.81334D+07
The 2s_3.2p_3.ct.lsj file has a different format. Here, the labels of the upper and lower states in the transition are in -notation. The J quantum number (multiplied by 2) is written to the left. The transition energy is given in cm and the wavelengths (vacuum and air) in angstrom (ANGS) where 1 ANGS m. The line strength S, the weighted oscillator strength and the transition rates in emission A (AKI) are given on two lines, where the upper line corresponds to the Babushkin (length) gauge and the lower line to the Coulomb (velocity) gauge. Finally,
is a parameter that can be related to the estimated uncertainty of the transition rates [
43].
8.2. Output Files from the Third Example
The third example case, see
Section 6.3, was calculations of the states belonging to the
and
configurations in Si VIII.
The jj2lsj program transforms from to coupling and gives the composition of the states. Below is the output file 2s22p3_2p5_3.lsj.lbl from the jj2lsj run of the rci wave functions given in the 2s22p3_2p5_3.c, 2s22p3_2p5_3.w, 2s22p3_2p5_3.cm files. For each case, the first line gives the position (number) of the eigenstate in the interaction matrix, parity, total energy and the percentage of the ASF that has been transformed. Thus, 99.907 % implies that 0.093 % has not been transformed.
Pos J Parity Energy Total Comp. of ASF
1 1/2 - -262.790633876 99.907%
0.98656029 0.97330122 1s(2).2s(2).2p(3)2P1_2P
0.15010905 0.02253273 1s(2).2p(5)_2P
-0.03364614 0.00113206 1s(2).2s_2S.2p(3)2P1_1P.3d_2P
2 1/2 - -259.497939898 99.123%
0.98251140 0.96532866 1s(2).2p(5)_2P
-0.14989835 0.02246951 1s(2).2s(2).2p(3)2P1_2P
-0.03674439 0.00135015 1s(2).2s_2S.2p(3)2D3_1D.3d_2P
0.03527443 0.00124429 1s(2).2s_2S.2p(3)2P1_3P.3d_2P
1 3/2 - -263.279784072 99.550%
0.99652486 0.99306180 1s(2).2s(2).2p(3)4S3_4S
0.03703202 0.00137137 1s(2).2s(2).2p(3)2P1_2P
2 3/2 - -262.955055547 99.670%
0.98954100 0.97919139 1s(2).2s(2).2p(3)2D3_2D
-0.12139907 0.01473773 1s(2).2s(2).2p(3)2P1_2P
-0.03690740 0.00136216 1s(2).2s_2S.2p(3)4S3_3S.3d_2D
3 3/2 - -262.788274233 99.912%
0.97818840 0.95685254 1s(2).2s(2).2p(3)2P1_2P
0.15010551 0.02253166 1s(2).2p(5)_2P
0.12276473 0.01507118 1s(2).2s(2).2p(3)2D3_2D
-0.03672205 0.00134851 1s(2).2s(2).2p(3)4S3_4S
-0.03335975 0.00111287 1s(2).2s_2S.2p(3)2P1_1P.3d_2P
4 3/2 - -259.524117905 99.004%
0.98234136 0.96499455 1s(2).2p(5)_2P
-0.15102023 0.02280711 1s(2).2s(2).2p(3)2P1_2P
0.03537700 0.00125153 1s(2).2s_2S.2p(3)2P1_3P.3d_2P
1 5/2 - -262.953820595 99.429%
0.99713868 0.99428554 1s(2).2s(2).2p(3)2D3_2D
There is a total of seven states. For each state, the file gives the -expansion. The lowest state (pos 1) with negative parity and energy a.u. has the -expansion
0.98656029 0.97330122 1s(2).2s(2).2p(3)2P1_2P
0.15010905 0.02253273 1s(2).2p(5)_2P
-0.03364614 0.00113206 1s(2).2s_2S.2p(3)2P1_1P.3d_2P
The second-lowest state (pos 2) with negative parity and energy a.u. has the -expansion
0.98251140 0.96532866 1s(2).2p(5)_2P
-0.14989835 0.02246951 1s(2).2s(2).2p(3)2P1_2P
-0.03674439 0.00135015 1s(2).2s_2S.2p(3)2D3_1D.3d_2P
0.03527443 0.00124429 1s(2).2s_2S.2p(3)2P1_3P.3d_2P
We see that the states are close to pure -coupling and the file provides meaningful labels that match labels given in, for example, the NIST data tables. The second column in the table gives the -composition, i.e., the squared expansion coefficients.
Finally, a few words about how to interpret the notation in the composition of the ASF.
Each subshell in the configuration is given with occupation, term designation and seniority. When the subshell is singly or fully occupied, the term designation and seniority are not written out. The term for each subshell is the coupled from left to right. Intermediate couplings are given after the underscore sign _.
In
Table 7 below, there is a list of possible terms and their seniority for commonly occurring subshells.
As a specific example of how to interpret the notation, we look at
1s(2).2s_2S.2p(3)2P1_3P.3d_2P
The first subshell 1s(2) is fully occupied and have only one term 1S0 that is not written out explicitly. The second subshell 2s is singly occupied and has only one term 2S1 that is not written out explicitly. The third subshell 2p(3) is coupled to an term 2P1. The fourth subshell 3d is singly occupied and has only one term 2D1 that is not written out explicitly. Coupling 1S0 and 2S1 of subshells one and two leads to an intermediate term _2S. Coupling _2S with 2P1 of the third subshell leads to the intermediate term _3P. Finally, coupling _3P with 2D1 of the fourth subshells gives the final term 2P.
The programs rtablevels and rtabtransE1 implement a LaTeX translation of the ASCII notation. In the LaTeX translation, the term and seniority of a subshell are given in parenthesis just after the subshell. For the intermediate terms, the underscore of the ASCII notation has been replaced by a space. In the LaTeX translation, the user also has a choice to omit the closed core. Translating the above example to LaTeX and omitting the 1s(2) we get
Please note how the seniority enters as a subscript.
The rtransition program computes transition data. Below is the output file
2s22p3_2p5_3.2s22p3_2p5_3.ct from the rtransition magnetic dipole M1 run of the rci wave functions given in the 2s22p3_2p5_3.c, 2s22p3_2p5_3.w, 2s22p3_2p5_3.cm files giving the states belonging to the and configurations
Transition in file:
f = 2s22p3_2p5_3
Magnetic 2**( 1)-pole transitions
=================================
Upper Lower
Lev J P Lev J P E (Kays) A (s-1) gf S
f 2 1/2 - f 1 1/2 - 722662.80 M 6.00621D-05 3.44839D-16 1.18001D-11
f 3 3/2 - f 1 1/2 - 517.88 M 1.22716D-03 2.74383D-08 1.31018D+00
f 4 3/2 - f 1 1/2 - 716917.39 M 4.29992D+00 5.01695D-11 1.73052D-06
f 1 1/2 - f 1 3/2 - 107356.06 M 3.11300D+01 8.09867D-09 1.86549D-03
f 2 1/2 - f 1 3/2 - 830018.86 M 4.56677D+00 1.98756D-11 5.92158D-07
f 1 1/2 - f 2 3/2 - 36086.39 M 1.27613D+01 2.93830D-08 2.01353D-02
f 2 1/2 - f 2 3/2 - 758749.18 M 4.24418D+00 2.21047D-11 7.20430D-07
f 2 1/2 - f 3 3/2 - 722144.92 M 1.09837D+01 6.31521D-11 2.16256D-06
f 2 1/2 - f 4 3/2 - 5745.41 M 3.40764D+00 3.09528D-07 1.33224D+00
f 2 3/2 - f 1 3/2 - 71269.67 M 1.41118D+00 1.66606D-09 5.78084D-04
f 3 3/2 - f 1 3/2 - 107873.94 M 7.52312D+01 3.87688D-08 8.88733D-03
f 4 3/2 - f 1 3/2 - 824273.45 M 1.25260D+01 1.10557D-10 3.31682D-06
f 3 3/2 - f 2 3/2 - 36604.27 M 2.11594D+01 9.47019D-08 6.39782D-02
f 4 3/2 - f 2 3/2 - 753003.77 M 9.62931D+00 1.01840D-10 3.34446D-06
f 4 3/2 - f 3 3/2 - 716399.51 M 7.21346D-02 8.42851D-13 2.90938D-08
f 1 5/2 - f 1 3/2 - 71540.71 M 1.99970D-02 3.51453D-11 1.21484D-05
f 1 5/2 - f 2 3/2 - 271.04 M 2.11526D-04 2.59002D-08 2.36306D+00
f 3 3/2 - f 1 5/2 - 36333.23 M 1.17593D+01 5.34186D-08 3.63575D-02
f 4 3/2 - f 1 5/2 - 752732.73 M 5.41422D+00 5.73023D-11 1.88251D-06
If the information of -coupling is available from a jj2lsj run, rtransition also produces a file
2s22p3_2p5_3.2s22p3_2p5_3.ct.lsj
Transition between files:
2s22p3_2p5_3
2s22p3_2p5_3
1 -262.79063388 1s(2).2s(2).2p(3)2P1_2P
1 -259.49793990 1s(2).2p(5)_2P
722662.80 CM-1 138.38 ANGS(VAC) 138.38 ANGS(AIR)
M1 S = 1.18001D-11 GF = 3.44839D-16 AKI = 6.00621D-05
1 -262.79063388 1s(2).2s(2).2p(3)2P1_2P
3 -262.78827423 1s(2).2s(2).2p(3)2P1_2P
517.88 CM-1 193094.26 ANGS(VAC) 193074.30 ANGS(AIR)
M1 S = 1.31018D+00 GF = 2.74383D-08 AKI = 1.22716D-03
1 -262.79063388 1s(2).2s(2).2p(3)2P1_2P
3 -259.52411791 1s(2).2p(5)_2P
716917.39 CM-1 139.49 ANGS(VAC) 139.49 ANGS(AIR)
M1 S = 1.73052D-06 GF = 5.01695D-11 AKI = 4.29992D+00
3 -263.27978407 1s(2).2s(2).2p(3)4S3_4S
1 -262.79063388 1s(2).2s(2).2p(3)2P1_2P
107356.06 CM-1 931.48 ANGS(VAC) 931.48 ANGS(AIR)
M1 S = 1.86549D-03 GF = 8.09867D-09 AKI = 3.11300D+01
3 -263.27978407 1s(2).2s(2).2p(3)4S3_4S
1 -259.49793990 1s(2).2p(5)_2P
830018.86 CM-1 120.48 ANGS(VAC) 120.48 ANGS(AIR)
M1 S = 5.92158D-07 GF = 1.98756D-11 AKI = 4.56677D+00
3 -262.95505555 1s(2).2s(2).2p(3)2D3_2D
1 -262.79063388 1s(2).2s(2).2p(3)2P1_2P
36086.39 CM-1 2771.13 ANGS(VAC) 2770.83 ANGS(AIR)
M1 S = 2.01353D-02 GF = 2.93830D-08 AKI = 1.27613D+01
3 -262.95505555 1s(2).2s(2).2p(3)2D3_2D
1 -259.49793990 1s(2).2p(5)_2P
758749.18 CM-1 131.80 ANGS(VAC) 131.80 ANGS(AIR)
M1 S = 7.20430D-07 GF = 2.21047D-11 AKI = 4.24418D+00
3 -262.78827423 1s(2).2s(2).2p(3)2P1_2P
1 -259.49793990 1s(2).2p(5)_2P
722144.92 CM-1 138.48 ANGS(VAC) 138.48 ANGS(AIR)
M1 S = 2.16256D-06 GF = 6.31521D-11 AKI = 1.09837D+01
3 -259.52411791 1s(2).2p(5)_2P
1 -259.49793990 1s(2).2p(5)_2P
5745.41 CM-1 17405.20 ANGS(VAC) 17403.40 ANGS(AIR)
M1 S = 1.33224D+00 GF = 3.09528D-07 AKI = 3.40764D+00
3 -263.27978407 1s(2).2s(2).2p(3)4S3_4S
3 -262.95505555 1s(2).2s(2).2p(3)2D3_2D
71269.67 CM-1 1403.12 ANGS(VAC) 1403.12 ANGS(AIR)
M1 S = 5.78084D-04 GF = 1.66606D-09 AKI = 1.41118D+00
3 -263.27978407 1s(2).2s(2).2p(3)4S3_4S
3 -262.78827423 1s(2).2s(2).2p(3)2P1_2P
107873.94 CM-1 927.01 ANGS(VAC) 927.01 ANGS(AIR)
M1 S = 8.88733D-03 GF = 3.87688D-08 AKI = 7.52312D+01
3 -263.27978407 1s(2).2s(2).2p(3)4S3_4S
3 -259.52411791 1s(2).2p(5)_2P
824273.45 CM-1 121.32 ANGS(VAC) 121.32 ANGS(AIR)
M1 S = 3.31682D-06 GF = 1.10557D-10 AKI = 1.25260D+01
3 -262.95505555 1s(2).2s(2).2p(3)2D3_2D
3 -262.78827423 1s(2).2s(2).2p(3)2P1_2P
36604.27 CM-1 2731.92 ANGS(VAC) 2731.63 ANGS(AIR)
M1 S = 6.39782D-02 GF = 9.47019D-08 AKI = 2.11594D+01
3 -262.95505555 1s(2).2s(2).2p(3)2D3_2D
3 -259.52411791 1s(2).2p(5)_2P
753003.77 CM-1 132.80 ANGS(VAC) 132.80 ANGS(AIR)
M1 S = 3.34446D-06 GF = 1.01840D-10 AKI = 9.62931D+00
3 -262.78827423 1s(2).2s(2).2p(3)2P1_2P
3 -259.52411791 1s(2).2p(5)_2P
716399.51 CM-1 139.59 ANGS(VAC) 139.59 ANGS(AIR)
M1 S = 2.90938D-08 GF = 8.42851D-13 AKI = 7.21346D-02
3 -263.27978407 1s(2).2s(2).2p(3)4S3_4S
5 -262.95382060 1s(2).2s(2).2p(3)2D3_2D
71540.71 CM-1 1397.81 ANGS(VAC) 1397.81 ANGS(AIR)
M1 S = 1.21484D-05 GF = 3.51453D-11 AKI = 1.99970D-02
3 -262.95505555 1s(2).2s(2).2p(3)2D3_2D
5 -262.95382060 1s(2).2s(2).2p(3)2D3_2D
271.04 CM-1 368948.37 ANGS(VAC) 368910.23 ANGS(AIR)
M1 S = 2.36306D+00 GF = 2.59002D-08 AKI = 2.11526D-04
5 -262.95382060 1s(2).2s(2).2p(3)2D3_2D
3 -262.78827423 1s(2).2s(2).2p(3)2P1_2P
36333.23 CM-1 2752.30 ANGS(VAC) 2752.01 ANGS(AIR)
M1 S = 3.63575D-02 GF = 5.34186D-08 AKI = 1.17593D+01
5 -262.95382060 1s(2).2s(2).2p(3)2D3_2D
3 -259.52411791 1s(2).2p(5)_2P
752732.73 CM-1 132.85 ANGS(VAC) 132.85 ANGS(AIR)
M1 S = 1.88251D-06 GF = 5.73023D-11 AKI = 5.41422D+00
Here, labels of the upper and lower states in the transition are in
-notation. In addition to transition energies in cm
also the wavelengths (vacuum and air) are given in angstrom (ANGS). On the next line the line strength
S, the weighted oscillator strength
and the transition rate
A (AKI) are given. The format is the same as the one produced by the transition program of
ATSP2K [
1]
In this section, we discuss contents of the files:
2s2p_DF.coup3.LK3.lbl
2s2p_DF.coup3.JK3.lbl
2s2p_DF.coup3.LS.lbl
2s2p_DF.coup3.LS3.lbl
2s2p_DF.coup3.LSJ3.lbl
2s2p_DF.coup3.jj.lbl
2s2p_DF.coup3.cLSJ3.lbl
These files are from the Coupling run of the rci/rmcdhf programs. The input files 2s2p_DF.lsj.c and 2s2p_DF.lsj.j were created by the program jj2lsj in non-default mode.
The 2s2p_DF.coup3.LK3.lbl file
The Coupling program transforms from to coupling and gives the composition of the states.
Pos J Parity Energy Total Comp. of ASF
1 0 -24.127087737 100.000%
-1.00000000 1.00000000 1s2_ 2s_2p_(3P) P_3[1]<0>
1 1 -24.127040409 100.000%
0.99999995 0.99999990 1s2_ 2s_2p_(3P) P_3[1]<1>
0.00030652 0.00000009 1s2_ 2s_2p_(1P) P_1[1]<1>
2 1 -23.915406084 100.000%
0.99999995 0.99999990 1s2_ 2s_2p_(1P) P_1[1]<1>
-0.00030652 0.00000009 1s2_ 2s_2p_(3P) P_3[1]<1>
1 2 -24.126945696 100.000%
1.00000000 1.00000000 1s2_ 2s_2p_(3P) P_3[1]<2>
Let us explain how to interpret the notation of the following ASF
1s2_ 2s_2p_(3P) P_3[1]<0>
First, it should be noted that the spin multiplicity, , is used to represent the spin of individual and coupled shells in the output files. The first 1s(2) subshell is fully occupied, whereas the second 2s and the third 2p subshells are singly occupied. Therefore, they have only one term, , , and respectively, that are not written out explicitly. The result of the coupling of the second and third subshells, 2s and 2p, is written in parentheses, i.e., (ML) = (3P). Following the coupling scheme, the total orbital angular momentum L is obtained by coupling the 1s shell angular momentum, L=0, with L=1. This momentum appears as the first spectroscopic symbol, P, of the final term construction P_3[1]<0>. The number 3 preceding the "[1]" symbol represents M. Coupling L=1 with the spin S=0 leads to the term K=1 which is written in square brackets [ and ]. Coupling K=1 with the spin S=1 leads to the final J term, J=0, that is written in angle brackets < and >.
The <name>.coup3.JK3.lbl file
The Coupling program transforms from to coupling and gives the composition of the states.
Pos J Parity Energy Total Comp. of ASF
1 0 -24.127087737 100.000%
-1.00000000 1.00000000 1s2_<0>2s_2p_(3P) 3[1]<0>
1 1 -24.127040409 100.000%
0.99999995 0.99999990 1s2_<0>2s_2p_(3P) 3[1]<1>
0.00030652 0.00000009 1s2_<0>2s_2p_(1P) 1[1]<1>
2 1 -23.915406084 100.000%
0.99999995 0.99999990 1s2_<0>2s_2p_(1P) 1[1]<1>
-0.00030652 0.00000009 1s2_<0>2s_2p_(3P) 3[1]<1>
1 2 -24.126945696 100.000%
1.00000000 1.00000000 1s2_<0>2s_2p_(3P) 3[1]<2>
The notation for the following ASF
1s2_<0>2s_2p_(3P) 3[1]<0>
can be understood as follows. The first subshell 1s(2) is fully occupied and has only one term . This part of the term () is not mentioned and only is written in the first angle brackets < and >. The second subshell 2s is singly occupied and has only one term, , that is not written out explicitly. The third subshell 2p is singly occupied and has the term , also omitted in the notation. The result of the coupling of the second and third subshells, 2s and 2p, is written in parentheses, i.e., (ML) = (3P). The first number, 3, appearing in the final term construction 3[1]<0> represents M. Coupling J=0 with the orbital angular momentum L=1 leads to the term K=1 that is written in square brackets [ and ]. Coupling K=1 with the spin S=1 leads to the final J term J=0 that is written in the final angle brackets < and >.
The <name>.coup3.LS.lbl file
The Coupling program transforms from to coupling and gives the composition of the states.
Pos J Parity Energy Total Comp. of ASF
1 0 -24.127087737 100.000%
-1.00000000 1.00000000 1s2_.2s_2S.2p_ 3P<0>
1 1 -24.127040409 100.000%
0.99999995 0.99999990 1s2_.2s_2S.2p_ 3P<1>
0.00030652 0.00000009 1s2_.2s_2S.2p_ 1P<1>
2 1 -23.915406084 100.000%
0.99999995 0.99999990 1s2_.2s_2S.2p_ 1P<1>
-0.00030652 0.00000009 1s2_.2s_2S.2p_ 3P<1>
1 2 -24.126945696 100.000%
1.00000000 1.00000000 1s2_.2s_2S.2p_ 3P<2>
The following ASF notation
1s2_.2s_2S.2p_ 3P<0>
can be interpreted as follows.
The first subshell 1s(2) is fully occupied and has only one term, , that is not written out explicitly. The second and third subshells 2s and 2p are singly occupied and have only one terms, and respectively, not displayed in the notation. The result of the coupling of the first and second subshells, ML2S, is written without parentheses. Coupling the spins S and S leads to the total spin S. A similar coupling is done with the orbital angular momenta, leading to the total orbital angular momentum, L, that is written as P, adopting the spectroscopic notation. Coupling the orbital angular momentum L=1 with the spin S=1 leads to the final J term J=0 that is written in angle brackets < and > and presented in the final term 3P<0>.
The <name>.coup3.LS3.lbl file
The Coupling program transforms from to coupling and gives the composition of the states.
Pos J Parity Energy Total Comp. of ASF
1 0 -24.127087737 100.000%
-1.00000000 1.00000000 1s2_ 2s_2p_(3P) 3P<0>
1 1 -24.127040409 100.000%
0.99999995 0.99999990 1s2_ 2s_2p_(3P) 3P<1>
0.00030652 0.00000009 1s2_ 2s_2p_(1P) 1P<1>
2 1 -23.915406084 100.000%
0.99999995 0.99999990 1s2_ 2s_2p_(1P) 1P<1>
-0.00030652 0.00000009 1s2_ 2s_2p_(3P) 3P<1>
1 2 -24.126945696 100.000%
1.00000000 1.00000000 1s2_ 2s_2p_(3P) 3P<2>
The following ASF
1s2_ 2s_2p_(3P) 3P<0>
should be read as follows. The first subshell 1s(2) is fully occupied and has only one term, , that is not written out explicitly. The second and third subshells 2s and 2p are singly occupied and have only one terms, and respectively, not shown in the notation. The result of the coupling of the second and third subshells, 2s and 2p, is written in parentheses, i.e., (ML) = (3P). Coupling the spin of the first shell 1s(2) S=0 with the spin S=1 leads to the total spin multiplicity M=3, which is the first number of the final term 3P<0>. P is the total orbital angular momentum, obtained by coupling L and L. Coupling the latter, L=1, with the total spin S=1 leads to the final J term J=0 that is written in angle brackets < and >.
The <name>.coup3.LSJ3.lbl file
The Coupling program transforms from to coupling and gives the composition of the states.
Pos J Parity Energy Total Comp. of ASF
1 0 -24.127087737 100.000%
-1.00000000 1.00000000 1s2_ 2s_2p_(3P) (0,0)<0>
1 1 -24.127040409 100.000%
0.99999995 0.99999990 1s2_ 2s_2p_(3P) (0,1)<1>
0.00030652 0.00000009 1s2_ 2s_2p_(1P) (0,1)<1>
2 1 -23.915406084 100.000%
0.99999995 0.99999990 1s2_ 2s_2p_(1P) (0,1)<1>
-0.00030652 0.00000009 1s2_ 2s_2p_(3P) (0,1)<1>
1 2 -24.126945696 100.000%
1.00000000 1.00000000 1s2_ 2s_2p_(3P) (0,2)<2>
In this coupling scheme, the ASF
1s2_ 2s_2p_(3P) (0,0)<0>
is built as follows. The first subshell 1s(2) is fully occupied and has only one term that is not written out explicitly. The second and third subshells 2s and 2p are singly occupied and have only one terms, and respectively, not displayed in the notation. The result of the coupling of the second and third subshells, 2s and 2p, is written in parentheses, i.e., (ML) = (3P). Coupling S=1 with the angular momentum L=1 leads to angular momentum J=0. The J=0 of the first subshell 1s(2) and J=0 can be found in the round brackets (0,0) of the final term (0,0)<0>. Coupling the angular momentum J=0 with the angular momentum J=0 leads to the final J term J=0 that is written in angle brackets < and >.
The <name>.coup3.jj.lbl file
The Coupling program transforms from to coupling and gives the composition of the states.
Pos J Parity Energy Total Comp. of ASF
1 0 -24.127087737 100.000%
1.00000000 1.00000000 1s+2_2s+_<1/2>.2p-_(1/2) <0>
1 1 -24.127040409 100.000%
0.81667351 0.66695562 1s+2_2s+_<1/2>.2p-_(1/2) <1>
-0.57709997 0.33304437 1s+2_2s+_<1/2>.2p+_(3/2) <1>
2 1 -23.915406084 100.000%
0.81667351 0.66695562 1s+2_2s+_<1/2>.2p+_(3/2) <1>
0.57709997 0.33304437 1s+2_2s+_<1/2>.2p-_(1/2) <1>
1 2 -24.126945696 100.000%
1.00000000 1.00000000 1s+2_2s+_<1/2>.2p+_ <2>
Let us illustrate the notation for the following ASF
1s+2_2s+_<1/2>.2p-_(1/2) <0>
The first subshell 1s+(2) is fully occupied and has only one term, , that is not written out explicitly. The second subshell 2s+ is singly occupied and has only one term, , not reported in the notation. Coupling the angular momenta J=0 and J=1/2 leads to the term J=1/2 that is written in angle brackets < and >. Coupling the latter, J=1/2, with the angular momentum J=1/2 (written in parentheses) leads to the final J term J=0 that is written in angle brackets < and >.
The <name>.coup3.cLSJ3.lbl file
The Coupling program transforms from to coupling and gives the composition of the states.
Pos J Parity Energy Total Comp. of ASF
1 0 -24.127087737 100.000%
-1.00000000 1.00000000 1s+2_ (0,0)<0> 2s_2p_(3P)<0> (0,0)<0>
1 1 -24.127040409 100.000%
0.99999995 0.99999990 1s+2_ (0,0)<0> 2s_2p_(3P)<1> (0,1)<1>
0.00030652 0.00000009 1s+2_ (0,0)<0> 2s_2p_(1P)<1> (0,1)<1>
2 1 -23.915406084 100.000%
0.99999995 0.99999990 1s+2_ (0,0)<0> 2s_2p_(1P)<1> (0,1)<1>
-0.00030652 0.00000009 1s+2_ (0,0)<0> 2s_2p_(3P)<1> (0,1)<1>
1 2 -24.126945696 100.000%
1.00000000 1.00000000 1s+2_ (0,0)<0> 2s_2p_(3P)<2> (0,2)<2>
Lastly, let us consider the following ASF
1s+2_ (0,0)<0> 2s_2p_(3P)<0> (0,0)<0>
that should be read as follows. In the three shells coupling scheme
, the first non relativistic subshell,
, is split into two relativistic subshells,
and
, with
and
, while the two others are expressed in
coupling (see Equation (26) of [
14]). In the corresponding
notation of this first shell, the coupling is written as
(J,J)<J>. In the present case, the first subshell is a closed subshell
1s for which
is the only
j-value allowed in the relativistic splitting
. However, for coding commodity, the
s subshells are treated as all the others
, keeping an artificial
coupling notation
(0,J)<J>, with
and
. Reading the ASF from left to right, the two next shells,
2s and
2p, are singly occupied and have only one
terms,
and
respectively, that are not displayed in the notation. The result of the coupling of the second and third subshells,
2s and
2p, is written in parentheses, i.e., (
ML) =
(3P). The following angle brackets
<J> contain the result of the coupling between
S and
L. The angular momenta
J and
J are given in the round parentheses
(0,0) of the final term
(0,0)<0>. The very last angle brackets
< and
> contain the value of the total angular momentum,
J, resulting from the coupling of
J and
J.
8.3. Output Files from the Fifth Example
The fifth example, see
Section 6.5, was the study of energy spectra for Ni XIV, giving the unique labels.
The unique label summary file
Using the unique option of the jj2lsj program produces a summary name.uni.lsj.sum. Below is the summary file Ni_even_n4.uni.lsj.sum. In name.uni.lsj.sum information for the levels is given: Pos, composition of the level, serial number of composition, and the label of the level. From the Ni_even_n4.uni.lsj.sum file we see that the level with J = 1/2 and Pos = 2 has serial No of composition = 2 and the level with J = 1/2 and Pos = 5 has serial No of composition = 4. These levels were thus re-identified.
Composition Serial No. Coupling
of compos.
J = 1/2
--------------------------------------------------
Pos 4 0.941868580 1 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
Pos 1 0.860336790 1 2s(2).2p(6).3s_2S.3p(4)3P2_4P
Pos 8 0.664884270 1 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2S
Pos 7 0.554223930 1 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2P
Pos 6 0.550189830 1 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
Pos 3 0.487994450 1 2s(2).2p(6).3s_2S.3p(4)1S0_2S
Pos 2 0.301420530 2 2s(2).2p(6).3s_2S.3p(4)3P2_2P
Pos 5 0.112794340 4 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2P
--------------------------------------------------
...........
Composition Serial No. Coupling
of compos.
J = 9/2
--------------------------------------------------
Pos 1 0.937469590 1 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4F
Pos 2 0.936205640 1 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2G
The unique label composition file
The jj2lsj program also produces name.uni.lsj.lbl. Below is the output file Ni_even_n4.uni.lsj.lbl from jj2lsj with the unique option. This file has the same format as Ni_even_n4.lsj.lbl, except that the levels with the same labels were re-identified. As seen from the output file, for the level with J = 1/2 and Pos = 2 the largest expansion coefficient does not appear on the first line. This level was re-identified. The users should use name.lsj.uni.lbl file in further calculations (rtransition, rhfs, etc.) to obtain output with unique labels.
Pos J Parity Energy Total Comp. of ASF
1 1/2 + -1441.689593921 99.941%
-0.92754342 0.86033679 2s(2).2p(6).3s_2S.3p(4)3P2_4P
-0.31644623 0.10013822 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
-0.13107223 0.01717993 2s(2).2p(6).3s_2S.3p(4)1S0_2S
-0.06808224 0.00463519 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)1S0_4P
-0.06306024 0.00397659 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)1D2_4P
-0.06139607 0.00376948 2s(2).2p(6).3p(4)3P2_3P.3d_4P
-0.04384478 0.00192236 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)3P2_4P
0.04315453 0.00186231 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2S
0.04160917 0.00173132 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)3P2_4P
2 1/2 + -1441.146026942 99.870%
0.54901778 0.30142053 2s(2).2p(6).3s_2S.3p(4)3P2_2P
0.55236001 0.30510158 2s(2).2p(6).3s_2S.3p(4)1S0_2S
-0.51850029 0.26884256 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2P
-0.25241177 0.06371170 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2S
0.14974129 0.02242245 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2P
0.08843416 0.00782060 2s(2).2p(6).3p(4)1D2_1D.3d_2P
-0.07913818 0.00626285 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
0.06957348 0.00484047 2s(2).2p(6).3s_2S.3p(2)1S0_2S.3d(2)1S0_2S
-0.06792804 0.00461422 2s(2).2p(6).3s_2S.3p(4)3P2_4P
-0.04635416 0.00214871 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)1D2_2P
-0.04439733 0.00197112 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)3P2_2P
0.03795472 0.00144056 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)3P2_2P
-0.03450153 0.00119036 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)3P2_2S
-0.03371402 0.00113663 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
-0.03274764 0.00107241 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)1D2_2S
0.03171981 0.00100615 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)3F2_2P
3 1/2 + -1441.041027919 99.883%
0.69856599 0.48799445 2s(2).2p(6).3s_2S.3p(4)1S0_2S
0.44943909 0.20199550 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_2P
-0.37641525 0.14168844 2s(2).2p(6).3s_2S.3p(4)3P2_2P
-0.31029154 0.09628084 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2S
-0.14516094 0.02107170 2s(2).2p(6).3s(2).3p(2)1D2_1D.3d_2P
0.11017096 0.01213764 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4D
-0.10592606 0.01122033 2s(2).2p(6).3s_2S.3p(4)3P2_4P
0.08894930 0.00791198 2s(2).2p(6).3s_2S.3p(2)1S0_2S.3d(2)1S0_2S
-0.06646514 0.00441762 2s(2).2p(6).3p(4)1D2_1D.3d_2P
-0.04537257 0.00205867 2s(2).2p(6).3s(2).3p(2)3P2_3P.3d_4P
-0.04336944 0.00188091 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)3P2_2S
-0.04274245 0.00182692 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)1D2_2S
0.04115897 0.00169406 2s(2).2p(6).3s_2S.3p(2)1D2_2D.3d(2)1D2_2P
0.03553871 0.00126300 2s(2).2p(6).3s_2S.3p(2)3P2_4P.3d(2)3P2_2P
...........
8.4. Output Files from the Eighth Example
The eighth example, see
Section 6.8, was the calculation of the radial density distribution
for Be ground state and the transformation to natural orbitals. The file
n4.cd contains three columns with the radial grid, the radial density distribution
and the spherical electron density function
as shown below. Using Matlab, GNU Octave or Python the distribution is readily plotted.
r [au] D(r)=4\pi*r^2*rho(r) rho(r)
1 0 +
0.0000000000D+00 0.0000000000D+00 3.5595248135D+01
2.5635548188D-08 2.9395876524D-13 3.5595191429D+01
5.2585459038D-08 1.2368931242D-12 3.5595161894D+01
8.0917121364D-08 2.9287460232D-12 3.5595164086D+01
1.1070137908D-07 5.4815947105D-12 3.5595163799D+01
1.4201270834D-07 9.0210123946D-12 3.5595163778D+01
1.7492940379D-07 1.3687574330D-11 3.5595163713D+01
2.0953377430D-07 1.9638527873D-11 3.5595163617D+01
2.4591234882D-07 2.7049642650D-11 3.5595163498D+01
2.8415609275D-07 3.6117260296D-11 3.5595163346D+01
3.2436063535D-07 4.7060564994D-11 3.5595163160D+01
3.6662650893D-07 6.0124098290D-11 3.5595162934D+01
4.1105940020D-07 7.5580544151D-11 3.5595162664D+01
......
2.2006596267D+01 1.4378899592D-12 2.3627079091D-16
2.3134898611D+01 2.4605656113D-13 3.6583890201D-17
2.4321050253D+01 3.8221221504D-14 5.1419760355D-18
2.5568017190D+01 5.3624070241D-15 6.5276342428D-19
2.6878917489D+01 6.7510077496D-16 7.4359283127D-20
2.8257029084D+01 7.6386795748D-17 7.6129930176D-21
2.9705797971D+01 5.9008393899D-18 5.3213458250D-22
3.1228846828D+01 3.8138158242D-19 3.1119883125D-23
3.2829984069D+01 1.9628254786D-22 1.4492071857D-26
8.5. Output Files from the Ninth Example
The ninth example was for the unexpected transition
in Ni XXV, see
Section 6.9. The file
odd_n3.cgjhfs is shown below. First, the
J quantum numbers, the parities, and the energies are shown for the computed states. Next come the reduced matrix elements
for the magnetic (Zeeman) interaction, see [
11] Equations (34), (35), (44) and (45). This is followed by the reduced electronic matrix elements
for the magnetic dipole interaction, see [
11] Equations (13) and (15). Finally, the reduced electronic matrix elements
for the electric quadrupole interaction, see [
11] Equations (14) and (16). The reduced matrix elements adhere to the Brink and Satchler definition of the Wigner-Eckart theorem and they are not symmetric, see [
11] Equation (57).
Number of relativistic eigenvalues
4
Lev J Parity E
1 2.0 - -944.099455445
1 1.0 - -944.694852121
2 1.0 - -942.723282825
1 0.0 - -944.877056498
Zeeman interaction matrix
0.18322E+01 -0.34691E+00 0.68227E-01 0.00000E+00
0.44786E+00 0.10439E+01 -0.67174E-01 0.40125E+00
-0.88081E-01 -0.67174E-01 0.71718E+00 -0.78350E-01
0.00000E+00 -0.69499E+00 0.13571E+00 0.00000E+00
HFI-matrix for the magnetic dipole operator
0.36369E+02 -0.10509E+02 0.27002E+02 0.00000E+00
0.13567E+02 0.36113E+02 0.22641E+02 0.15293E+02
-0.34859E+02 0.22641E+02 -0.18146E+01 0.81436E+01
0.00000E+00 -0.26488E+02 -0.14105E+02 0.00000E+00
HFI-matrix for the electric quadrupole operator
0.28620E+03 0.32475E+03 -0.59600E+02 -0.22196E+03
-0.41925E+03 -0.22145E+03 0.14396E+03 0.00000E+00
0.76944E+02 0.14396E+03 0.46833E+03 0.00000E+00
-0.49633E+03 -0.00000E+00 -0.00000E+00 0.00000E+00


 ,
, 



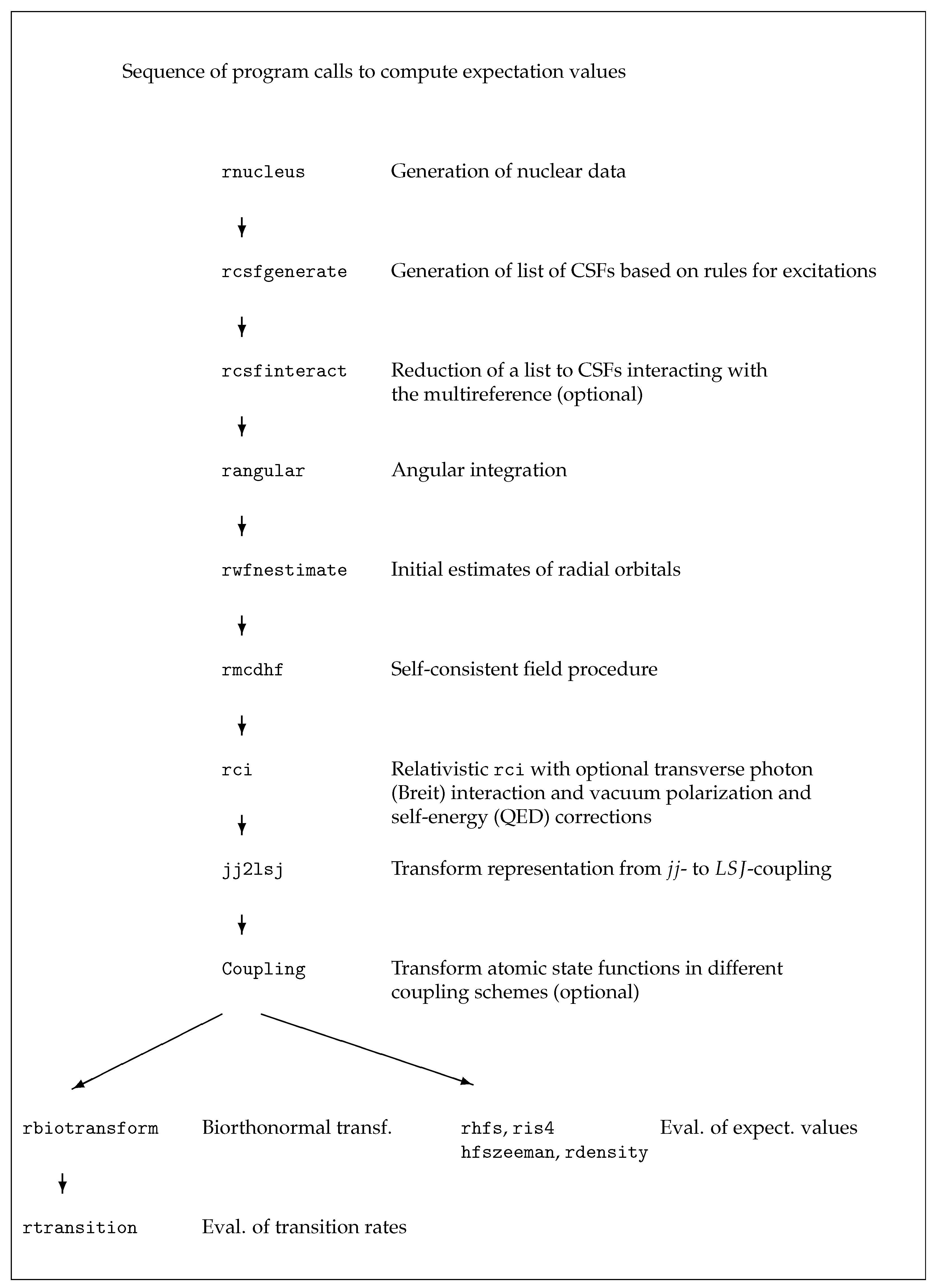












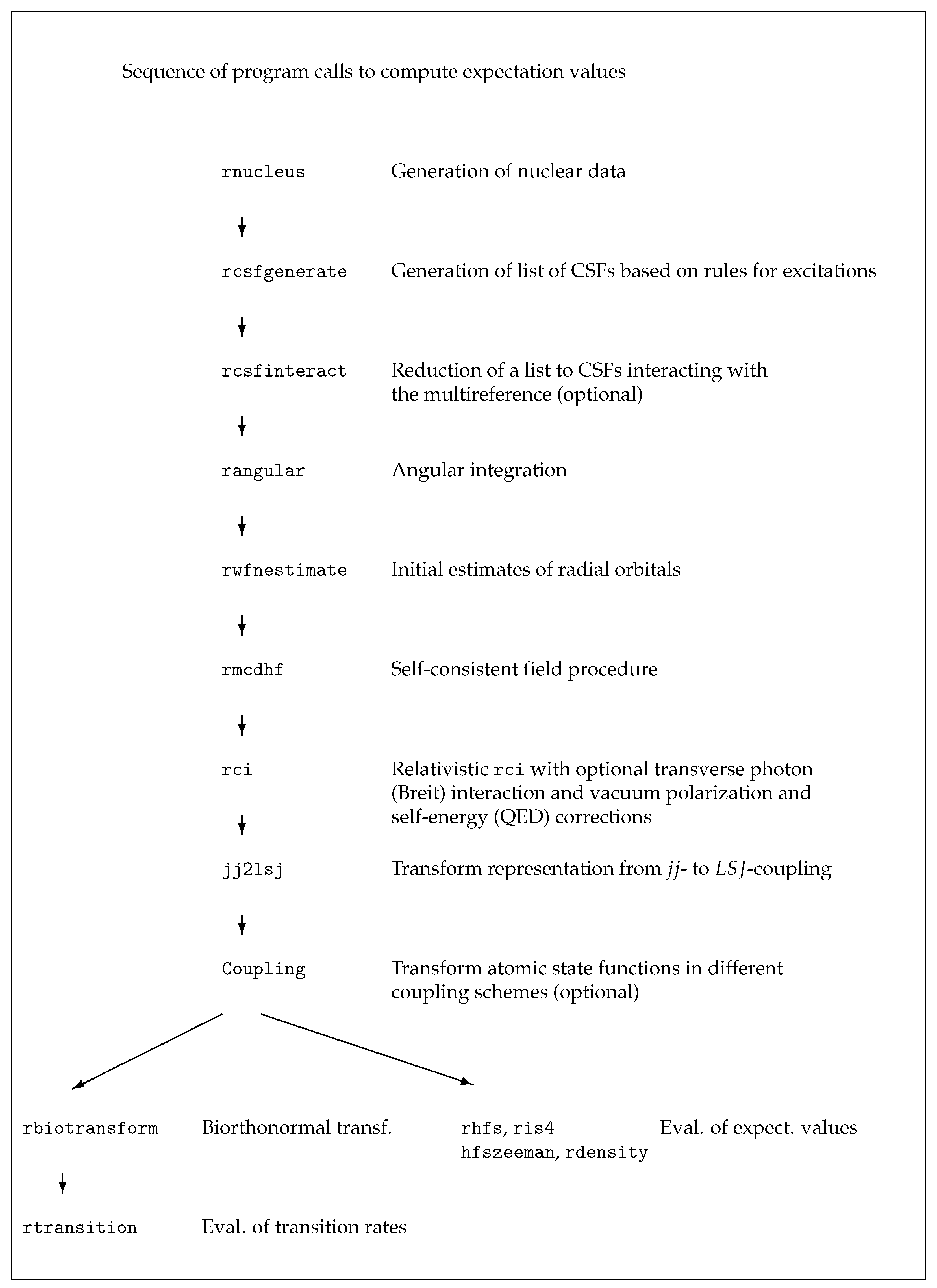{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}