Two-Photon Vibrational Transitions in 16O2+ as Probes of Variation of the Proton-to-Electron Mass Ratio


Abstract
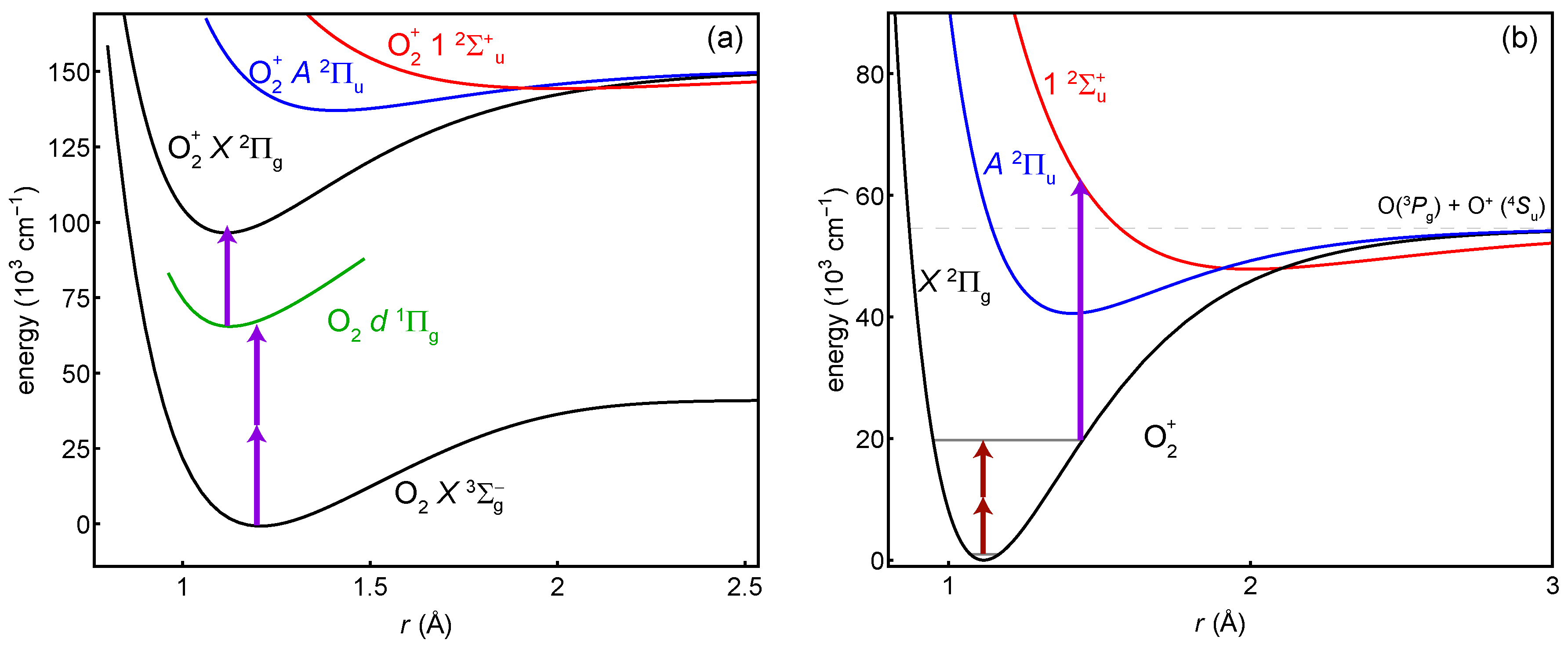
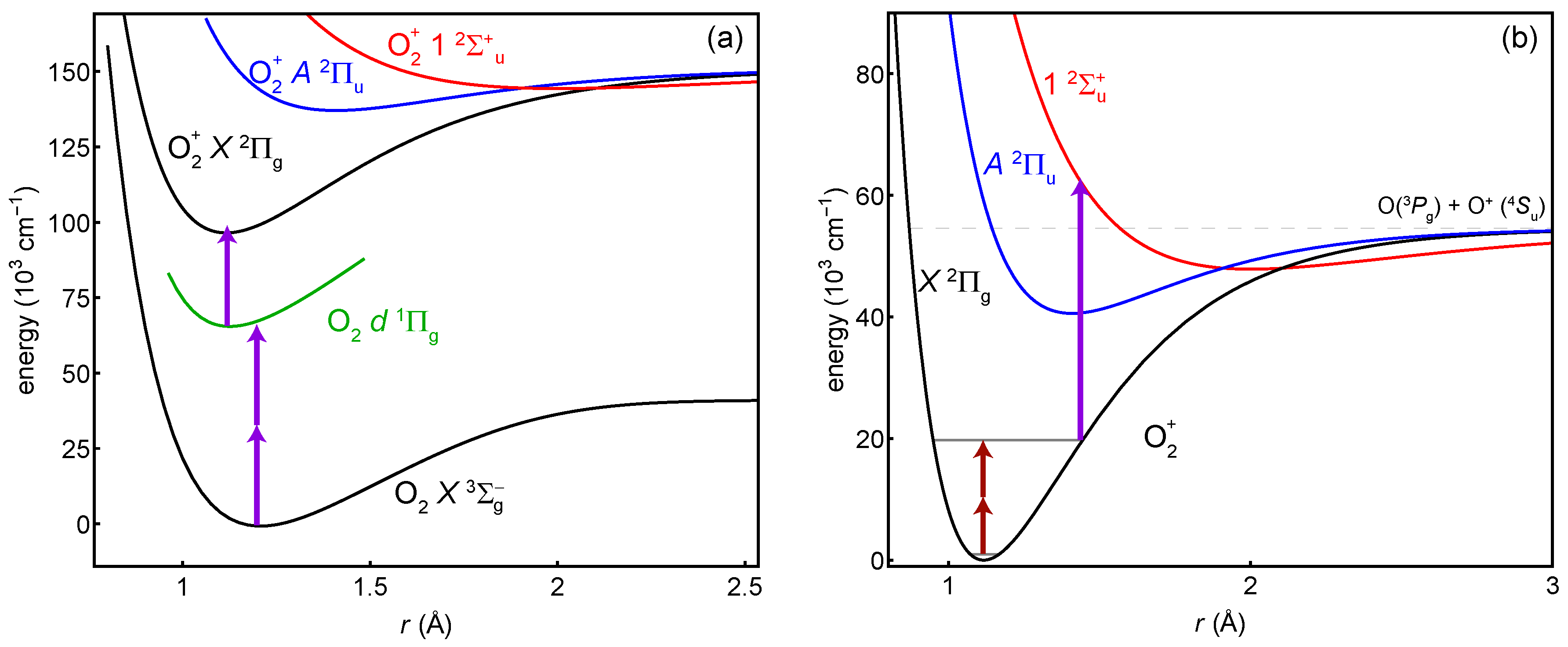
:1. Introduction
2. Experimental Procedures
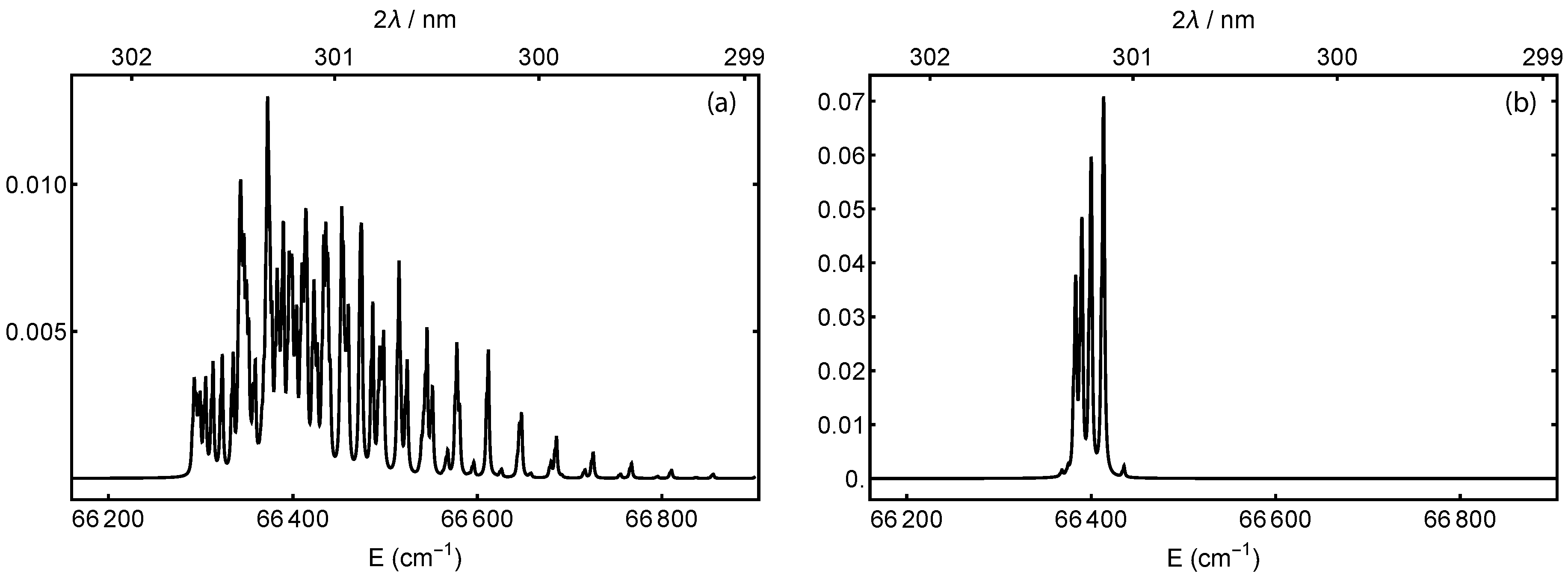
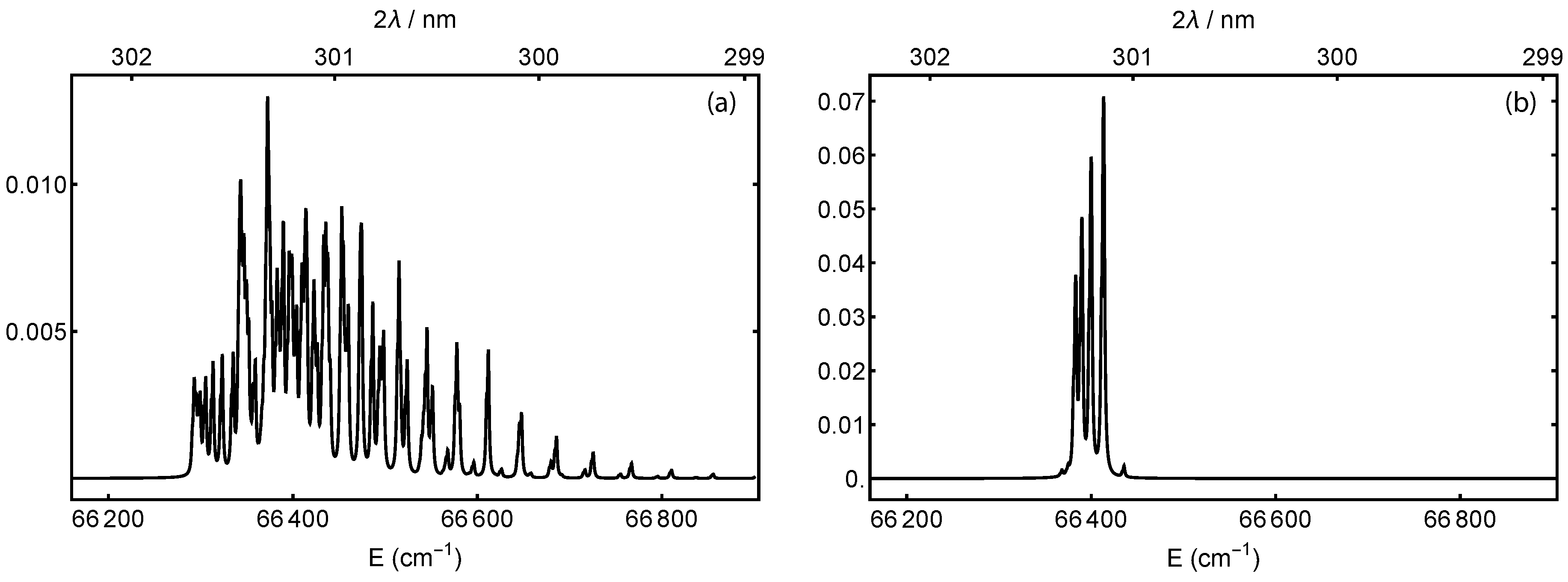
2.1. State Preparation: Resonance-Enhanced Multi-Photon Ionization
2.2. Probe: Two-Photon Transition
2.3. Detection: Selective Dissociation
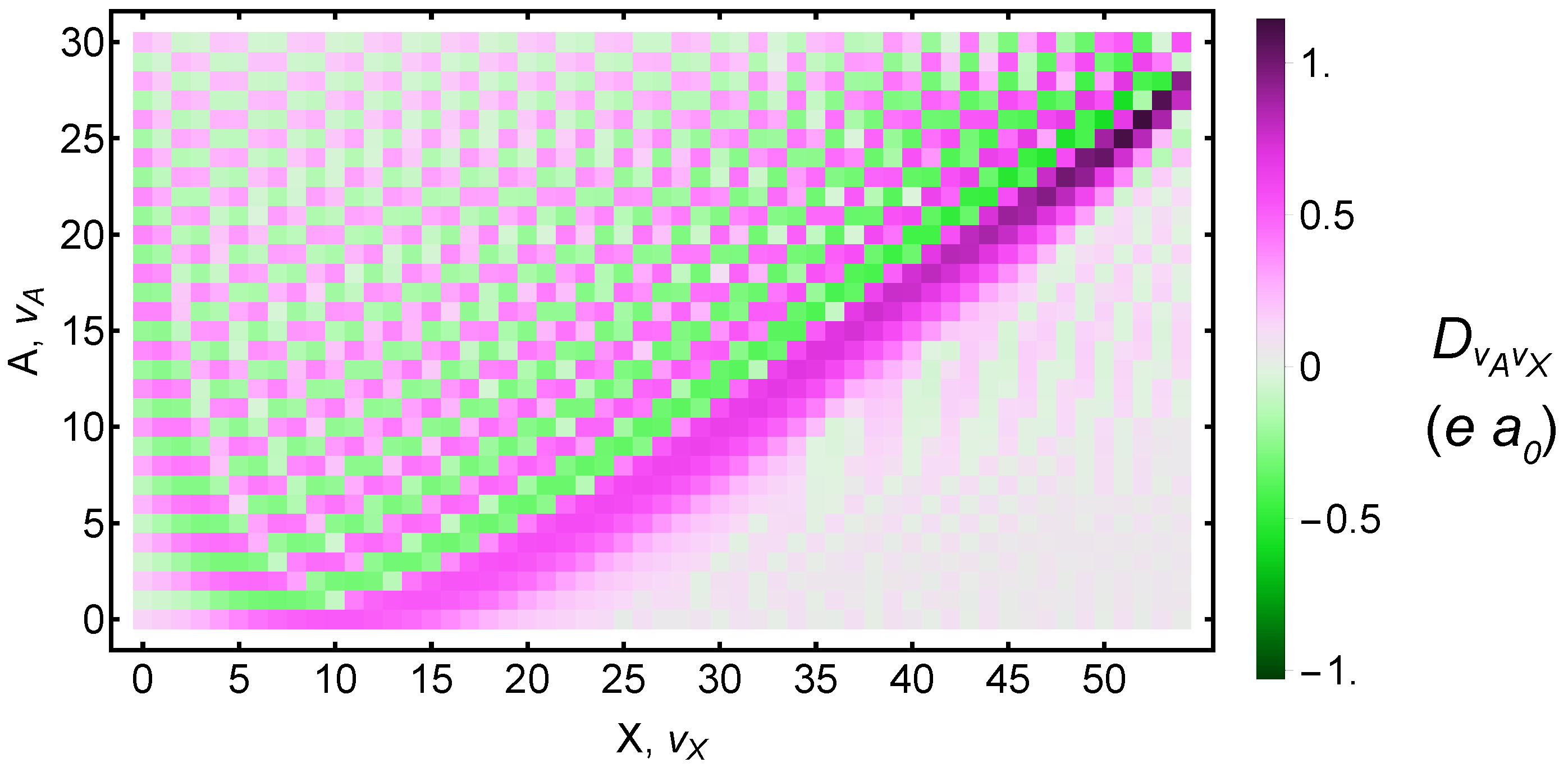
3. Transition Rates and Electric-Dipole-Related Systematics
3.1. Calculating the Transition Dipole Moments
3.2. Transition Rate
3.3. Stark Shifts
3.3.1. Probe Laser
3.3.2. Trapping Fields
3.3.3. Blackbody Radiation
4. Additional Systematic Effects
4.1. Doppler Shifts
4.2. Electric Quadrupole Shift
4.3. Zeeman Shift
5. Prospects
5.1. Choice of State and Techniques
5.2. Reference Transitions
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- DeMille, D.; Doyle, J.M.; Sushkov, A.O. Probing the frontiers of particle physics with tabletop-scale experiments. Science 2017, 357, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Safronova, M.S.; Budker, D.; DeMille, D.; Jackson Kimball, D.F.; Derevianko, A.; Clark, C.W. Search for new physics with atoms and molecules. Rev. Mod. Phys. 2018, 90, 025008. [Google Scholar] [CrossRef]
- Salumbides, E.J.; Koelemeij, J.C.J.; Komasa, J.; Pachucki, K.; Eikema, K.S.E.; Ubachs, W. Bounds on fifth forces from precision measurements on molecules. Phys. Rev. D 2013, 87, 112008. [Google Scholar] [CrossRef]
- DeMille, D.; Cahn, S.B.; Murphree, D.; Rahmlow, D.A.; Kozlov, M.G. Using Molecules to Measure Nuclear Spin-Dependent Parity Violation. Phys. Rev. Lett. 2008, 100, 023003. [Google Scholar] [CrossRef]
- Quack, M.; Stohner, J.; Willeke, M. High-Resolution Spectroscopic Studies and Theory of Parity Violation in Chiral Molecules. Ann. Rev. Phys. Chem. 2008, 59, 741–769. [Google Scholar] [CrossRef] [PubMed]
- Cairncross, W.B.; Gresh, D.N.; Grau, M.; Cossel, K.C.; Roussy, T.S.; Ni, Y.; Zhou, Y.; Ye, J.; Cornell, E.A. Precision Measurement of the Electron’s Electric Dipole Moment Using Trapped Molecular Ions. Phys. Rev. Lett. 2017, 119, 153001. [Google Scholar] [CrossRef]
- ACME Collaboration; Andreev, V.; Ang, D.G.; DeMille, D.; Doyle, J.M.; Gabrielse, G.; Haefner, J.; Hutzler, N.R.; Lasner, Z.; Meisenhelder, C.; et al. Improved limit on the electric dipole moment of the electron. Nature 2018, 562, 355–360. [Google Scholar] [CrossRef]
- Carr, L.D.; DeMille, D.; Krems, R.V.; Ye, J. Cold and ultracold molecules: Science, technology, and applications. New J. Phys. 2009, 11, 055049. [Google Scholar] [CrossRef]
- Chin, C.; Flambaum, V.V.; Kozlov, M.G. Ultracold molecules: New probes on the variation of fundamental constants. New J. Phys. 2009, 11, 055048. [Google Scholar] [CrossRef]
- Jansen, P.; Bethlem, H.L.; Ubachs, W. Perspective: Tipping the scales: Search for drifting constants from molecular spectra. J. Chem. Phys. 2014, 140, 010901. [Google Scholar] [CrossRef] [Green Version]
- Bressel, U.; Borodin, A.; Shen, J.; Hansen, M.; Ernsting, I.; Schiller, S. Manipulation of Individual Hyperfine States in Cold Trapped Molecular Ions and Application to HD+ Frequency Metrology. Phys. Rev. Lett. 2012, 108, 183003. [Google Scholar] [CrossRef] [PubMed]
- Germann, M.; Tong, X.; Willitsch, S. Observation of electric-dipole-forbidden infrared transitions in cold molecular ions. Nat. Phys. 2014, 10, 820–824. [Google Scholar] [CrossRef] [Green Version]
- Biesheuvel, J.; Karr, J.; Hilico, L.; Eikema, K.S.E.; Ubachs, W.; Koelemeij, J.C.J. Probing QED and fundamental constants through laser spectroscopy of vibrational transitions in HD+. Nat. Commun. 2016, 7, 10385. [Google Scholar] [CrossRef] [PubMed]
- Calvin, A.T.; Brown, K.R. Spectroscopy of Molecular Ions in Coulomb Crystals. J. Phys. Chem. Lett. 2018, 9, 5797–5804. [Google Scholar] [CrossRef]
- Uzan, J.P. The fundamental constants and their variation: Observational and theoretical status. Rev. Mod. Phys. 2003, 75, 403–455. [Google Scholar] [CrossRef]
- Calmet, X.; Keller, M. Cosmological evolution of fundamental constants: From theory to experiment. Mod. Phys. Lett. A 2015, 30, 1540028. [Google Scholar] [CrossRef] [Green Version]
- Stadnik, Y.V.; Flambaum, V.V. Searching for Dark Matter and Variation of Fundamental Constants with Laser and Maser Interferometry. Phys. Rev. Lett. 2015, 114, 161301. [Google Scholar] [CrossRef]
- Arvanitaki, A.; Huang, J.; Van Tilburg, K. Searching for dilaton dark matter with atomic clocks. Phys. Rev. D 2015, 91, 015015. [Google Scholar] [CrossRef]
- Derevianko, A.; Pospelov, M. Hunting for topological dark matter with atomic clocks. Nat. Phys. 2014, 10, 933–936. [Google Scholar] [CrossRef] [Green Version]
- McGrew, W.F.; Zhang, X.; Leopardi, H.; Fasano, R.J.; Nicolodi, D.; Beloy, K.; Yao, J.; Sherman, J.A.; Schäffer, S.A.; Savory, J.; et al. Towards Adoption of an Optical Second: Verifying Optical Clocks at the SI Limit. arXiv, 2018; arXiv:1811.05885. [Google Scholar]
- Flambaum, V.V.; Tedesco, A.F. Dependence of nuclear magnetic moments on quark masses and limits on temporal variation of fundamental constants from atomic clock experiments. Phys. Rev. C 2006, 73, 055501. [Google Scholar] [CrossRef] [Green Version]
- Ludlow, A.D.; Boyd, M.M.; Ye, J.; Peik, E.; Schmidt, P.O. Optical atomic clocks. Rev. Mod. Phys. 2015, 87, 637–701. [Google Scholar] [CrossRef] [Green Version]
- Schiller, S.; Korobov, V. Tests of time independence of the electron and nuclear masses with ultracold molecules. Phys. Rev. A 2005, 71, 032505. [Google Scholar] [CrossRef]
- Chin, C.; Flambaum, V.V. Enhanced Sensitivity to Fundamental Constants in Ultracold Atomic and Molecular Systems near Feshbach Resonances. Phys. Rev. Lett. 2006, 96, 230801. [Google Scholar] [CrossRef]
- Flambaum, V.V.; Kozlov, M.G. Enhanced Sensitivity to the Time Variation of the Fine-Structure Constant and mp/me in Diatomic Molecules. Phys. Rev. Lett. 2007, 99, 150801. [Google Scholar] [CrossRef] [PubMed]
- Shelkovnikov, A.; Butcher, R.J.; Chardonnet, C.; Amy-Klein, A. Stability of the Proton-to-Electron Mass Ratio. Phys. Rev. Lett. 2008, 100, 150801. [Google Scholar] [CrossRef] [PubMed]
- DeMille, D.; Sainis, S.; Sage, J.; Bergeman, T.; Kotochigova, S.; Tiesinga, E. Enhanced Sensitivity to Variation of me/mp in Molecular Spectra. Phys. Rev. Lett. 2008, 100, 043202. [Google Scholar] [CrossRef] [PubMed]
- Zelevinsky, T.; Kotochigova, S.; Ye, J. Precision Test of Mass-Ratio Variations with Lattice-Confined Ultracold Molecules. Phys. Rev. Lett. 2008, 100, 043201. [Google Scholar] [CrossRef]
- Kajita, M.; Abe, M.; Hada, M.; Moriwaki, Y. Estimated accuracies of pure XH+ (X: Even isotopes of group II atoms) vibrational transition frequencies: Toward the test of the variance in mp/me. J. Phys. B 2011, 44, 025402. [Google Scholar] [CrossRef]
- Kajita, M.; Gopakumar, G.; Abe, M.; Hada, M.; Keller, M. Test of mp/me changes using vibrational transitions in . Phys. Rev. A 2014, 89, 032509. [Google Scholar] [CrossRef]
- Hanneke, D.; Carollo, R.A.; Lane, D.A. High sensitivity to variation in the proton-to-electron mass ratio in . Phys. Rev. A 2016, 94, 050101. [Google Scholar] [CrossRef]
- Kajita, M. Accuracy estimation for the transition frequencies targeting the search for the variation in the proton-electron mass ratio. Phys. Rev. A 2017, 95, 023418. [Google Scholar] [CrossRef]
- Kokish, M.G.; Stollenwerk, P.R.; Kajita, M.; Odom, B.C. Prospects for Polar Molecular Ion Optical Probe of Varying Proton-Electron Mass Ratio. arXiv, 2017; arXiv:1710.08589v3. [Google Scholar] [CrossRef]
- Stollenwerk, P.R.; Kokish, M.G.; de Oliveira-Filho, A.G.S.; Ornellas, F.R.; Odom, B.C. Optical Pumping of TeH+: Implications for the Search for Varying mp/me. Atoms 2018, 6, 53. [Google Scholar] [CrossRef]
- Herzberg, G. Molecular Spectra and Molecular Structure, Vol. I: Spectra of Diatomic Molecules; D. Van Nostrand Co.: New York, NY, USA, 1950. [Google Scholar]
- Beloy, K.; Kozlov, M.G.; Borschevsky, A.; Hauser, A.W.; Flambaum, V.V.; Schwerdtfeger, P. Rotational spectrum of the molecular ion NH+ as a probe for α and me/mp variation. Phys. Rev. A 2011, 83, 062514. [Google Scholar] [CrossRef]
- Pašteka, L.F.; Borschevsky, A.; Flambaum, V.V.; Schwerdtfeger, P. Search for the variation of fundamental constants: Strong enhancements in X2Π cations of dihalogens and hydrogen halides. Phys. Rev. A 2015, 92, 012103. [Google Scholar] [CrossRef]
- Schiller, S.; Bakalov, D.; Korobov, V.I. Simplest Molecules as Candidates for Precise Optical Clocks. Phys. Rev. Lett. 2014, 113, 023004. [Google Scholar] [CrossRef] [PubMed]
- Hilico, L.; Billy, N.; Grémaud, B.; Delande, D. Polarizabilities, light shifts and two-photon transition probabilities between J = 0 states of the and molecular ions. J. Phys. B 2001, 34, 491–507. [Google Scholar] [CrossRef]
- Karr, J.P. and HD+: Candidates for a molecular clock. J. Mol. Spectrosc. 2014, 300, 37–43. [Google Scholar] [CrossRef]
- Coxon, J.A.; Haley, M.P. Rotational Analysis of the A2Πu→X2Πg Second Negative Band System of . J. Mol. Spectrosc. 1984, 108, 119–136. [Google Scholar] [CrossRef]
- Song, Y.; Evans, M.; Ng, C.Y.; Hsu, C.W.; Jarvis, G.K. Rotationally resolved pulsed field ionization photoelectron bands of (X2Π1/2,3/2g,v+= 0–38) in the energy range of 12.05–18.15 eV. J. Chem. Phys. 1999, 111, 1905–1916. [Google Scholar] [CrossRef]
- Song, Y.; Evans, M.; Ng, C.Y.; Hsu, C.W.; Jarvis, G.K. Rotationally resolved pulsed-field ionization photoelectron bands of (A2Πu,v+= 0–12) in the energy range of 17.0–18.2 eV. J. Chem. Phys. 2000, 112, 1271–1278. [Google Scholar] [CrossRef]
- Staanum, P.F.; Højbjerre, K.; Skyt, P.S.; Hansen, A.K.; Drewsen, M. Rotational laser cooling of vibrationally and translationally cold molecular ions. Nat. Phys. 2010, 6, 271–274. [Google Scholar] [CrossRef]
- Schneider, T.; Roth, B.; Duncker, H.; Ernsting, I.; Schiller, S. All-optical preparation of molecular ions in the rovibrational ground state. Nat. Phys. 2010, 6, 275–278. [Google Scholar] [CrossRef] [Green Version]
- Lien, C.Y.; Seck, C.M.; Lin, Y.W.; Nguyen, J.H.; Tabor, D.A.; Odom, B.C. Broadband optical cooling of molecular rotors from room temperature to the ground state. Nat. Commun. 2014, 5, 4783. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.; Kurz, C.; Hume, D.B.; Plessow, P.N.; Leibrandt, D.R.; Leibfried, D. Preparation and coherent manipulation of pure quantum states of a single molecular ion. Nature 2017, 545, 203–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, P.O.; Rosenband, T.; Langer, C.; Itano, W.M.; Bergquist, J.C.; Wineland, D.J. Spectroscopy Using Quantum Logic. Science 2005, 309, 749–752. [Google Scholar] [CrossRef] [Green Version]
- Mur-Petit, J.; García-Ripoll, J.J.; Pérez-Ríos, J.; Campos-Martínez, J.; Hernández, M.I.; Willitsch, S. Temperature-independent quantum logic for molecular spectroscopy. Phys. Rev. A 2012, 85, 022308. [Google Scholar] [CrossRef]
- Wolf, F.; Wan, Y.; Heip, J.C.; Gerbert, F.; Shi, C.; Schmidt, P.O. Non-destructive state detection for quantum logic spectroscopy of molecular ions. Nature 2016, 530, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Kong, W.; Hepburn, J.W. Rotationally resolved threshold photoelectron spectroscopy of O2 using coherent XUV: Formation of vibrationally excited ions in the Franck–Condon gap. Can. J. Phys. 1994, 72, 1284–1293. [Google Scholar] [CrossRef]
- Merkt, F.; Signorell, R.; Palm, H.; Osterwalder, A.; Sommavilla, M. Towards resolving the hyperfine structure in ions by photoelectron spectroscopy. Mol. Phys. 1998, 95, 1045–1054. [Google Scholar] [CrossRef]
- Song, Y.; Evans, M.; Ng, C.Y.; Hsu, C.W.; Jarvis, G.K. Rotationally resolved pulsed field ionization photoelectron bands of (a4Πu,v+= 0–18) in the energy range of 16.0–18.0 eV. J. Chem. Phys. 2000, 112, 1306–1315. [Google Scholar] [CrossRef]
- Pratt, S.T. Excited-state molecular photoionization dynamics. Rep. Prog. Phys. 1995, 58, 821–883. [Google Scholar] [CrossRef]
- Morrill, J.S.; Ginter, M.L.; Lewis, B.R.; Gibson, S.T. The (X2Πg)nsσg1,3Πg Rydberg states of O2: Spectra, structures, and interactions. J. Chem. Phys. 1999, 111, 173–185. [Google Scholar] [CrossRef]
- Sur, A.; Ramana, C.V.; Colson, S.D. Optical spectra of the lowest Πg Rydberg states in O2. J. Chem. Phys. 1985, 83, 904–905. [Google Scholar] [CrossRef]
- Park, H.; Miller, P.J.; Chupka, W.A.; Colson, S.D. Production of vibrationally state-selected via newly discovered 4s–3d and 5s–4d Rydberg states of O2. J. Chem. Phys. 1988, 89, 3919–3921. [Google Scholar] [CrossRef]
- Ogorzalek Loo, R.; Marinelli, W.J.; Houston, P.L.; Arepalli, S.; Wiesenfeld, J.R.; Field, R.W. Multiphoton ionization of O2X3, a1Δg, and b1 via the two-photon resonant nsσg, ndσg, and ndπg Rydberg levels. J. Chem. Phys. 1989, 91, 5185–5200. [Google Scholar] [CrossRef]
- Sur, A.; Friedman, R.S.; Miller, P.J. Rotational dependence of the Rydberg-valence interactions in the 1Πg states of molecular oxygen. J. Chem. Phys. 1991, 94, 1705–1711. [Google Scholar] [CrossRef]
- Dochain, A.; Urbain, X. Production of a rovibrationally selected beam for dissociative recombination studies. EPJ Web Conf. 2015, 84, 05001. [Google Scholar] [CrossRef]
- Tong, X.; Winney, A.H.; Willitsch, S. Sympathetic Cooling of Molecular Ions in Selected Rotational and Vibrational States Produced by Threshold Photoionization. Phys. Rev. Lett. 2010, 105, 143001. [Google Scholar] [CrossRef]
- Tong, X.; Wild, D.; Willitsch, S. Collisional and radiative effects in the state-selective preparation of translationally cold molecular ions in ion traps. Phys. Rev. A 2011, 83, 023415. [Google Scholar] [CrossRef]
- Scoles, G.; Bassi, D.; Buck, U.; Laine, D.C. (Eds.) Atomic and Molecular Beam Methods; Oxford University Press: New York, NY, USA, 1988; Volume 1. [Google Scholar]
- Xie, J.; Zare, R.N. Selection rules for the photoionization of diatomic molecules. J. Chem. Phys. 1990, 93, 3033–3038. [Google Scholar] [CrossRef]
- Germann, M.; Willitsch, S. Fine- and hyperfine-structure effects in molecular photoionization. I. General theory and direct photoionization. J. Chem. Phys. 2016, 145, 044314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibfried, D. Quantum state preparation and control of single molecular ions. New J. Phys. 2012, 14, 023029. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Matsukevich, D.N. Quantum logic for the control and manipulation of molecular ions using a frequency comb. New J. Phys. 2012, 14, 023028. [Google Scholar] [CrossRef] [Green Version]
- Degen, C.L.; Reinhard, F.; Cappellaro, P. Quantum sensing. Rev. Mod. Phys. 2017, 89, 035002. [Google Scholar] [CrossRef]
- Bertelsen, A.; Jørgensen, S.; Drewsen, M. The rotational temperature of polar molecular ions in Coulomb crystals. J. Phys. B 2006, 39, L83–L89. [Google Scholar] [CrossRef]
- Roth, B.; Koelemeij, J.C.J.; Daerr, H.; Schiller, S. Rovibrational spectroscopy of trapped molecular hydrogen ions at millikelvin temperatures. Phys. Rev. A 2006, 74, 040501. [Google Scholar] [CrossRef]
- Seck, C.M.; Hohenstein, E.G.; Lien, C.Y.; Stollenwerk, P.R.; Odom, B.C. Rotational state analysis of AlH+ by two-photon dissociation. J. Mol. Spectrosc. 2014, 300, 108–111. [Google Scholar] [CrossRef]
- Ni, K.K.; Loh, H.; Grau, M.; Cossel, K.C.; Ye, J.; Cornell, E.A. State-specific detection of trapped HfF+ by photodissociation. J. Mol. Spectrosc. 2014, 300, 12–15. [Google Scholar] [CrossRef]
- Schlemmer, S.; Kuhn, T.; Lescop, E.; Gerlich, D. Laser excited in a 22-pole ion trap: Experimental studies of rotational relaxation processes. Int. J. Mass Spectrom. 1999, 185, 589–602. [Google Scholar] [CrossRef]
- Akahori, T.; Morioka, Y.; Tanaka, T.; Yoshii, H.; Hayaishi, T.; Ito, K. High resolution threshold photoelectron spectrum of oxygen in the 12–19 eV region. J. Chem. Phys. 1997, 107, 4875–4880. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Gordon, M.S.; Song, Y.; Ng, C.Y. Theoretical study of spin-orbit coupling constants for (A2Π3/2,1/2u,v+= 0–17 and a4Π5/2,3/2,1/2,-1/2u,v+= 0–25). J. Chem. Phys. 2001, 115, 7393–7400. [Google Scholar] [CrossRef]
- Liu, H.; Shi, D.; Sun, J.; Zhu, Z. Accurate theoretical investigations of the 20 Λ-S and 58 Ω states of cation including spin–orbit coupling effect. Mol. Phys. 2015, 113, 120–136. [Google Scholar] [CrossRef]
- Zhang, C.B.; Offenberg, D.; Roth, B.; Wilson, M.A.; Schiller, S. Molecular-dynamics simulations of cold single-species and multispecies ion ensembles in a linear Paul trap. Phys. Rev. A 2007, 76, 012719. [Google Scholar] [CrossRef]
- Baba, T.; Waki, I. Cooling and Mass-Analysis of Molecules Using Laser-Cooled Atoms. Jpn. J. Appl. Phys. 1996, 35, L1134–L1137. [Google Scholar] [CrossRef]
- Baba, T.; Waki, I. Spectral shape of in situ mass spectra of sympathetically cooled molecular ions. J. Appl. Phys. 2002, 92, 4109–4116. [Google Scholar] [CrossRef]
- Schowalter, S.J.; Chen, K.; Rellergert, W.G.; Sullivan, S.T.; Hudson, E.R. An integrated ion trap and time-of-flight mass spectrometer for chemical and photo- reaction dynamics studies. Rev. Sci. Instrum. 2012, 83, 043103. [Google Scholar] [CrossRef]
- Deb, N.; Pollum, L.L.; Smith, A.D.; Keller, M.; Rennick, C.J.; Heazlewood, B.R.; Softley, T.P. Coulomb crystal mass spectrometry in a digital ion trap. Phys. Rev. A 2015, 91, 033408. [Google Scholar] [CrossRef]
- Schneider, C.; Schowalter, S.J.; Yu, P.; Hudson, E.R. Electronics of an ion trap with integrated time-of-flight mass spectrometer. Int. J. Mass Spectrom. 2016, 394, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.C.; Greenberg, J.; Miller, M.I.; Loeffler, K.; Lewandowski, H.J. An ion trap time-of-flight mass spectrometer with high mass resolution for cold trapped ion experiments. Rev. Mod. Phys. 2017, 88, 123107. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre-Brion, H.; Field, R.W. The Spectra and Dynamics of Diatomic Molecules; Elsevier: San Diego, CA, USA, 2004. [Google Scholar]
- Gilmore, F.R.; Laher, R.R.; Espy, P.J. Franck–Condon Factors, r-Centroids, Electronic Transition Moments, and Einstein Coefficients for Many Nitrogen and Oxygen Band Systems. J. Phys. Chem. Ref. Data 1992, 21, 1005–1107. [Google Scholar] [CrossRef]
- Le Roy, R.J. RKR1: A computer program implementing the first-order RKR method for determining diatomic molecule potential energy functions. J. Quant. Spectrosc. Radiat. Transf. 2017, 186, 158–166. [Google Scholar] [CrossRef]
- Le Roy, R.J. LEVEL: A Computer Program for Solving the Radial Schrödinger Equation for Bound and Quasibound Levels. J. Quant. Spectrosc. Radiat. Transf. 2017, 186, 167–178. [Google Scholar] [CrossRef]
- Yudin, V.I.; Taichenachev, A.V.; Oates, C.W.; Barber, Z.W.; Lemke, N.D.; Ludlow, A.D.; Sterr, U.; Lisdat, C.; Riehle, F. Hyper-Ramsey spectroscopy of optical clock transitions. Phys. Rev. A 2010, 82, 011804. [Google Scholar] [CrossRef]
- Yudin, V.I.; Taichenachev, A.V.; Basalaev, M.Y.; Zanon-Willette, T.; Pollock, J.W.; Shuker, M.; Donley, E.A.; Kitching, J. Generalized Autobalanced Ramsey Spectroscopy of Clock Transitions. Phys. Rev. Appl. 2018, 9, 054034. [Google Scholar] [CrossRef] [Green Version]
- Sanner, C.; Huntemann, N.; Lange, R.; Tamm, C.; Peik, E. Autobalanced Ramsey Spectroscopy. Phys. Rev. Lett. 2018, 120, 053602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanon-Willette, T.; Lefevre, R.; Metzdorff, R.; Sillitoe, N.; Almonacil, S.; Minissale, M.; de Clercq, E.; Taichenachev, A.V.; Yudin, V.I.; Arimondo, E. Composite laser-pulses spectroscopy for high-accuracy optical clocks: A review of recent progress and perspectives. Rep. Prog. Phys. 2018, 81, 094401. [Google Scholar] [CrossRef]
- Huntemann, N.; Lipphardt, B.; Okhapkin, M.; Tamm, C.; Peik, E.; Taichenachev, A.V.; Yudin, V.I. Generalized Ramsey Excitation Scheme with Suppressed Light Shift. Phys. Rev. Lett. 2012, 109, 213002. [Google Scholar] [CrossRef]
- Huntemann, N.; Sanner, C.; Lipphardt, B.; Tamm, C.; Peik, E. Single-Ion Atomic Clock with 3 × 10-18 Systematic Uncertainty. Phys. Rev. Lett. 2016, 116, 063001. [Google Scholar] [CrossRef]
- Berkeland, D.J.; Miller, J.D.; Bergquist, J.C.; Itano, W.M.; Wineland, D.J. Minimization of ion micromotion in a Paul trap. J. Appl. Phys. 1998, 83, 5025–5033. [Google Scholar] [CrossRef]
- Wineland, D.J.; Monroe, C.; Itano, W.M.; Leibfried, D.; King, B.E.; Meekhof, D.M. Experimental Issues in Coherent Quantum-State Manipulation of Trapped Atomic Ions. J. Res. Natl. Inst. Stand. Technol. 1998, 103, 259–328. [Google Scholar] [CrossRef] [PubMed]
- Farley, J.W.; Wing, W.H. Accurate calculation of dynamic Stark shifts and depopulation rates of Rydberg energy levels induced by blackbody radiation. Hydrogen, helium, and alkali-metal atoms. Phys. Rev. A 1981, 23, 2397–2424. [Google Scholar] [CrossRef]
- Porsev, S.G.; Derevianko, A. Multipolar theory of blackbody radiation shift of atomic energy levels and its implications for optical lattice clocks. Phys. Rev. A 2006, 74, 020502. [Google Scholar] [CrossRef]
- Rosenband, T.; Itano, W.M.; Schmidt, P.O.; Hume, D.B.; Koelemeij, J.C.J.; Bergquist, J.C.; Wineland, D.J. Blackbody radiation shift of the 27Al+1S0→3P0 transition. arXiv, 2006; arXiv:physics/0611125. [Google Scholar]
- Arnold, K.J.; Kaewuam, R.; Roy, A.; Tan, T.R.; Barrett, M.D. Blackbody radiation shift assessment for a lutetium ion clock. Nat. Commun. 2018, 9, 1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicke, R.H. The Effect of Collisions upon the Doppler Width of Spectral Lines. Phys. Rev. 1953, 89, 472–473. [Google Scholar] [CrossRef]
- Rosenband, T.; Hume, D.B.; Schmidt, P.O.; Chou, C.W.; Brusch, A.; Lorini, L.; Oskay, W.H.; Drullinger, R.E.; Fortier, T.M.; Stalnaker, J.E.; et al. Frequency Ratio of Al+ and Hg+ Single-Ion Optical Clocks; Metrology at the 17th Decimal Place. Science 2008, 319, 1808–1812. [Google Scholar] [CrossRef]
- Chen, J.S.; Brewer, S.M.; Chou, C.W.; Wineland, D.J.; Leibrandt, D.R.; Hume, D.B. Sympathetic Ground State Cooling and Time-Dilation Shifts in an 27Al+ Optical Clock. Phys. Rev. Lett. 2017, 118, 053002. [Google Scholar] [CrossRef]
- Itano, W.M. External-Field Shifts of the 199Hg+ Optical Frequency Standard. J. Res. Natl. Inst. Stand. Technol. 2000, 105, 829–837. [Google Scholar] [CrossRef]
- Bakalov, D.; Schiller, S. The electric quadrupole moment of molecular hydrogen ions and their potential for a molecular ion clock. Appl. Phys. B 2014, 114, 213–230. [Google Scholar] [CrossRef]
- Schadee, A. On the Zeeman effect in electronic transitions of diatomic molecules. J. Quant. Spectrosc. Radiat. Transf. 1978, 19, 517–531. [Google Scholar] [CrossRef]
- Berdyugina, S.V.; Solanki, S.K. The molecular Zeeman effect and diagnostics of solar and stellar magnetic fields. Astron. Astrophys. 2002, 385, 701–715. [Google Scholar] [CrossRef] [Green Version]
- Schiff, L.I.; Snyder, H. Theory of the Quadratic Zeeman Effect. Phys. Rev. 1939, 55, 59–63. [Google Scholar] [CrossRef]
- Garstang, R.H. Atoms in high magnetic fields (white dwarfs). Rep. Prog. Phys. 1977, 40, 105–154. [Google Scholar] [CrossRef]
- Townes, C.H.; Schawlow, A.L. Microwave Spectroscopy; Dover: Mineola, NY, USA, 1975. [Google Scholar]
- Brown, J.; Carrington, A. Rotational Spectroscopy of Diatomic Molecules; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Itano, W.M.; Bergquist, J.C.; Bollinger, J.J.; Gilligan, J.M.; Heinzen, D.J.; Moore, F.L.; Raizen, M.G.; Wineland, D.J. Quantum projection noise: Population fluctuations in two-level systems. Phys. Rev. A 1993, 47, 3554–3570. [Google Scholar] [CrossRef] [PubMed]
- Kajita, M. Search for the Variation in (mp/me) Using Two Vibrational Transition Frequencies of Molecular Ions. J. Phys. Soc. Jpn. 2017, 86, 133301. [Google Scholar] [CrossRef]
- Gerstenkorn, S.; Luc, P. Atlas du Spectre d’Absorption de la Molecule d’Iode, 14 800–20 000 cm−1; CNRS: Paris, France, 1978. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
(nm) | (THz) | (nm) | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 10,614 | −28 | 113 | 4.94 | −0.59 | −0.12 | −0.16 | −3.59 |
| 2 | 5386 | −54 | 123 | 1.24 | −0.40 | −0.32 | −0.11 | −2.43 |
| 3 | 3617 | −80 | 134 | 1.56 | −0.47 | −0.30 | −0.12 | −2.86 |
| 4 | 2738 | −104 | 144 | 0.91 | −0.45 | −0.49 | −0.12 | −2.69 |
| 5 | 2211 | −128 | 155 | 0.99 | −0.51 | −0.52 | −0.13 | −3.07 |
| 6 | 1859 | −151 | 166 | 0.91 | −0.53 | −0.59 | −0.14 | −3.14 |
| 7 | 1609 | −172 | 179 | 0.68 | −0.62 | −0.90 | −0.15 | −3.56 |
| 8 | 1421 | −193 | 192 | 0.91 | −0.67 | −0.73 | −0.16 | −3.79 |
| 9 | 1275 | −213 | 206 | 0.51 | −0.78 | −1.53 | −0.18 | −4.26 |
| 10 | 1158 | −231 | 221 | 0.92 | −0.88 | −0.95 | −0.20 | −4.59 |
| 11 | 1063 | −249 | 238 | 0.40 | −1.03 | −2.56 | −0.22 | −5.14 |
| 12 | 984 | −266 | 256 | 0.93 | −1.22 | −1.31 | −0.24 | −5.65 |
| 13 | 917 | −281 | 276 | 0.33 | −1.53 | −4.57 | −0.28 | −6.39 |
| 14 | 860 | −296 | 299 | 0.94 | −1.97 | −2.09 | −0.31 | −7.14 |
| 15 | 810 | −310 | 323 | 0.30 | −2.79 | −9.20 | −0.35 | −8.14 |
| 16 | 767 | −323 | 351 | 0.96 | −4.72 | −4.91 | −0.40 | −9.20 |
| 17 | 730 | −334 | − | 0.31 | −1.48 | −4.77 | −0.46 | −10.58 |
| 18 | 696 | −345 | − | 0.98 | 9.10 | 9.29 | −0.52 | −12.06 |
| 19 | 667 | −355 | − | 0.35 | 17.52 | 49.40 | −0.60 | −13.95 |
| 20 | 640 | −364 | − | 1.01 | 1.79 | 1.77 | −0.69 | −16.06 |
| 21 | 616 | −371 | − | 0.43 | −6.69 | −15.59 | −0.81 | −18.81 |
| 22 | 594 | −378 | − | 1.01 | 5.88 | 5.79 | −0.95 | −21.97 |
| 23 | 575 | −384 | − | 0.56 | 3.55 | 6.30 | −1.12 | −26.10 |
| 24 | 557 | −389 | − | 1.01 | 3.34 | 3.29 | −1.32 | −30.82 |
| 25 | 541 | −393 | − | 0.75 | 2.66 | 3.55 | −1.58 | −36.94 |
| 26 | 526 | −395 | − | 0.99 | 2.18 | 2.21 | −1.88 | −43.99 |
| 27 | 513 | −397 | − | 0.99 | 2.04 | 2.06 | −2.27 | −53.31 |
| 28 | 500 | −398 | − | 0.86 | 1.85 | 2.14 | −2.74 | −64.36 |
| 29 | 489 | −398 | − | 1.28 | 1.80 | 1.40 | −3.35 | −79.02 |
| 30 | 478 | −397 | − | 0.59 | 1.71 | 2.88 | −4.09 | −96.75 |
| 31 | 468 | −395 | − | 1.58 | 1.69 | 1.07 | −5.06 | −120.31 |
| 32 | 459 | −392 | − | 0.22 | 1.66 | 7.62 | −6.29 | −150.64 |
| 33 | 451 | −388 | − | 1.84 | 1.68 | 0.91 | −7.92 | −191.28 |
| 34 | 444 | −383 | − | 0.38 | 1.72 | 4.47 | −9.98 | −242.92 |
| 35 | 437 | −377 | − | 1.91 | 1.73 | 0.91 | −12.32 | −303.11 |
| 36 | 430 | −369 | − | 1.21 | 1.78 | 1.47 | −15.20 | −379.12 |
| 37 | 424 | −361 | − | 1.69 | 1.77 | 1.05 | −18.99 | −483.29 |
| 38 | 418 | −352 | − | 2.28 | 1.86 | 0.82 | −24.72 | −646.53 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carollo, R.; Frenett, A.; Hanneke, D. Two-Photon Vibrational Transitions in 16O2+ as Probes of Variation of the Proton-to-Electron Mass Ratio. Atoms 2019, 7, 1. https://doi.org/10.3390/atoms7010001
Carollo R, Frenett A, Hanneke D. Two-Photon Vibrational Transitions in 16O2+ as Probes of Variation of the Proton-to-Electron Mass Ratio. Atoms. 2019; 7(1):1. https://doi.org/10.3390/atoms7010001
Chicago/Turabian StyleCarollo, Ryan, Alexander Frenett, and David Hanneke. 2019. "Two-Photon Vibrational Transitions in 16O2+ as Probes of Variation of the Proton-to-Electron Mass Ratio" Atoms 7, no. 1: 1. https://doi.org/10.3390/atoms7010001
APA StyleCarollo, R., Frenett, A., & Hanneke, D. (2019). Two-Photon Vibrational Transitions in 16O2+ as Probes of Variation of the Proton-to-Electron Mass Ratio. Atoms, 7(1), 1. https://doi.org/10.3390/atoms7010001