1. Introduction
Spectroscopic techniques can provide useful quantitative measurements across a variety of scientific disciplines. While broadly applicable, spectroscopic methods are often tailored to achieve a niche measurement. Therefore, data can be collected across a wide range of spectral resolutions, instrument parameters, excitation sources, temporal sampling rates, optical depths, and temperature variations. The high dimensional nature of this measurement space presents a challenge for implementing generalized analysis and forward modeling capability that is effective across all disciplines and experimental methods.
To augment expensive quantitative measurements, methods for simulating optical spectra are well documented in the scientific literature [
1,
2]. Utilizing forward modeling allows a simulated spectrum to be parametrically fit to measured data, thereby accounting for temperature and instrument effects using closed form equations and modest computational resources. In many cases, the primary limitation of forward spectral modeling is a lack of spectral constants available in the literature or spectral databases. These constants may not be available due to a lack of resources in the community, complexity in theoretical calculations, or the sheer volume of experiments required to produce the needed fundamental parameters.
Integral to any quantitative optical spectral model is the transition probability. The transition probability (a.k.a. Einstein coefficient, A-coefficient, oscillator strength, gf-value) is a temperature independent property representing the spontaneous emission rate in a two-level energy model. The pedagogy of both theoretical and experimental determination of transition probabilities is very rich, as the preferred methods of both areas have changed over time [
3,
4,
5]. For the simplest nuclei, complete quantum mechanical calculations can yield nearly exact values more precise than any experiment [
6,
7,
8]. For light elements, Hartree–Fock calculations are widely accepted theoretical treatments and yield accuracies comparable with experimental measurements [
9,
10,
11,
12,
13,
14]. Transition probabilities for heavy nuclei are derived almost entirely from experimental data and can have the largest uncertainties [
15,
16,
17].
Machine learning (ML) has recently gained traction as a potential method to perform a generalized analysis of spectroscopic data [
18,
19,
20,
21,
22] and there is some work published on predicting spectra using these methods [
23]. Although efforts are being made to generalize spectral analysis with artificial intelligence [
19,
24], many approaches still implicitly reduce the dimensionality. For example, reduced generalization occurs for a model which only analyzes data collected with a single type of instrument and spectral resolution. Additionally, any variation in temperature or optical depth during an experiment can significantly alter an optical emission or absorption spectrum which may limit analytical performance of ML when applied more broadly than the specific training conditions. To overcome these hindrances to generalization, we hypothesize that neural network (NN) architectures can predict transition probabilities and can be coupled with closed-form, forward spectral modeling to more generically analyze and simulate optical spectra.
Contributions
This work examines a novel application of machine learning to spectroscopic data by implementing NN architectures trained on fundamental spectroscopic information to predict Einstein A-coefficients. We investigate if NNs can provide a method to estimate Einstein A-coefficient constants at a usable accuracy on a significantly shorter time scale relative to theoretical calculation or direct measurement. The general approach in this first-of-its-kind efficacy study is to predict Einstein A-coefficients for electronic transitions in atomic spectra by training NNs on published values of known spectral constants. In this way, the predictions of the neural network can be directly compared to data that are widely used by the community. This effort demonstrates a numeric encoding to represent spectroscopic transitions to be used by machine learning models followed by predictions of Einstein A-coefficients for various elements on the periodic table. The numeric dataset is built from the NIST Atomic Spectral Database (ASD) [
25] as it is the paragon for tabulations of transition probabilities with bounded uncertainty, which allows us to assess the variance in the predictions produced by the neural network. In
Section 2, we detail how NIST data are transformed into a machine-learnable format, followed by
Section 3 where we describe experiment design and metrics used. A discussion of results including intraelement and interelement experiments, a direct comparison to previous theoretical work and model feature importance is presented in
Section 4, followed by conclusions in
Section 5.
2. Data Representation
Data representation in any machine learning model is arguably one of the most important design criteria. The data which is used to train the machine learning model must be provided to the model in a way that accurately preserves the most relevant information within the data [
26]. Significant work in the field of cheminformatics has provided groundwork for presenting chemical and physical structure in representations interpretable by a NN [
27,
28,
29]. Care must be taken in order to preserve the statistical characteristics (e.g., ordinal, categorical, boundedness) of each feature, or model input dimension, while providing a feature that can be interpreted by predictive models. The best possible set of features is the subset which preserves the most statistical information in the lowest possible dimension while contributing to model learning [
30,
31]. It is assumed in this focused work that feature representation of spectroscopic transitions would be intimately aligned with nuclear and electronic structural parameters as these are the fundamentals informing theoretical calculations [
32]. In this section, we describe how the NIST tables of spectroscopic transitions are transformed into a ML-ready format.
The NIST ASD [
25] contains a tabulated list of known, element-specific spectral transitions and transition probabilities per each element. For each tabulated spectral transition for a given element, we extracted from the NIST ASD the transition wavelength, the upper and lower state energy, the upper and lower state term symbol, the upper and lower electron configuration, the upper and lower degeneracy, the transition type (i.e., allowed or forbidden), and the transition probability. For a more detailed description of these parameters, the reader is directed to the NIST Atomic Spectroscopy compendium by Martin and Wiese [
33].
As a data pre-processing step, we first strip away all transitions that do not have published A-coefficients since we cannot use them to train and evaluate our models. It is worth noting that a large set of transitions within each element do not have published Einstein coefficients but may be directly modeled using our approach subsequent to model training.
As it is standard in most machine learning pre-processing pipelines, we perform transformations on the data to create features and regressands that are well distributed [
26]. Einstein A-coefficients, transition wavelength, and upper and lower energies typically range orders of magnitude across the various datasets. Such large variations in model features can create learning instabilities in the model. One mitigation strategy we employ is to transform these values with large dynamic ranges using
. In this way, the widely ranging values are transformed to a scale that is more amenable to training while also avoiding undefined instances in the data. We additionally scale features by standardizing. Standardization scales model features to provide a data distribution with a mean of zero and a unit standard deviation. Standardization is a common ML practice as it is useful for improving algorithm stability [
34]. However, standardizing data that is non-continuous (e.g., binary or categorical, such as the categories of “allowed” or “forbidden” transitions) must be performed with care. We encode these variables with a one-hot schema (−1/+1) to encourage symmetry about zero during rescaling. In this way, one category is designated numerically with a value of −1, while the other category is designated with a value of +1.
During our pre-processing, the type of transition (e.g., electric dipole, magnetic dipole, etc.) intuitively represents a valuable feature strongly influencing the transition probability. We initially labeled each transition type with a one-hot encoding scheme representing the type of transition covering all of the NIST-reported designations [
35]. The NIST datasets are dominated by electronic dipole transitions to the point where most other transitions showed up as outliers in our trained models. Because of this, we elected to drop transitions other than electronic dipoles from the scope of this paper. This is also important as it removes the differing wavelength dependencies between line strength and A-coefficient across the various transition types (E1, M1, E2, etc.) [
36,
37]. We discuss how these transitions could be more accurately modeled in
Section 4.
In the case of multiplet transitions with unresolved fine structure, tabulations include all of the allowable total angular momentum (J) values. From a data representation standpoint, those transitions are split up into otherwise identical transitions, each one having one of the allowable J values in the multiplet. This maintains a constant distribution over J values instead of introducing outlier features.
2.1. Electron Configuration
Quantum energy states with a defined electron configuration provide an opportunity to succinctly inform the model regarding wave function of the upper and lower states. This is arguably the most important feature as the configuration describes the wave functions which subsequently provide the overlap integral for the transition probability between two states [
11,
33,
38]. We refer the reader to the text of Martin and Wiese [
33] for more rigorous discussion of atomic states, quantum numbers, and multi-electron configurations.
Our encoding scheme for the electron configuration follows nl
nomenclature and represents each subshell with the principle quantum number (n) as well as the occupation number (k). For context in the present work, allowable values of n are positive integers, and l are integer values spaced by 1, ranging from 0 to n − 1. The variable l is represented in configurations with letters s, p, d, etc. denoting l = 0, 1, 2, etc. [
33]. The reader is encouraged to find further information regarding electron configurations in the following references [
33,
38]. The orbital angular momentum quantum number (l) is represented through the feature’s location in the final array. That is to say, the total configuration, once encoded, is a fixed length array and the first eight entries are reserved for s-type subshells, the second eight for p-type subshells, and so on. Our representation accommodates multiple subshells of the same orbital angular momentum quantum number (i.e., s
, s
, etc.). An example configuration where this is needed is the 3d
(
D)4s(
D)4d state of neutral iron when there are two d-type subshells that need to be accommodated. An abbreviated example encoding of the LS-coupled 19,350.891 cm
level of neutral iron is shown in
Table 1. The representation simply illustrates the rules followed in our schema. Actual encoding schema allows up to four of each subshell type (e.g., s
through s
), and orbital angular momentum quantum numbers up to 7 (s-type through k-type subshells). The complete encoded feature vector including configuration, coupling scheme, and other parameters for this energy level as an example can be found in
Appendix A.
Coupling terms in the configuration were not included in this feature. Term symbol coupling, however, was included and is discussed in the following section. Additional functionality was built in to allow selection of filled subshells, or strictly valence shells.
2.2. Term Symbol
NIST ASD contains transitions of several different coupling schemes which can be inferred from the term symbol notation. The physical meaning of the coupling is summarized by Martin and Wiese of NIST [
33] and comprehensively dissected by Cowan [
38]. Our representation of the information contained in a term symbol for a given energy state is reduced to four numerically-encoded features that accommodate
(Russel–Saunders),
,
→
, and
→
coupling. Inferred from the term symbol notation, we assign the first feature for the coupling scheme: [1, −1, −1] for
, [−1, 1, −1] for
, and [−1, −1, 1] for
→
and
→
as the latter share the same notation. The choice of a −1 or +1 value for these coupling schemes is simply another example of the use of categorical schema for representing transition data in our framework.
The second and third features become the two quantum numbers for the vectors that couple to give the total angular momentum quantum number (J). For example, in the case of LS coupling, these two numbers are the orbital (L) and spin (S) angular momentum quantum numbers. The fourth feature extracted from the term symbol is the parity. We assign a value of −1 for odd parity and +1 for even parity. Examples of each term symbol representation are shown in
Table 2.
In addition to the previous parameters that describe the electron and orbital in a radiative transition, we include several features that describe the nucleus to which each transition belongs. These include protons, neutrons, electrons, nuclear spin, molar mass, period/group on the periodic table, and ionization state. There is redundancy in some of these features if all of them are included in a single experiment; however, additional experiments were conducted with subsets of features focused on optimizing results while minimizing inputs. In general, we discard columns where features have zero variance. All the data were originally collected but selectively culled on a per-experiment basis. The primary motivation behind including features of nuclear properties is to provide context for the predictive model, such that multi-material experiments can be conducted to study relational learning ability across different elements and ions thereof.
3. Experiments
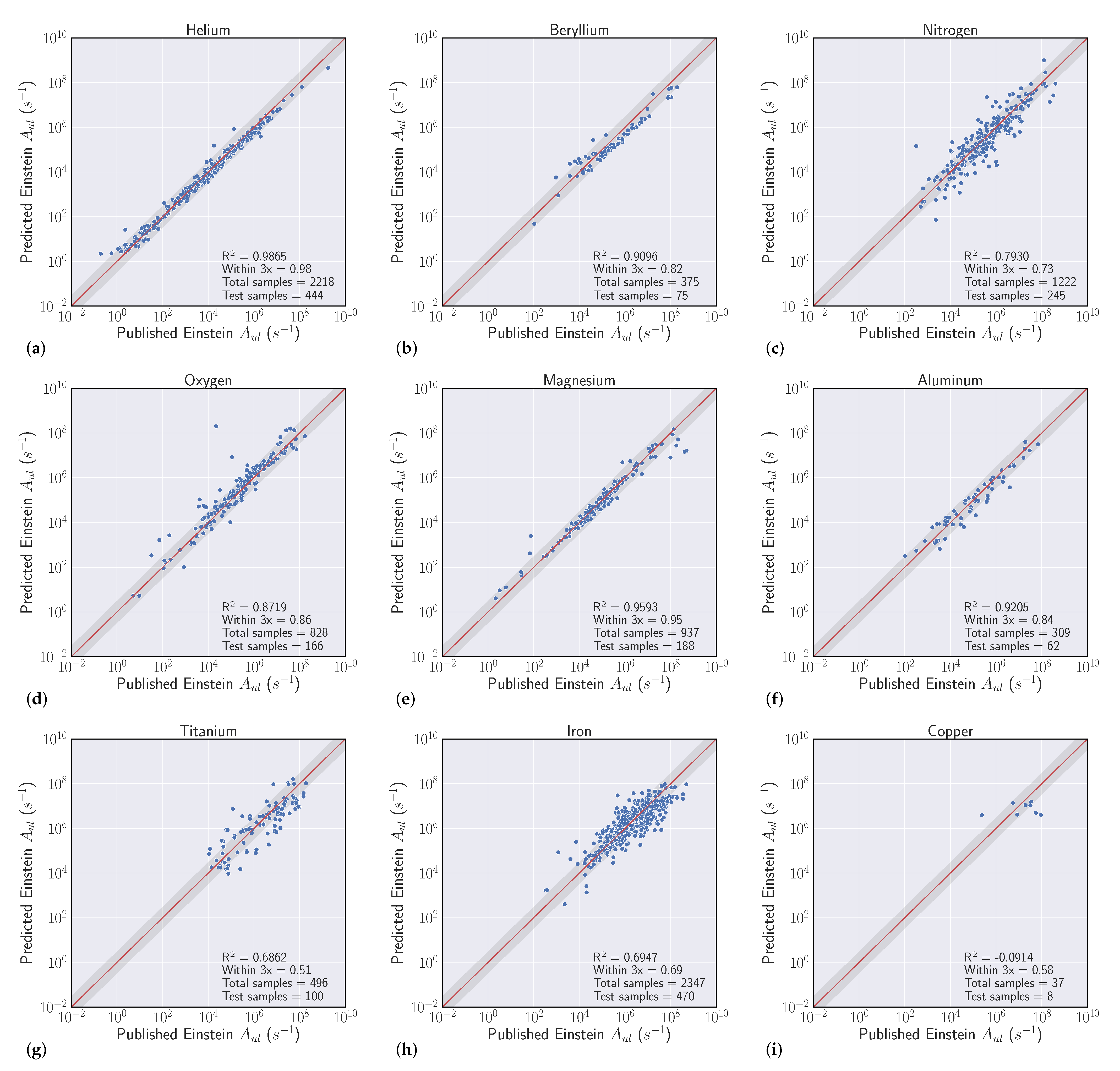
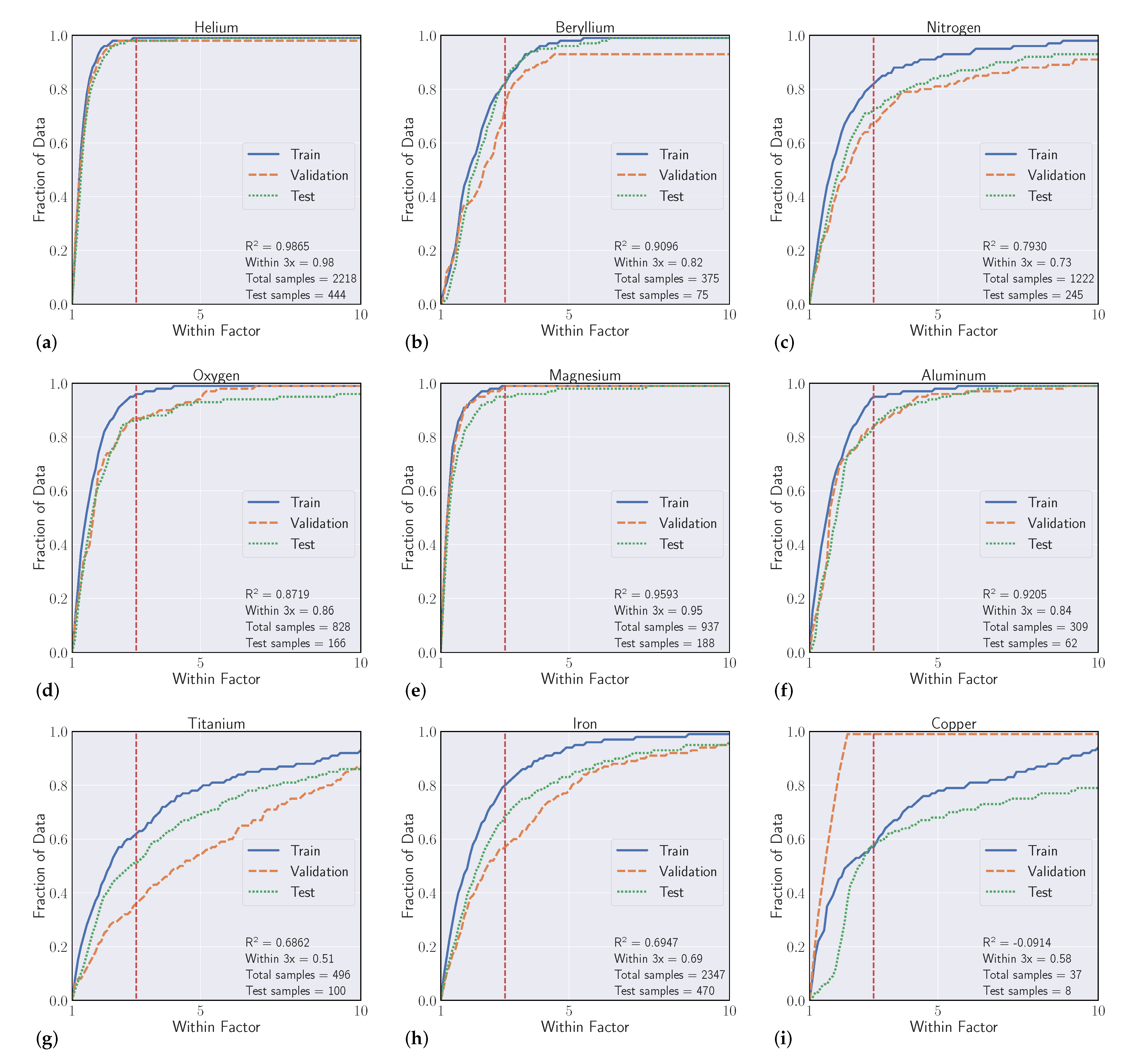
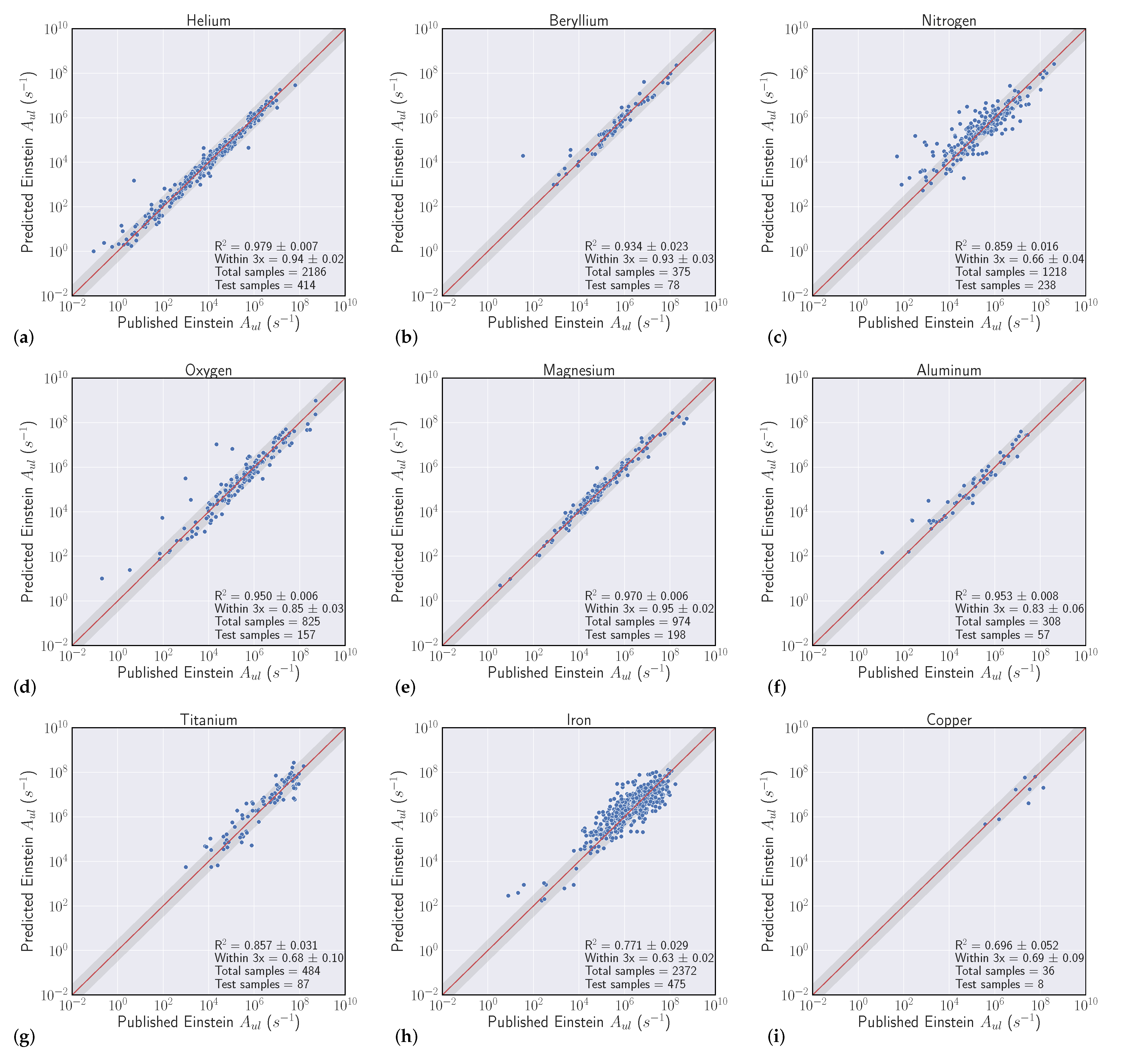
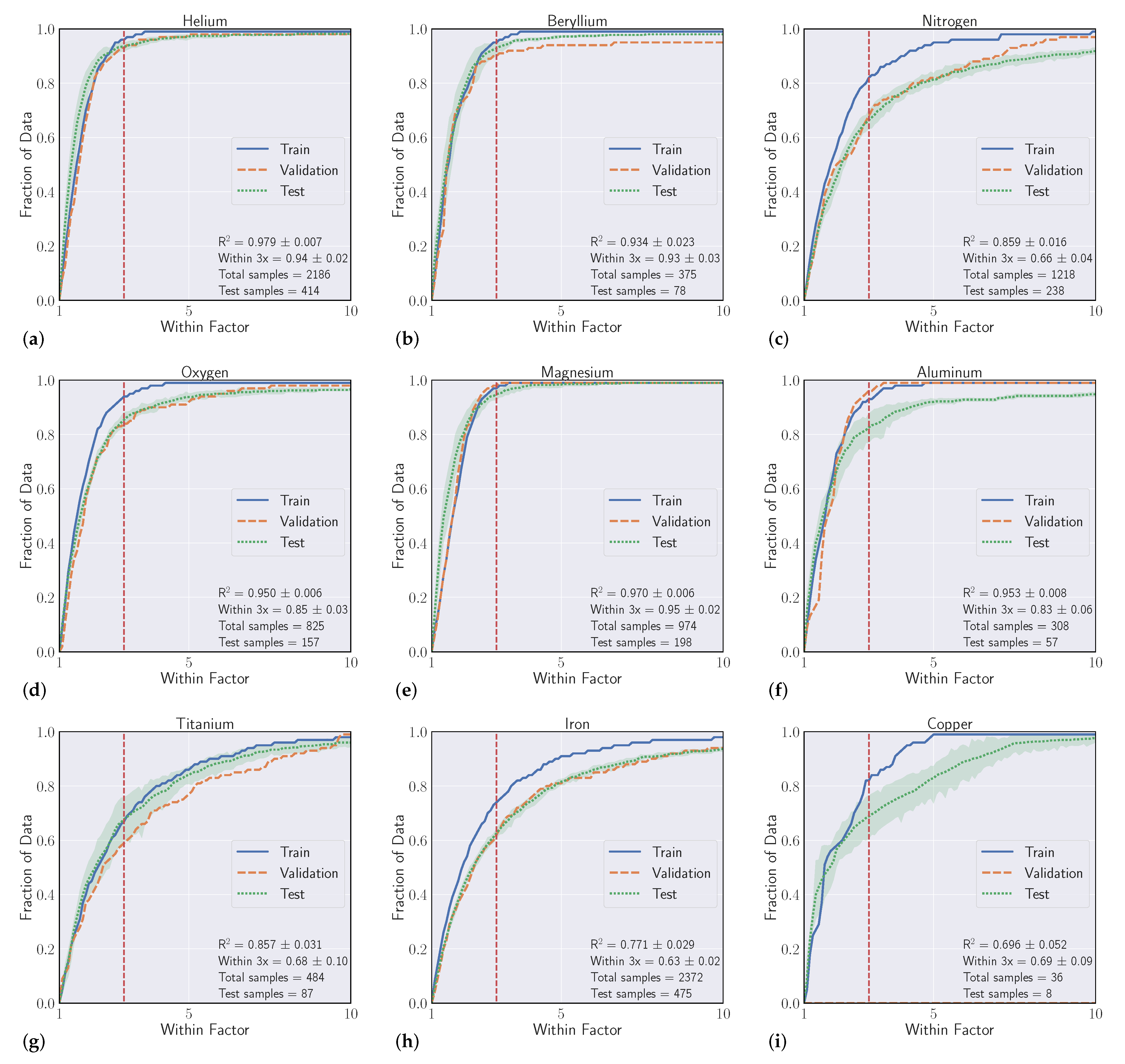
We conduct a series of experiments to show the efficacy of using machine learning models to regress Einstein A-coefficients directly from spectroscopic transition data. The first set of experiments are labeled as ’intraelement’ and the second we denote as ’interelement’ models. Intraelement models are single element, meaning the training, validation, and test sets all come from the same element. Interelement models extend a single model to predict coefficients from multiple elements. That is, the training and validation sets are a combination of multiple elements, and the model is tested on single element test sets. We describe the datasets, metrics, and model selection process with more detail in the following section.
3.1. Datasets
Our experiments were guided by the availability of data within the NIST ASD, where transitions from elements with atomic number Z > 50 quickly become sparse. We curated datasets from the first five rows of the periodic table, excluding arsenic (Z = 33), selenium (Z = 34), zirconium (Z = 40), niobium (Z = 41), and iodine (Z = 53) solely based on data availability. With a large set of spectroscopic transitions spanning nearly 50 elements, we intended to compile interesting findings and correlations that could help inform when models are high and low performing.
More concretely, let
be the set of 49 elements we were able to acquire data from and can be seen in
Table A1. For
, intraelement experiments are based on a data matrix
and vector
for a single element (
) described by the encoding scheme in
Section 2.
n is defined as the number of transitions with published A-coefficients and let
p be the number of features describing each spectroscopic transition of
E. We randomly subset
into train (70%), validation (10%), and test (20%) sets and train a variety of models for the regression task. The training set is the data used to fit the model, the validation set is the data used to evaluate the parameters found during training, and the test set is the data held out to evaluate the model performance on never before seen data.
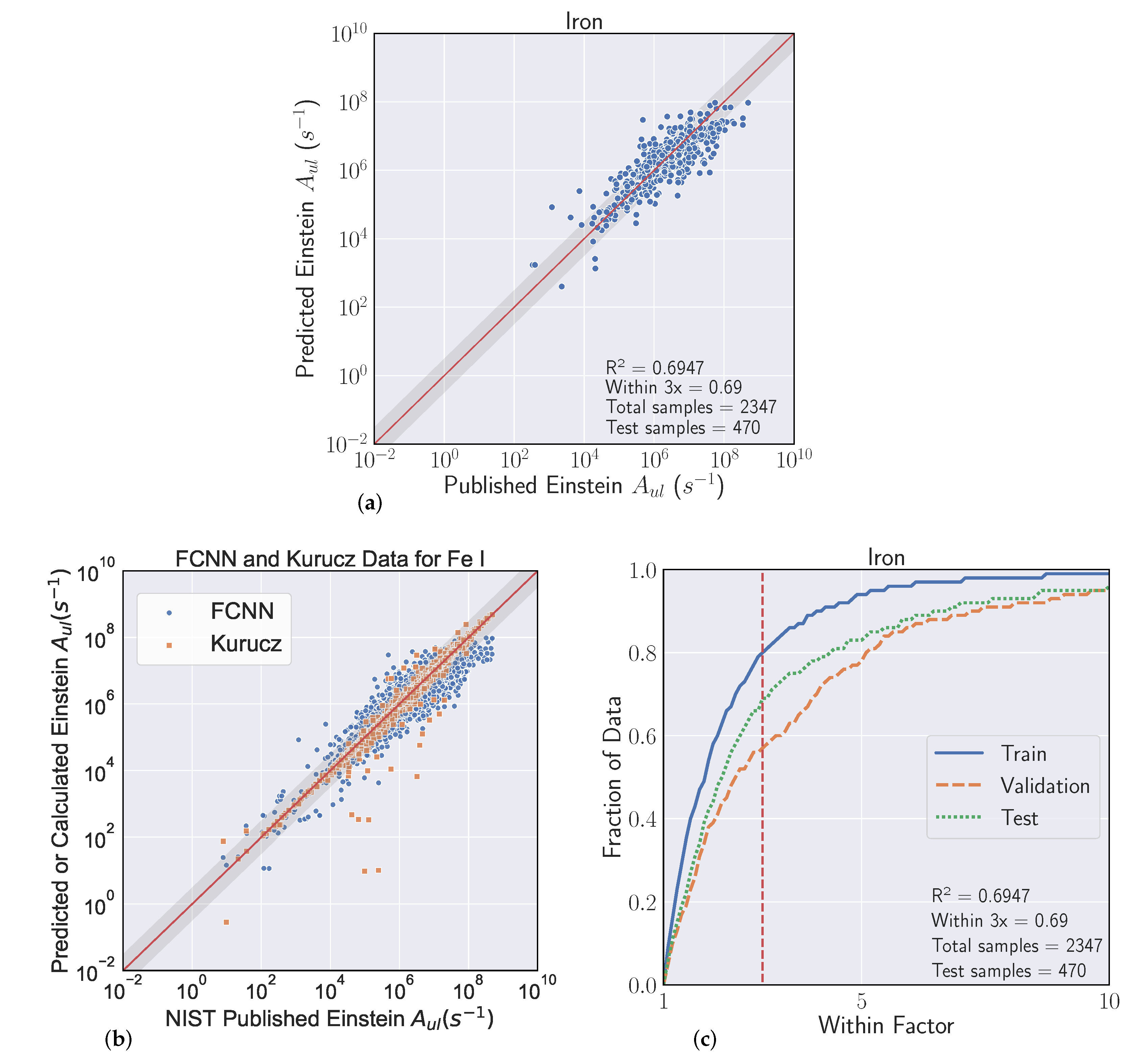
3.2. Metrics
Our goal in these experiments is to find a regressive function
which achieves the best fit to a held out validation set which generalizes to unforeseen data (the test set). We evaluated model fit with two separate scores. The first is to use the typical
metric for regression, which is the square of the Pearson correlation coefficient between predicted
and actual
A-coefficients (note that
). Even though we optimize our models to reduce Mean Squared Error (MSE) [
34], where
is a standard metric, the second score is more relevant to our particular task, which we refer to as the ’within-3x’ score. Within-3x, or
refers to the percentage of predicted transitions that fall within a factor of three of the predicted value and is defined by Equation (
1), where
is the indicator function and
is the indices of the data set. The within-3x score was used in previous work [
9] when comparing experimental data to the published values in the Kurucz Atomic Spectral Line Database [
10].
Interelement experiments are similarly constructed to intraelement models. The major difference being that the set of elements chosen for a particular dataset is greater than one . In this experimental setup, we experience the case where the length of features differs as a function of element . To mitigate, we constrain all data to include the largest common subset of features between all elements of E.
3.3. Model Selection
Our model search spanned the typical set of supervised regression methods found in most machine learning textbooks [
34,
37]. Namely, linear methods for regression such as least squares, ridge regression, and lasso regression, tree methods such as random forests, support vector machines, and nonlinear fully connected neural networks (FCNN). During an initial method selection phase, we evaluated these separate methods on a small set of intraelement datasets. Our model evaluation showed that FCNNs with rectified linear unit activation functions consistently outperformed the other models in
and
regardless of feature engineering for nearly every element tested. After this initial candidate model phase, we used extensive hyperparameter optimization over the FCNN architectures, permuting the number of neurons, layers, epochs, batch sizes, optimizers, and dropout. We randomly sample 1000 model configurations for each intra- and interelement model and optimize each FCNN to minimize a MSE loss function with respect to the training set using gradient descent based methods. Each model is evaluated against the validation set using MSE and the lowest error model is selected as our optimal model. Most of the selected model architectures are 3–5 layers deep with 50 hidden units in each layer. Because the models architectures are relatively small (low memory usage), fast to train (less than 2 min), and are constrained to FCNNs, we argue that there is room to improve model performance with additional architecture complexity.
5. Conclusions
As machine learning becomes more prevalent in physical science, it is critical that communities investigate which problem types are amenable to its application and the accuracy machine learning tools offer in these problem spaces. In this investigation, we tested the feasibility of using machine learning and ultimately FCNNs to predict fundamental spectroscopic constants based on the electronic structure of atoms, particularly prediction of transition probabilities. In contrast to analyzing raw spectral data with machine learning, our approach implemented neural networks to predict broadly applicable spectral constants which inform forward models removing the temperature and instrument dependence of spectral information that may otherwise limit the scope of machine learning analysis.
Our results show that NNs are capable of predicting atomic transition probabilities and learning from the feature set of novel electronic orbital encodings we developed. The absolute accuracy of the predicted transition probabilities is typically observed to be lower than can be calculated with modern theoretical methods or experiments for elements with lower atomic numbers (see
Section 4.1). However, the value proposition of increased speed (a few minutes for training and seconds for inference) and reduced resources to acquire transition probability values via neural network prediction is appealing for many applications based on the accuracy that was achieved.
Overall, our experiments showed that S-Block elements are typically higher performing than higher periods of the periodic table. Intuitively, elements that have a small number of atomic transitions perform worse than elements with a larger amount of data. Though this poor performance can typically be augmented with data from other elements to improve overall performance. Additionally, model performance is heavily dependent upon feature representation of each atomic transition and we see model performance gains to be had in this space. For example, our
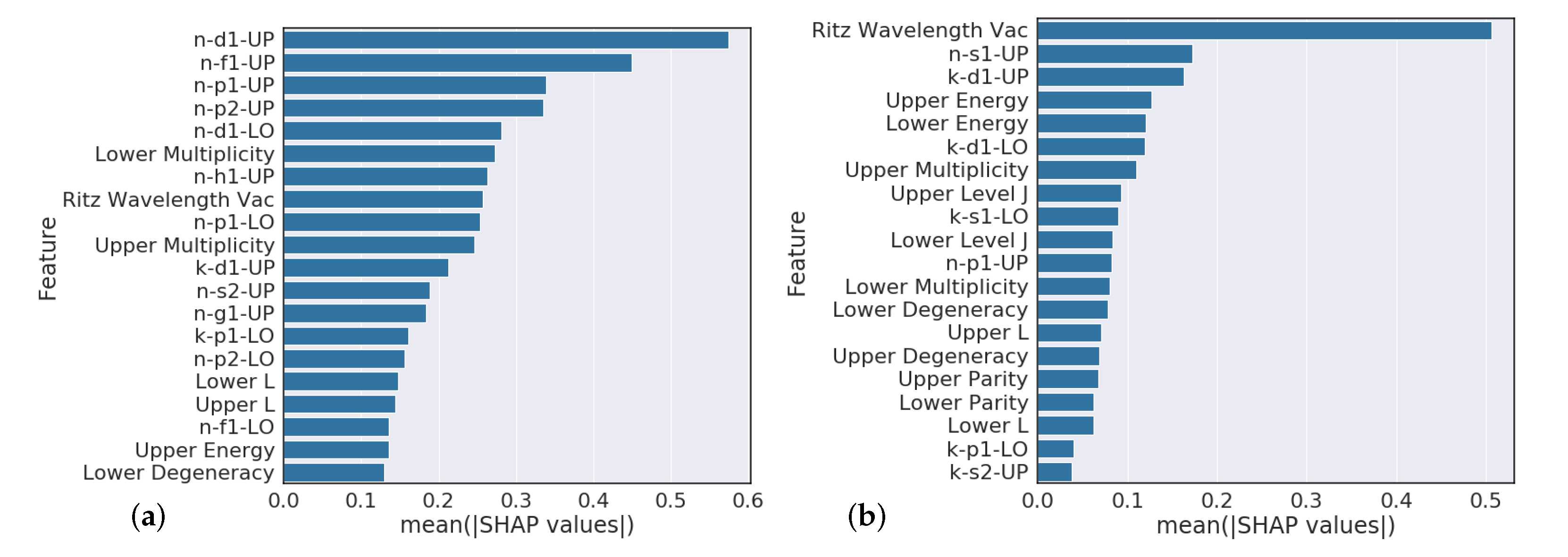
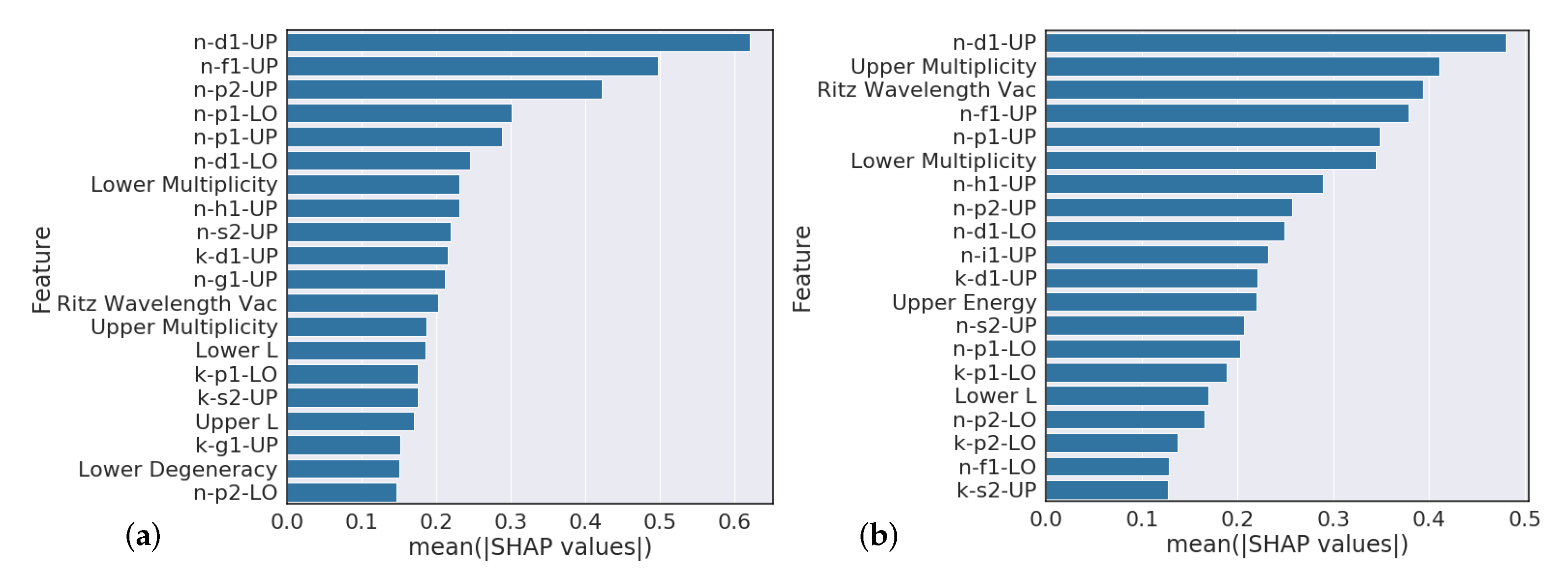
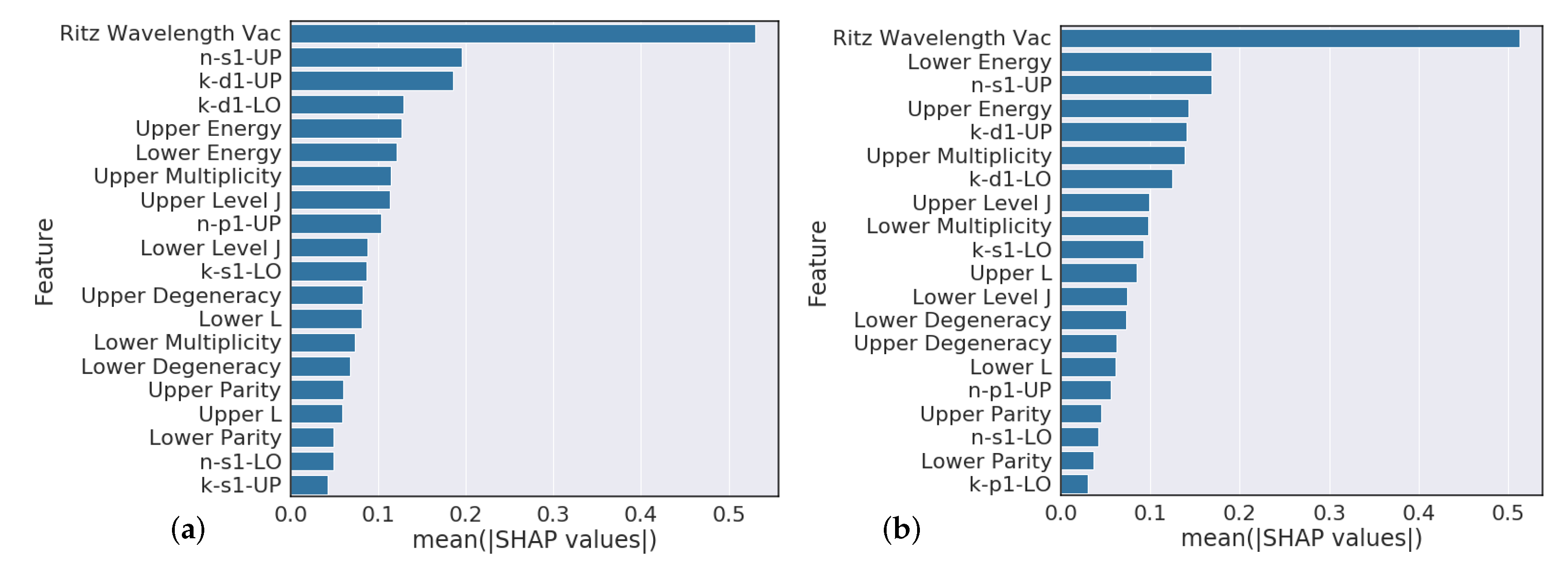
Section 4.3 analysis suggests that model predictions that heavily rely on the Ritz wavelength are indicators of poor performance while models that heavily rely on orbital information are typically higher performing. Further feature engineering to reflect this finding could give modest performance gains.
Significant potential in this technique still remains if the accuracy and dynamic range of neural network predictions are improved on in the future. This technique offers orders of magnitude speed up compared to traditional methods. The technique would allow not only new spectra to be explored, but it would also enable quality checking of previously reported values and allow modeling of transitions that are known, but have not been measured yet. As theoretical transition probability calculations and experimental accuracy are improved over time, the inputs to the machine learning models will also improve, thereby potentially enhancing the value of the NN approach for non-measured or non-calculated transitions.
Our future efforts in this area will focus on improving optimization of neural network models, determining the minimum and most important subsets of features required for accurate predictions, and attempting to extend this technique to higher Z elements on the periodic table as well as ions. Additionally, it is of interest to determine if training on specific periodic table trends (e.g., only transition metals) increases the accuracy for elements in that trend.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













