1. Introduction
In 1996 Oleg Zatsarinny published a program for computing matrix elements in a non-orthogonal radial basis in which configuration state functions (CSFs) were expanded in determinants [
1]. A non-orthogonal basis can greatly reduce the size of multiconfiguration wave function expansions. The orthogonal versus non-orthogonal issue is a complex one, so I invited Oleg for a three-month visit, with the National Science Foundation support for members of the former Soviet Union. Before he left, I shared with him the B-spline library I had developed for calculations of high accuracy “good to the last bit”. Oleg improved and extended the library, which became the basis for the BSR code he published in 2006 [
2] for both non-relativistic and the Breit–Pauli R-matrix theory. It was not until 2011 [
3] that I published a non-relativistic B-spline Hartree-Fock program for bound states using the extended library. In the mean time, Oleg extended BSR to Dirac relativistic theory where the R-matrix approach relies on eigenvectors of a B-spline matrix at the boundary. However with the traditional Galerkin method applied to the pair of first-order differential equations, spurious solutions appeared for the R-matrix.
The presence of spurious solutions from the application of the Galerkin methods is well known—a Google search for “Galerkin spurious” has 165,000 responses from areas of theoretical physics, applied mathematics, and engineering. Inspired by the work of Igarashi [
4,
5], I had the idea that the problem was related to the fact that Dirac equations are a system of first-order equations. I showed that the problem also occurs for
, with boundary conditions
and
, when the single second-order equation is replaced by a pair of first-order equations. Igarashi was focusing mostly on kinetically-balanced basis sets but tried also some other ideas like using B-splines of a different order. If we think of the Galerkin method approximating the small component by say,
, where
is a B-spline of order
(or piecewise polynomial of degree
), then the large component is proportional to
, namely piecewise polynomials of degree
with a B-spline basis of order
. Representing these functions by a higher-order polynomial presents difficulties that would not arise if a lower order of splines were used. In my test case, the eigenvalues from using different orders for pairs of equations were the same as those from a single second-order equation, indicating that the problem was not just a Dirac equation-related problem. Splines of different orders worked beautifully for the R-matrix approach as well as the Thomas–Reiche–Kuhn sum rule and all our other tests. I convinced Oleg that we should prepare a paper for Physical Review Letters, but the referee was not as excited by this result as I was. Oleg was not prepared to argue so the paper was published in Computer Physics Communication [
6].
The present paper is dedicated to the memory of Oleg Zatsarinny who extended the bound state B-spline codes to the fully relativistic
dbsr-hf version [
7].
2. In the Beginning ...
In the early days of computing, atomic and molecular calculations relied either on finite-difference methods based on numerical procedures developed by Hartree [
8], or analytic methods based on basis sets such as Slater-type orbitals [
9]. Bachau et al. [
10] in their excellent review on B-splines, refer to these methods as
local versus
global methods, respectively, in that finite difference methods relied on only a few values at adjacent points of a grid in specifying an approximation. Much changed when de Boor [
11] published his book in 1985 about B-splines with a
local but complete basis for piece-wise polynomial approximations along with Fortran programs for standard spline procedures. Among the first to use his codes were Johnson and Sapirstein [
12], who used B-splines to generate their numerical basis set for perturbation theory that greatly advanced the theory. However B-splines also offer advantages for a wide range of applications as Bachau et al., show in their extensive review.
By the 1990s, the non-relativistic Hartree–Fock variational method had evolved into a multiconfiguration theory as implemented in the
mchf77 [
13] program. Later this theory was extended to include relativistic corrections through a Breit–Pauli approximation and, together with programs for atomic properties (hyperfine, isotope shifts, and transition probabilities), became an atomic structure package
atsp2K [
14]. All calculations were based on numerical procedures and supported a limited amount of non-orthogonality of the radial basis [
15] of one or at most two overlap integrals.
With the development of faster computers along with considerable more memory, an interesting question arose—what is the best method for solving the mchf equations? The latter required node counting to control the convergence of an integro-differential equation with many solutions, dealing also with Lagrange multipliers for satisfying orthonormality.
This paper reviews the development of spline methods for variational approaches to solutions of the wave equation for atoms with the assumption that the reader is familiar with the basic properties of splines as presented by Bachau et al. [
10]. Unlike the latter, the emphasis here is more on the computational methods and the programs that have emerged rather than their application.
3. The B-Spline Basics
A spline approximation of order is an approximation that is a piecewise polynomial of degree (with coefficients) in intervals defined by “knots” that define a grid. In applications, the grid itself, in a sense, is arbitrary but the choice of grid can have a large influence on the accuracy of an approximation.
3.1. Spline Grid for Radial Functions
In atomic structure applications, it is convenient to define the grid in terms of the variable
(the hydrogenic case) and then transform to the current value of Z. Let
. This is not strictly necessary, but it ensures that the arithmetic is exact in binary arithmetic. Then:
Thus the grid is linear for m intervals near the origin after the knots of multiplicity , then linear in . If continuum calculations are involved, the range may be extended to include equally-spaced values at the end of the exponential range, followed by final knots of of multiplicity . The above grid is most appropriate for a finite nucleus. The number of non-zero intervals between knots is where is the number of basis states. Note also that, the end of the exponential region, defines the maximum range R of the bound-state orbitals, that are assumed to be zero for .
In an early B-spline study, Froese Fischer and Parpia [
16] studied the accuracy of the application of B-splines to the Dirac equation for He with a grid over the range
, comparing a grid for
with a grid for
. The former was considerably more accurate but is rarely used other than for plotting since
transforms to
in the first transformation, whereas in the latter it transforms to
. However, an advantage of splines is that points need not be equally spaced so
can be chosen as a grid-point (as in Equation (
1)) even when the remaining grid-points are on an exponential grid.
3.2. Integration Methods
The piecewise polynomial values are represented by
values at the Gaussian points for Gauss–Legendre integration. Then,
where
are the Gaussian weights at the Gaussian points
. This formula is exact for integrands
that are polynomials of degree
.
3.3. Slater Integrals
A detailed analysis of algorithms for the calculation of Slater integrals was reported by Qiu and Froese Fischer [
17], and will be summarized here.
Let
denote a set of
quantum numbers associated with a radial function:
with similar expansions for
and
d. Then,
where,
will be referred to as B-spline Slater integrals. Many symmetries exist. Furthermore,
Symmetry and the “local” property of these integrals, greatly reduce the number of B-spline Slater integrals included in the summation.
The spline-approximation divides the integration range into sub-intervals and the two-dimensional integration into patches or “cells”. Thus the fundamental process is one of integration over a cell,
and
, namely,
However for an off-diagonal cell, the above two-dimensional integral is separable and equal to (for
):
where
and
are referred to as moments.
The diagonal cells reduce to integrations over upper and lower triangles or, through an interchange of arguments, as:
and
Thus the computational complexity of an integration over a diagonal cell is
and for the
array, it is
. Furthermore, when all B-splines are defined on an exponential grid where
, (as in the middle region of the logarithmic grid) scaling laws can be applied, namely:
With such a grid, some Slater integrals would be computed using the basic integration procedures and others could be scaled.
4. Tensor Products of B-Splines as a Basis
Traditionally, the wave functions for multi-electron systems are expanded in terms of configuration state functions (CSFs) that are products of one-electron wave functions (or orbitals) where, for bound-state problems, the latter provides an orthonormal basis. An early spline study of the
,
or
, and
or
states of Helium [
18], took a different approach and explored the direct use of a tensor product of B-splines as a non-orthogonal basis.
For a two-electron system, a wave function for a state
, where
denotes the configuration, can be expressed in terms of pair functions, identified by their angular symmetry, namely:
Here,
where
is a normalisation factor that may depend on symmetry,
is a permutation operator,
a is spin-angular factor that identifies the pair function, and
and
are radial functions. In other words, the pair function is a linear combination of configuration state functions of the same symmetry. Substituting into Equation (
16) and noting that each term has the same spin-angular factor, that the double sum is an expansion in a basis of a function of two variables, we get a pair function
, such that:
In essence, is a general two-dimensional function, usually expanded in terms of orbitals but one that could also be expanded in terms of tensor products of B-splines, namely . The advantage of this basis is the local support (non-zero region) compared with products of orbitals which extend over the entire two-dimensional region.
In the case of
state, the possible orbital symmetries (
) can be represented as (
) where
, with
. Then the
state can be expressed in terms of partial waves as:
where,
The application of the Galerkin method leads to a system linear equations, where the equations for a specific basis, identified by the parameters
,
and
, are:
In the above,
and
are matrices with:
In the B-spline basis, the one-electron operators ( and S matrices) connect the coefficients within a pair function ( for a given l) whereas the Slater integrals in the B-spline basis, connect the different pair functions. The coefficients of the Slater integrals arise from angular integrals related to the Slater integrals. Both can be treated as data for the calculation and depend only on the underlying grid. The equations themselves were solved iteratively with the energy E determined as a Rayleigh quotient. The solution is numerically intensive and has many options for parallelism.
Of interest in 1991 was the performance of the algorithm on parallel vector processors. An accuracy of a micro-Hartree was achieved for the energy.
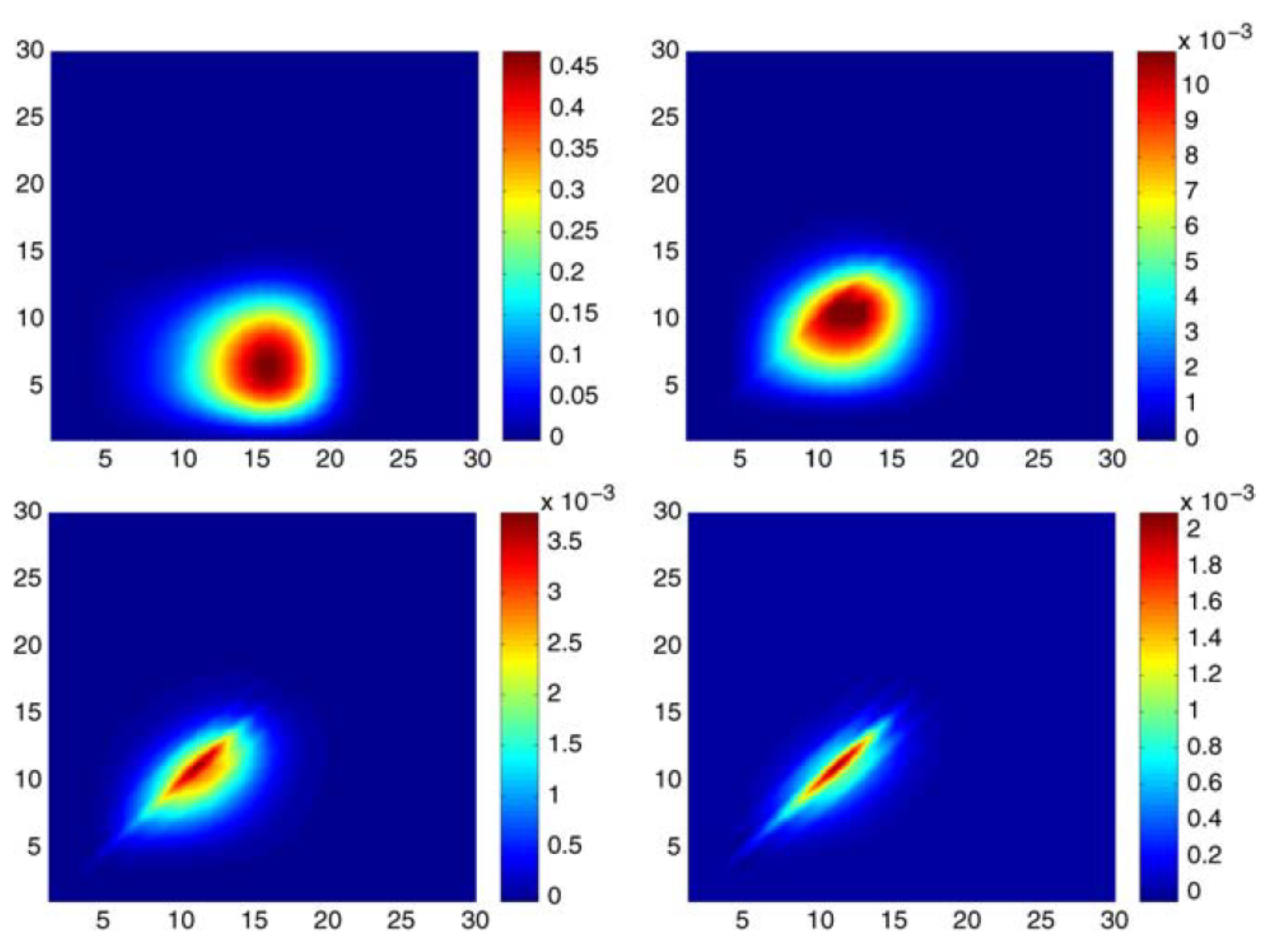
Figure 1 shows the matrix of coefficients
for B-spline tensor-product expansions for
(or
) pair functions from left to right and top to bottom, respectively. Notice that each subplot has a different scale with the maximum value decreasing. For
symmetry, the matrix is approximately the cross-product of the expansion coefficients for the
and
Hartree–Fock orbitals in a B-spline basis. As
l increases, the maximum coefficient (dark red in colour) moves closer to the diagonal
region. However more importantly, the significant components are concentrated in a smaller and smaller region. What this calculation clearly shows is that correlation is a “local” correction and that, as the orbital symmetry
l increases, an oscillating orthonormal basis would not be an efficient basis.
However the example is also interesting from other perspectives. Like the Hylleraas method, there are no orbitals, nor are there orthonormality constraints and hence, no Lagrange multipliers. The wave function is a sum of pair functions rather than a linear combination of configuration state functions, and there is no matrix diagonalisation. In present
grasp [
20] or
atsp calculations, the usual first step is to compute all angular data, which then needs to be read and stored in memory when needed. With pair functions the amount of angular data is greatly reduced and often could be computed as needed. It should also be remembered that orbitals are a theoretical concept.
5. Spline Galerkin and Inverse Iteration Methods
The development of B-splines (of an order greater than that of cubic splines) began before the LAPACK [
21] routines were released. Available instead, were LINPACK [
22] routines for solving systems of equations. Some early papers by Froese Fischer and Idrees [
23,
24], described a spline algorithm for solving the continuum functions that used the spline Galerkin method for deriving linear equations of the form:
and inverse iteration for solving the equation for a given energy. The latter was similar to the power method for finding the eigenvector associated with the largest eigenvalue. For continuum solutions, the energy
E is specified and what is needed is the eigenvector of the nearest (or smallest) eigenvalue to
E. Like the power method, the method is iterative.
Let the matrix
A be an
matrix and
L and
U lower and upper triangular matrices, respectively, of similar dimension, such that
. and
,
is a vector. Then, starting with
and incrementing by 1, until the vector
c has converged (i.e.,
), let:
where
x and
y are vectors of length
N. The method was tested for the hydrogen scattering problem and then photoionisation in He. It was also determined how orthogonality conditions could be included by extending the definition of
and the vector
c for the case,
as an example. Resonances, phase shifts, or photoionisation cross-sections were extracted from the results. A more extensive investigation of resonance positions and widths for H
and He was reported by Brage et al. [
25,
26], adding to the rich ‘flora’ of results by many different methods for these cases. Xi and Froese Fischer extended these results to three-electron He
system in the investigation of cross-section and angular-distribution for the photodetachment of He
below the He (
) threshold [
27] and also below the
detachment level [
28]. A multichannel theory was developed. Among the resonances found was a
resonance state immediately below the
threshold. A similar theory was applied to Be
[
29].
The above methods had only one region. These simple approaches were extended by Oleg Zatsarinny to numerous continuum processes based on the R-matrix method [
2,
30] with its inner and outer regions and non-orthogonal orbitals. He referred to one region methods as “straight forward” methods.
6. Spline Methods for Bound State Problems
Several options are available for B-spline solutions that were not feasible for finite difference methods. Consider the simple equation for
that was studied by Froese Fischer and Guo [
31]. The differential equation that needs to be solved is:
where
when
or
and the radial function
is expanded in a B-spline basis so that:
This problem can be linearised by computing
from current estimates, and then solved as a generalised eigenvalue problem for an improved estimate, with at best a linear rate of convergence. Or, we can think of the equations as non-linear equations, and solve for changes in the expansion coefficients and energy parameters, using the Newton–Raphson (NR) method with a quadratic rate of convergence (see Ref. [
32] for details).
The many-electron variational methods are extensions of the Hartree–Fock methods [
3,
19] for which three categories of methods have been implemented and evaluated.
6.1. Generalised Eigenvalue Problem for a Single Orbital
In this approach, orbitals are improved one at a time according to a generalized eigenvalue problem,
When two orbitals are constrained through orthogonality, as in the case of
, then the iterative process will not converge without first rotating the two orbitals, say
a and
b for a stationary energy, a process implemented in the
sphf program [
3]. Projection operators may then be applied to eliminate the off-diagonal Lagrange multiplier
The matrix
is then a full matrix when exchange contributions are present.
6.2. Multiple Orbitals and SVD
Another possibility is to solve for a set of orbitals at the same time using singular value decomposition (SVD) which is closely related to inverse iteration and is included in LAPACK [
21].
Let
be the expansion vector for orbital
i. Then the system of equations can be written as:
where
contains the contributions from one-electron integrals, the direct Slater integrals for orbital
i, as well as
, and
(
) contains the contribution from exchange integrals between orbitals
i and
j and possible orthogonality constraints. In this case, all matrices are banded. In this approach, the energy parameters are computed from current estimates of the orbitals.
The earlier SVD study [
19] showed poor convergence in the case of
but
converged linearly. For the latter, the off-diagonal energy parameter is zero and rotation of orbitals is not important. Applying a projection operator to a system of equations is equivalent to using current estimates of a radial function to determine off-diagonal energy parameters. It is possible that SVD equations should be extended to include the energy parameters (diagonal
) as well as off-diagonal (
) unknowns so that an effective rotation of orbitals is part of the SVD solution.
6.3. Newton–Raphson with Quadratic Rate of Convergence
Consider the case of two orbitals
a and
b, with an orthogonality constraint between them. Then the unknowns are
. The equations to be solved are the two orbital equations along with the three orthonormality conditions that are part of the energy functional. Let
be the current estimates that are used to evaluate the
-matrix. If symmetry conditions are not satisfied exactly, we can use the average value,
Then, by the Newton–Raphson method [
3].
and
are solutions of:
In the above, , namely the amount by which the current estimates do not satisfy the equations.
7. The sphf and spmchf Programs
The B-spline methods clearly offer some advantages, even when they are more computationally intensive than finite difference methods. The eigenvalue method is the preferred method for singly occupied orbitals, particularly with large principal quantum numbers. However for a multiply occupied shell, the Newton–Raphson method is efficient in that the “self-energy” is readily accounted for which greatly improves the rate of convergence. When many subshells are present with different principal quantum numbers, convergence can be improved with the simultaneous improvement of orbitals. For this reason, both
sphf [
3] and
spmchf (available at GitHub [
33]) have “phases” and different “levels” of accuracy. All iterative methods need initial estimates. The first phase is stable even when estimates are of a poor accuracy and the grid that defines the B-spline basis is relatively coarse. Orbitals are updated sequentially using the generalised eigenvalue method on the first iteration but, in other iterations use NR for a single orbital when a subshell is multiply occupied. This first phase is referred to as the “SCF” phase. The codes also have the initial level of accuracy (with a coarse grid) and less accurate tests for convergence and a higher level of accuracy. When initial estimates are for the refined grid, the program assumes there is only one level of accuracy. In any event, the convergence tests may be reset by the user, over-riding program defaults. Unlike the
sphf program, the
spmchf program does not include orbital rotation in this phase. The next phase is thought of as a “clean-up” phase that raises the level of accuracy and improves the relationship of one subshell to the other. In the Be
case, if the
orbital is too contracted the
subshell will be too expanded. The NR phase deals with this issue that greatly affects the Virial Theorem (V/T) ratio, as well as orbital rotation when off-diagonal energy parameters are important.
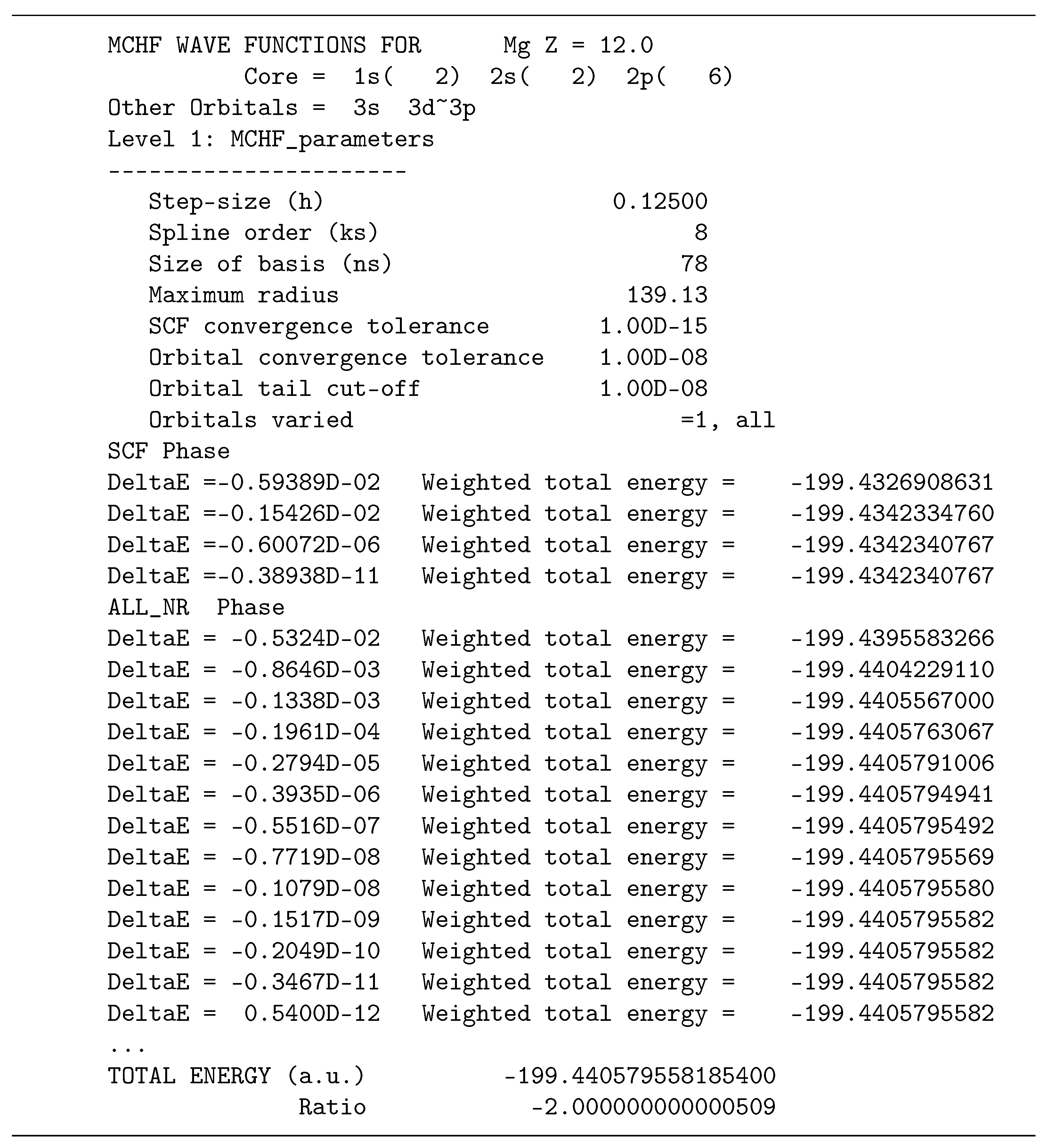
Figure 2 shows the parameters for two levels in the calculation for Be
. The default grids and convergence parameters are displayed for both accuracy levels. No initial estimates were provided in this case so the initial range was estimated to be fairly large. At the second level of accuracy, the range is known much more precisely. Notice the improved VT in going from the first to the second level of accuracy. The
case is very similar except now off-diagonal Lagrange multipliers are needed for a stationary energy with respect to rotation and the requirement that
.
Table 1 shows how during the SCF iterations, the values are opposite in sign and the calculations do not converge, but everything changes during the NR iterations and iterations converge rapidly. The default options of
dbsr-hf do not include orbital rotations. For the
case, it converges but to an incorrect value, made evident only through the Virial theorem (VT). Thus VT is an important check for this code.
The
spmchf method differs not only in that expansion coefficients for configuration states (CSFs) need to be determined but also in that the the energy expression is no longer limited to direct (
) and exchange (
) Slater integrals. The process by which the energy expression relates to the matrix form
requires that two occurrences of the orbital
a be present in the matrix element [
19]. In the MCHF approximation:
the interaction matrix element is
. The
spmchf treats the contribution to Equation (
27) for
or
as a “residual” term (not included in the matrix), but Zatsarinny [
34] pointed out that if the matrix element was treated as:
then the matrix form could be retained. The factors,
and
are normalisation integrals. This modification has not yet been implemented in
spmchf.
Figure 3 and
Figure 4 show the
spmchf solution for this example. Neither
sphf nor
spmchf require initial estimates so the first calculation was for a Hartree Fock calculation for Mg
. The output was then used in the second multiconfiguration run as input to serve as initial estimates and the first level varied only the
orbital, not present in the input. Then the second varied all orbitals with excellent results as reported.
At this time, the spmchf program for multiconfiguration wave functions has not yet been been fully tested. Through the use of the term=jj command-line option, the fully relativistic dbsr-hf code can be used as an average energy level (EAL) approximation.
8. Concluding Remarks
The spline codes reviewed in this article were developed in the last 20 or so years. For bound state problems, the
GRASP code, for example [
20], based on finite difference methods and an orthonormal orbital basis, is extensively used for complete spectra including relatively highly excited levels [
35] and certain heavy elements, primarily for highly ionised systems where valence correlation can be dealt with and the effect of core correlation on spectra and other properties is limited.
spmchf with its greater flexibility still needs to be tested on lanthanides and actinides with two open shells (
for lanthanides and
for actinides) where natural orbital transformation could prove useful in connection with orthonormal radial functions [
36]. Multiple
f-orbitals could also be a challenge for
bsr in that the number of determinants can increase rapidly.
In 1984, it took 254 seconds to execute a numerical HF program for Ra on a VAX 11/780; in 1987, 2 seconds on a Cray X-MP; and in 1993, 1 s on a Dec Alpha, which was a popular computer at its time. Thus time (cost) is no longer an important factor but rather ease of use and reliability. The same is not true when accurate wave functions are needed for many-electron systems for different atomic properties. Important now is how well an algorithm can be made parallel, and how efficiently memory usage can be managed.
The present paper has been about non-relativistic calculations. Relativistic methods are similar, except that the decision to represent large and small components of radial functions by splines of different order, automatically implies that every Slater integral is the sum of four integrals. This decision was important in
bsr [
2] in that it eliminated spurious solutions in the R-matrix and was also used for
dbsr-hf. However, is it the most efficient solution of the problem? For a grid of
intervals, the number of independent basis functions is
. Thus, by going from
to
, the number of independent basis states decreases. The spurious solutions generally are higher in the spectrum. For bound state solutions, the asymptotic conditions are such that both the large and small components and their derivatives go to zero. In fact, in non-relativistic calculations, applying both conditions stabilises the solution at large
r. Further study might be appropriate.








{kind=link}
{kind=link}
{kind=link}
{kind=link}