Drosophila as a Model for Assessing the Function of RNA-Binding Proteins during Neurogenesis and Neurological Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. RBP Dysfunction Underlies Myriad Neurological Disorders
2. Drosophila Models of Neurological Disease
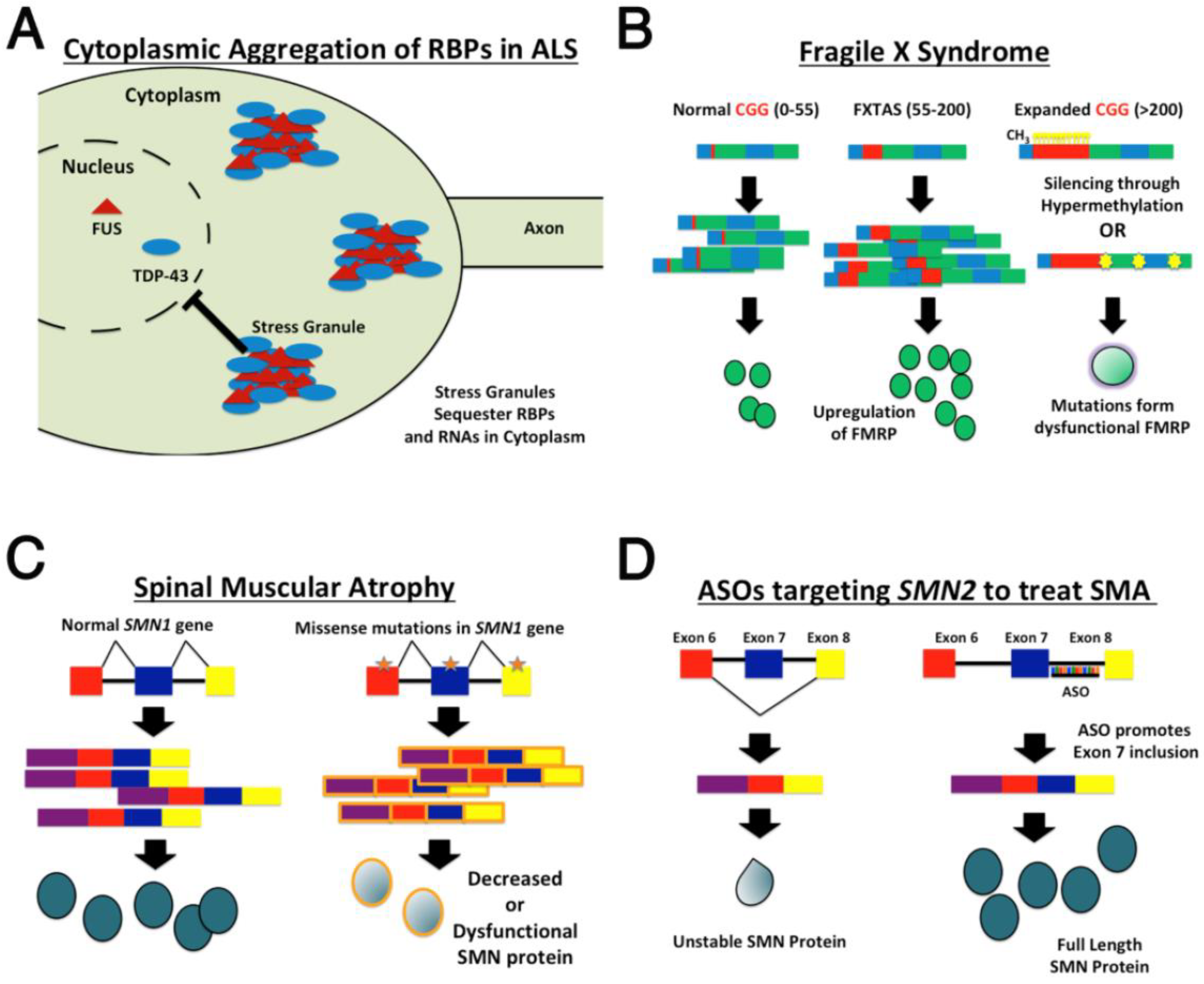
2.1. Drosophila Models of Amyotrophic Lateral Sclerosis (ALS)
2.2. Modeling TAR DNA Binding Protein 43 (TDP-43) Mutations in Drosophila
2.3. Modeling Fused in Sarcoma (FUS) Mutations in Drosophila
2.4. Modeling C9orf72 Aberrations in Drosophila
2.5. Modeling Sporadic ALS in Drosophila
3. Modeling Fragile X Syndrome in Drosophila
3.1. Genetic Models of FXS
3.2. Assessing Drug Efficacy in Treating Specific FXS Phenotypes
3.3. FMRP Function in Neuronal Remodeling
3.4. Modeling Fragile X-Associated Tremor Ataxia Syndrome FXTAS
4. Spinal Muscular Atrophy
4.1. Drosophila Models of Spinal Muscular Atrophy
4.2. Determining the Efficacy of SMA Therapeutics Using Model Organisms
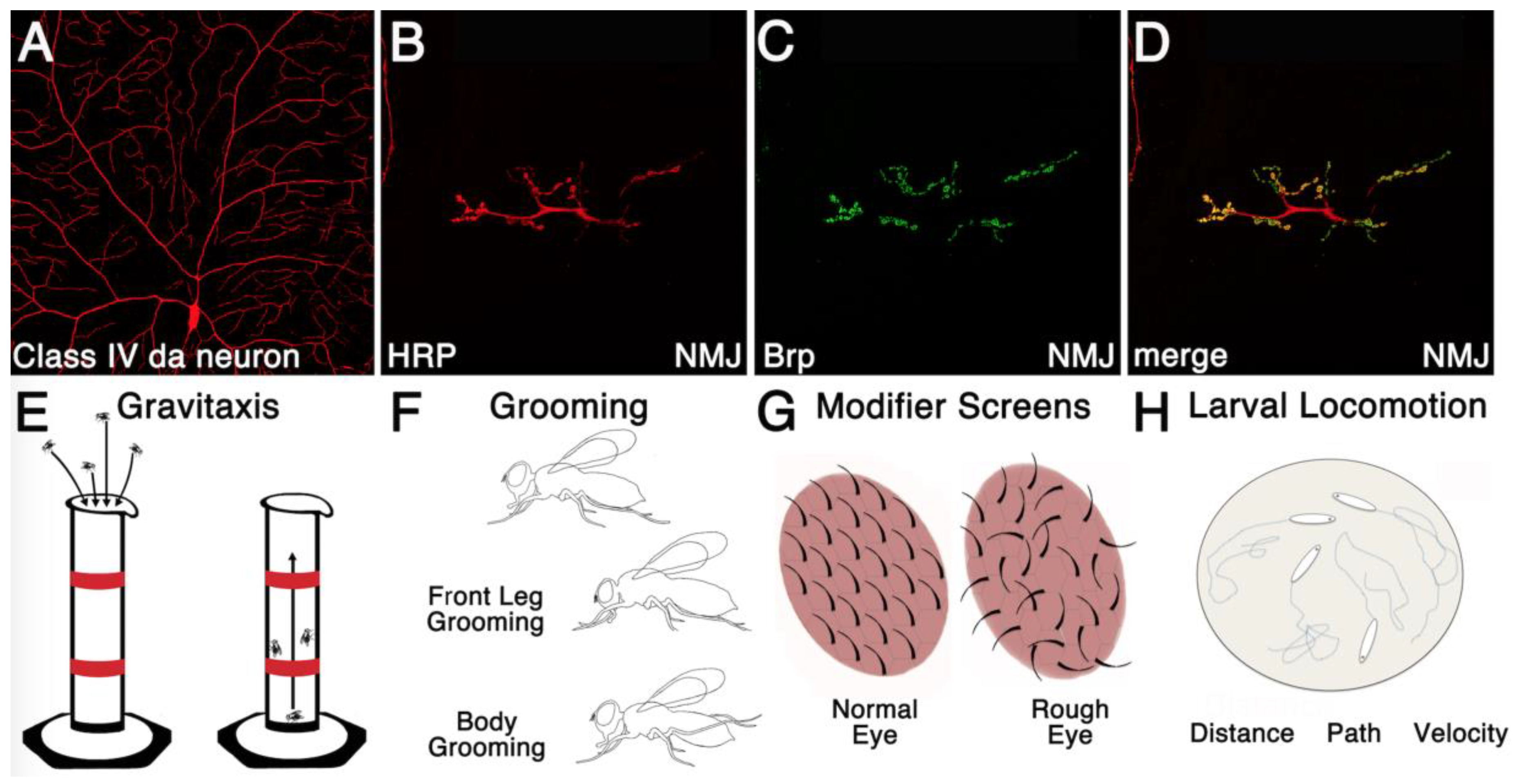
5. Additional Roles for RBPs throughout Drosophila Neurogenesis
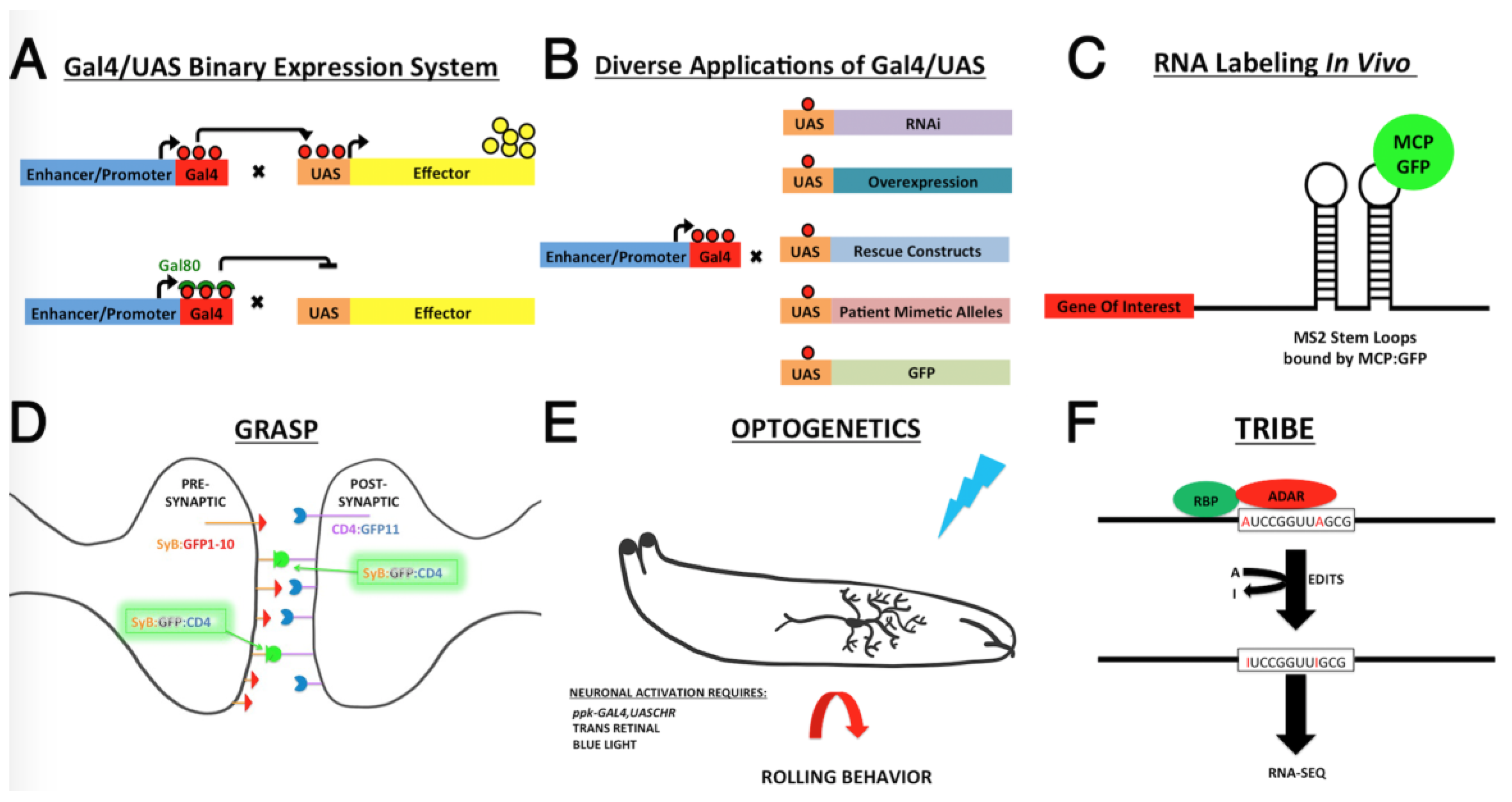
6. A Unique Toolkit for Investigating RBP Function within Neurons
6.1. Precise Spatiotemporal Manipulation of RBP Function
6.2. RNA Editing Tools to Identify Tissue Specific RBP Target RNAs
6.3. Emerging Tools for Mapping Neural Circuitry
7. Outstanding Questions and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Anthony, K.; Gallo, J.M. Aberrant RNA processing events in neurological disorders. Brain Res. 2010, 1338, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Doxakis, E. RNA binding proteins: A common denominator of neuronal function and dysfunction. Neurosci. Bull. 2014, 30, 610–626. [Google Scholar] [CrossRef] [PubMed]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta 2009, 1792, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmens, R.; Moore, M.J.; Al-Chalabi, A.; Brown, R.H.; Robberecht, W. RNA metabolism and the pathogenesis of motor neuron diseases. Trends Neurosci. 2010, 33, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Darnell, R.B. Splicing regulation in neurologic disease. Neuron 2006, 52, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.D.; Janitz, M. Alternative splicing of mRNA in the molecular pathology of neurodegenerative diseases. Neurobiol. Aging 2012, 33, 1012.e11–1012.e24. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J. The evidence for altered RNA metabolism in amyotrophic lateral sclerosis (ALS). J. Neurol. Sci. 2010, 288, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chai, A.; Pennetta, G. Insights into ALS pathomechanisms: from flies to humans. Fly (Austin) 2015, 9, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casci, I.; Pandey, U.B. A fruitful endeavor: Modeling ALS in the fruit fly. Brain Res. 2015, 1607, 47–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero, E.N.; Wang, H.; Mitra, J.; Hegde, P.M.; Stowell, S.E.; Liachko, N.F.; Kraemer, B.C.; Garruto, R.M.; Rao, K.S.; Hegde, M.L. TDP-43/FUS in motor neuron disease: Complexity and challenges. Prog. Neurobiol. 2016, 145–146, 78–97. [Google Scholar] [CrossRef] [PubMed]
- Vanden Broeck, L.; Kleinberger, G.; Chapuis, J.; Gistelinck, M.; Amouyel, P.; Van Broeckhoven, C.; Lambert, J.C.; Callaerts, P.; Dermaut, B. Functional complementation in Drosophila to predict the pathogenicity of TARDBP variants: evidence for a loss-of-function mechanism. Neurobiol. Aging 2015, 36, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Miguel, L.; Frébourg, T.; Campion, D.; Lecourtois, M. Both cytoplasmic and nuclear accumulations of the protein are neurotoxic in Drosophila models of TDP-43 proteinopathies. Neurobiol. Dis. 2011, 41, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Avendaño-Vázquez, S.E.; Dhir, A.; Bembich, S.; Buratti, E.; Proudfoot, N.; Baralle, F.E. Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 2012, 26, 1679–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Pons, M.; Miguel, L.; Miel, C.; Avequin, T.; Juge, F.; Frebourg, T.; Campion, D.; Lecourtois, M. Splicing factors act as genetic modulators of TDP-43 production in a new autoregulatory TDP-43 Drosophila model. Hum. Mol. Genet. 2017, 26, 3396–3408. [Google Scholar] [CrossRef] [PubMed]
- Coyne, A.N.; Siddegowda, B.B.; Estes, P.S.; Johannesmeyer, J.; Kovalik, T.; Daniel, S.G.; Pearson, A.; Bowser, R.; Zarnescu, D.C. Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 15962–15974. [Google Scholar] [CrossRef] [PubMed]
- Coyne, A.N.; Lorenzini, I.; Chou, C.C.; Torvund, M.; Rogers, R.S.; Starr, A.; Zaepfel, B.L.; Levy, J.; Johannesmeyer, J.; Schwartz, J.C.; et al. Post-transcriptional Inhibition of Hsc70-4/HSPA8 Expression Leads to Synaptic Vesicle Cycling Defects in Multiple Models of ALS. Cell Rep. 2017, 21, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Sartoris, A.; Nativio, R.; Van Deerlin, V.; Toledo, J.B.; Porta, S.; Liu, S.; Chung, C.Y.; Garcia, B.A.; Lee, V.M.; et al. TDP-43 Promotes Neurodegeneration by Impairing Chromatin Remodeling. Curr. Biol. 2017, 27, 3579–3590. [Google Scholar] [CrossRef] [PubMed]
- Lo Piccolo, L.; Bonaccorso, R.; Attardi, A.; Li Greci, L.; Romano, G.; Sollazzo, M.; Giurato, G.; Ingrassia, A.M.R.; Feiguin, F.; Corona, D.F.V.; et al. Loss of ISWI Function in Drosophila Nuclear Bodies Drives Cytoplasmic Redistribution of Drosophila TDP-43. Int. J. Mol. Sci. 2018, 19, 1082. [Google Scholar] [CrossRef] [PubMed]
- Sasayama, H.; Shimamura, M.; Tokuda, T.; Azuma, Y.; Yoshida, T.; Mizuno, T.; Nakagawa, M.; Fujikake, N.; Nagai, Y.; Yamaguchi, M. Knockdown of the Drosophila fused in sarcoma (FUS) homologue causes deficient locomotive behavior and shortening of motoneuron terminal branches. PLoS ONE 2012, 7, e39483. [Google Scholar] [CrossRef] [PubMed]
- Machamer, J.B.; Woolums, B.M.; Fuller, G.G.; Lloyd, T.E. FUS causes synaptic hyperexcitability in Drosophila dendritic arborization neurons. Brain Res. 2018, 1693, 55–56. [Google Scholar] [CrossRef] [PubMed]
- Kanai, K.; Kuwabara, S.; Misawa, S.; Tamura, N.; Ogawara, K.; Nakata, M.; Sawai, S.; Hattori, T.; Bostock, H. Altered axonal excitability properties in amyotrophic lateral sclerosis: impaired potassium channel function related to disease stage. Brain 2006, 129, 953–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celona, B.; Dollen, J.V.; Vatsavayai, S.C.; Kashima, R.; Johnson, J.R.; Tang, A.A.; Hata, A.; Miller, B.L.; Huang, E.J.; Krogan, N.J.; et al. Suppression of C9orf72RNA repeat-induced neurotoxicity by the ALS-associated RNA-binding protein Zfp106. Elife 2017, 6, e19023. [Google Scholar] [CrossRef] [PubMed]
- Hentati, A.; Ouahchi, K.; Pericak-Vance, M.A.; Nijhawan, D.; Ahmad, A.; Yang, Y.; Rimmler, J.; Hung, W.; Schlotter, B.; Ahmed, A.; et al. Severe muscle wasting and denervation in mice lacking the RNA-binding protein ZFP106. Neurogenetics 1998, 2, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Cannavino, J.; Li, H.; Anderson, K.M.; Nelson, B.R.; McAnally, J.; Bezprozvannaya, S.; Liu, Y.; Lin, W.; Liu, N.; et al. Severe muscle wasting and denervation in mice lacking the RNA-binding protein ZFP106. Proc. Natl. Acad. Sci. USA 2016, 113, E4494–E4503. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, S.; Bogaert, E.; Michiels, E.; Gijselinck, I.; Sieben, A.; Jovičić, A.; De Baets, G.; Scheveneels, W.; Steyaert, J.; Cuijt, I.; et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 2016, 6, 20877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moens, T.G.; Mizielinska, S.; Niccoli, T.; Mitchell, J.S.; Thoeng, A.; Ridler, C.E.; Grönke, S.; Esser, J.; Heslegrave, A.; Zetterberg, H.; et al. Sense and antisense RNA are not toxic in Drosophila models of C9orf72-associated ALS/FTD. Acta Neuropathol. 2018, 135, 445–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.; Almeida, S.; Moore, J.; Gendron, T.F.; Chalasani, U.; Lu, Y.; Du, X.; Nickerson, J.A.; Petrucelli, L.; Weng, Z.; et al. Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron 2015, 87, 1207–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, J.G.; Bogdanik, L.; Muhammad, A.K.M.G.; Gendron, T.F.; Kim, K.J.; Austin, A.; Cady, J.; Liu, E.Y.; J Zarrow, J.; Grant, S.; et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 2015, 88, 892–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, O.M.; Cabrera, G.T.; Tran, H.; Gendron, T.F.; McKeon, J.E.; Metterville, J.; Weiss, A.; Wightman, N.; Salameh, J.; Kim, J.; et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 2015, 88, 902–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burguete, A.S.; Almeida, S.; Gao, F.B.; Kalb, R.; Akins, M.R.; Bonini, N.M. GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. Elife 2015, 4, e08881. [Google Scholar] [CrossRef] [PubMed]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.N.; Gochenaur, L.; Singh, A.; Grant, R.; Patel, K.; Watkins, S.; Wu, J.Y.; Pandey, U.B. Traumatic injury induces stress granule formation and enhances motor dysfunctions in ALS/FTD models. Hum. Mol. Genet. 2018, 27, 1366–1381. [Google Scholar] [CrossRef] [PubMed]
- Weisz, E.D.; Monyak, R.E.; Jongens, T.A. Deciphering discord: How Drosophila research has enhanced our understanding of the importance of FMRP in different spatial and temporal contexts. Exp. Neurol. 2015, 274, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Chang, C.W.; Kelly, S.M.; Bhattacharya, A.; McBride, S.M.; Danielson, S.W.; Jiang, M.Q.; Chan, C.B.; Ye, K.; Gibson, J.R.; et al. Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep. 2015, 11, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Okray, Z.; de Esch, C.E.; Van Esch, H.; Devriendt, K.; Claeys, A.; Yan, J.; Verbeeck, J.; Froyen, G.; Willemsen, R.; de Vrij, F.M.; et al. A novel fragile X syndrome mutation reveals a conserved role for the carboxy-terminus in FMRP localization and function. EMBO Mol. Med. 2015, 7, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Myrick, L.K.; Deng, P.Y.; Hashimoto, H.; Oh, Y.M.; Cho, Y.; Poidevin, M.J.; Suhl, J.A.; Visootsak, J.; Cavalli, V.; Jin, P.; et al. Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc. Natl. Acad. Sci. USA 2015, 112, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Tauber, J.M.; Vanlandingham, P.A.; Zhang, B. Elevated levels of the vesicular monoamine transporter and a novel repetitive behavior in the Drosophila model of fragile X syndrome. PLoS ONE 2011, 6, e27100. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Fragile X disappointments upset autism ambitions. Nat. Rev. Drug Discov. 2015, 14, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Kashima, R.; Redmond, P.L.; Ghatpande, P.; Roy, S.; Kornberg, T.B.; Hanke, T.; Knapp, S.; Lagna, G.; Hata, A. Hyperactive locomotion in a Drosophila model is a functional readout for the synaptic abnormalities underlying fragile X syndrome. Sci. Signal. 2017, 10, eaai8133. [Google Scholar] [CrossRef] [PubMed]
- Hutson, R.L.; Thompson, R.L.; Bantel, A.P.; Tessier, C.R. Acamprosate rescues neuronal defects in the Drosophila model of Fragile X Syndrome. Life Sci. 2018, 195, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Franco, L.M.; Okray, Z.; Linneweber, G.A.; Hassan, B.A.; Yaksi, E. Reduced Lateral Inhibition Impairs Olfactory Computations and Behaviors in a Drosophila Model of Fragile X Syndrome. Curr. Biol. 2017, 27, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Doll, C.A.; Broadie, K. Activity-dependent FMRP requirements in development of the neural circuitry of learning and memory. Development 2015, 142, 1346–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, C.A.; Broadie, K. Neuron class-specific requirements for Fragile X Mental Retardation Protein in critical period development of calcium signaling in learning and memory circuitry. Neurobiol. Dis. 2016, 89, 76–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, R.M.; Stone, E.F.; Wayne, C.W.; Marcinkevicius, E.M.; Ulherait, M.; Delventhal, R.; Pantalia, M.M.; Hill, V.M.; Zhou, C.G.; McAllister, S.; et al. A Drosophilamodel of Fragile X syndrome exhibits defects in phagocytosis by innate immune cells. J. Cell Sci. 2017, 216, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; He, F.; Krans, A.; Frazer, M.; Taylor, J.P.; Paulson, H.L.; Todd, P.K. RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X-associated tremor ataxia syndrome. Hum. Mol. Genet. 2015, 24, 4317–4326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burghes, A.H.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Edens, B.M.; Ajroud-Driss, S.; Ma, L.; Ma, Y.C. Molecular mechanisms and animal models of spinal muscular atrophy. Biochim. Biophys. Acta 2015, 1852, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Praveen, K.; Wen, Y.; Matera, A.G. A Drosophila model of spinal muscular atrophy uncouples snRNP biogenesis functions of survival motor neuron from locomotion and viability defects. Cell Rep. 2012, 1, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Imlach, W.L.; Beck, E.S.; Choi, B.J.; Lotti, F.; Pellizzoni, L.; McCabe, B.D. SMN is required for sensory-motor circuit function in Drosophila. Cell 2012, 151, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, M.L.; Esposito, A.; Maccallini, P.; Micheli, E.; Bavasso, F.; Gallotta, I.; Vernì, F.; Feiguin, F.; Cacchione, S.; McCabe, B.D.; et al. WDR79/TCAB1 plays a conserved role in the control of locomotion and ameliorates phenotypic defects in SMA models. Neurobiol. Dis. 2017, 105, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Praveen, K.; Wen, Y.; Gray, K.M.; Noto, J.J.; Patlolla, A.R.; Van Duyne, G.D.; Matera, A.G. A Drosophila model of spinal muscular atrophy uncouples snRNP biogenesis functions of survival motor neuron from locomotion and viability defects. PLoS Genet. 2014, 10, e1004489. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, C.; Sanyal, S. Behavioral and electrophysiological outcomes of tissue-specific Smn knockdown in Drosophila melanogaster. Brain Res. 2012, 1489, 66–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.C.; Dimlich, D.N.; Yokokura, T.; Mukherjee, A.; Kankel, M.W.; Sen, A.; Sridhar, V.; Fulga, T.A.; Hart, A.C.; Van Vactor, D.; et al. Modeling spinal muscular atrophy in Drosophila. PLoS ONE 2008, 3, e3209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borg, R.M.; Bordonne, R.; Vassallo, N.; Cauchi, R.J. Genetic Interactions between the Members of the SMN-Gemins Complex in Drosophila. PLoS ONE. 2015, 10, e0130974. [Google Scholar] [CrossRef] [PubMed]
- Mirra, A.; Rossi, S.; Scaricamazza, S.; Di Salvio, M.; Salvatori, I.; Valle, C.; Rusmini, P.; Poletti, A.; Cestra, G.; Carrì, M.T.; et al. Functional interaction between FUS and SMN underlies SMA-like splicing changes in wild-type hFUS mice. Sci. Rep. 2017, 7, 2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, A.; Dimlich, D.N.; Guruharsha, K.G.; Kankel, M.W.; Hori, K.; Yokokura, T.; Brachat, S.; Richardson, D.; Loureiro, J.; Sivasankaran, R.; et al. Genetic circuitry of Survival motor neuron, the gene underlying spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2013, 110, E2371–E2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, M.J.A.; Talbot, K.; Bowerman, M. Spinal muscular atrophy: antisense oligonucleotide therapy opens the door to an integrated therapeutic landscape. Hum. Mol. Genet. 2017, 26, R151–R159. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerrity, M.S.; Prasad, V.; Obley, A.J. Concerns About the Approval of Nusinersen Sodium by the US Food and Drug Administration. JAMA Intern. Med. 2018, 178, 743–744. [Google Scholar] [CrossRef] [PubMed]
- Wishart, T.M.; Mutsaers, C.A.; Riessland, M.; Reimer, M.M.; Hunter, G.; Hannam, M.L.; Eaton, S.L.; Fuller, H.R.; Roche, S.L.; Somers, E.; et al. Dysregulation of ubiquitin homeostasis and β-catenin signaling promote spinal muscular atrophy. J. Clin. Investig. 2014, 124, 1821–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohr, C.; Hartmann, B. Alternative splicing in Drosophila neuronal development. J. Neurogenet. 2014, 28, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Carmena, A. Compromising asymmetric stem cell division in Drosophila central brain: Revisiting the connections with tumorigenesis. Fly (Austin) 2018, 12, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Hayakawa-Yano, Y.; Okano, H. RNA regulation went wrong in neurodevelopmental disorders: The example of Msi/Elavl RNA binding proteins. Int. J. Dev. Neurosci. 2016, 55, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Zalfa, F.; Achsel, T.; Bagni, C. mRNPs, polysomes or granules: FMRP in neuronal protein synthesis. Curr. Opin. Neurobiol. 2006, 16, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Hillebrand, J.; Barbee, S.A.; Ramaswami, M. P-body components, microRNA regulation, and synaptic plasticity. Sci. World J. 2007, 7, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Vicente, M.; Monferrer, L.; Artero, R. The Muscleblind family of proteins: An emerging class of regulators of developmentally programmed alternative splicing. Differentiation 2006, 74, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Baines, R.A. Neuronal homeostasis through translational control. Mol. Neurobiol. 2005, 32, 113–121. [Google Scholar] [CrossRef]
- Froldi, F.; Cheng, L.Y. Understanding how differentiation is maintained: Lessons from the Drosophila brain. Cell. Mol. Life Sci. 2016, 73, 1641–1644. [Google Scholar] [CrossRef] [PubMed]
- Spana, E.P.; Doe, C.Q. The prospero transcription factor is asymmetrically localized to the cell cortex during neuroblast mitosis in Drosophila. Development 1995, 121, 3187–3195. [Google Scholar] [PubMed]
- Hirata, J.; Nakagoshi, H.; Nabeshima, Y.; Matsuzaki, F. Asymmetric segregation of the homeodomain protein Prospero during Drosophila development. Nature 1995, 377, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Schuldt, A.J.; Adams, J.H.; Davidson, C.M.; Micklem, D.R.; Haseloff, J.; St Johnston, D.; Brand, A.H. Miranda mediates asymmetric protein and RNA localization in the developing nervous system. Genes Dev. 1998, 12, 1847–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broadus, J.; Fuerstenberg, S.; Doe, C.Q. Staufen-dependent localization of prospero mRNA contributes to neuroblast daughter-cell fate. Nature 1998, 391, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Shan, Z.; Yang, Y.; Liu, C.; Li, J.; Luo, Z.G.; Zhang, M.; Cai, Y.; Wen, W.; Wang, W. The structural basis of Miranda-mediated Staufen localization during Drosophila neuroblast asymmetric division. Nat. Commun. 2015, 6, 8381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Yang, C.P.; Sugino, K.; Fu, C.C.; Liu, L.Y.; Yao, X.; Lee, L.P.; Lee, T. Opposing intrinsic temporal gradients guide neural stem cell production of varied neuronal fates. Science 2015, 350, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Yang, C.P.; Liu, Z.; Sugino, K.; Mok, K.; He, Y.; Ito, M.; Nern, A.; Otsuna, H.; Lee, T. Stem Cell-Intrinsic, Seven-up-Triggered Temporal Factor Gradients Diversify Intermediate Neural Progenitors. Curr. Biol. 2017, 27, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.P.; Samuels, T.J.; Huang, Y.; Yang, L.; Ish-Horowicz, D.; Davis, I.; Lee, T. Imp and Syp RNA-binding proteins govern decommissioning of Drosophilaneural stem cells. Development 2017, 144, 3454–3464. [Google Scholar] [CrossRef] [PubMed]
- Abramczuk, M.K.; Burkard, T.R.; Rolland, V.; Steinmann, V.; Duchek, P.; Jiang, Y.; Wissel, S.; Reichert, H.; Knoblich, J.A. The splicing co-factor Barricade/Tat-SF1 is required for cell cycle and lineage progression in Drosophilaneural stem cells. Development 2017, 144, 3932–3945. [Google Scholar] [CrossRef] [PubMed]
- Arama, E.; Dickman, D.; Kimchie, Z.; Shearn, A.; Lev, Z. Mutations in the beta-propeller domain of the Drosophila brain tumor (brat) protein induce neoplasm in the larval brain. Oncogene 2000, 19, 3706–3716. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Tucker-Burden, C.; Zhang, C.; Moberg, K.; Read, R.; Hadjipanayis, C.; Brat, D.J. Drosophila Brat and Human Ortholog TRIM3 Maintain Stem Cell Equilibrium and Suppress Brain Tumorigenesis by Attenuating Notch Nuclear Transport. Cancer Res. 2016, 76, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, I.; Bonnay, F.; Steinmann, V.; Loedige, I.; Burkard, T.R.; Meister, G.; Knoblich, J.A. The tumor suppressor Brat controls neuronal stem cell lineages by inhibiting Deadpan and Zelda. EMBO Rep. 2018, 19, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Narbonne-Reveau, K.; Lanet, E.; Dillard, C.; Foppolo, S.; Chen, C.H.; Parrinello, H.; Rialle, S.; Sokol, N.S.; Maurange, C. Neural stem cell-encoded temporal patterning delineates an early window of malignant susceptibility in Drosophila. Elife 2016, 5, e13463. [Google Scholar] [CrossRef] [PubMed]
- Landskron, L.; Steinmann, V.; Bonnay, F.; Burkard, T.R.; Steinmann, J.; Reichardt, I.; Harzer, H.; Laurenson, A.S.; Reichert, H.; Knoblich, J.A. The asymmetrically segregating lncRNA cherub is required for transforming stem cells into malignant cells. Elife. 2018, 7, e31347. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Qu, C.; Hewes, R.S. Neuronal remodeling during metamorphosis is regulated by the alan shepard (shep) gene in Drosophila melanogaster. Genetics 2014, 197, 1267–1283. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Petritsch, C.; Clark, I.E.; Gavis, E.R.; Jan, L.Y.; Jan, Y.N. Nanos and Pumilio are essential for dendrite morphogenesis in Drosophila peripheral neurons. Curr. Biol. 2004, 14, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Olesnicky, E.C.; Bhogal, B.; Gavis, E.R. Combinatorial use of translational co-factors for cell type-specific regulation during neuronal morphogenesis in Drosophila. Dev. Biol. 2012, 365, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, B.; Plaza-Jennings, A.; Gavis, E.R. Nanos-mediated repression of hid protects larval sensory neurons after a global switch in sensitivity to apoptotic signals. Development 2016, 143, 2147–2159. [Google Scholar] [CrossRef] [PubMed]
- Brechbiel, J.L.; Gavis, E.R. Spatial regulation of nanos is required for its function in dendrite morphogenesis. Curr. Biol. 2008, 18, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Brechbiel, J.L.; Gavis, E.R. Dynein-dependent transport of nanos RNA in Drosophila sensory neurons requires Rumpelstiltskin and the germ plasm organizer Oskar. J. Neurosci. 2013, 33, 14791–14800. [Google Scholar] [CrossRef] [PubMed]
- Olesnicky, E.C.; Killian, D.J.; Garcia, E.; Morton, M.C.; Rathjen, A.R.; Sola, I.E.; Gavis, E.R. Extensive use of RNA-binding proteins in Drosophila sensory neuron dendrite morphogenesis. G3 (Bethesda) 2014, 4, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Schachtner, L.T.; Sola, I.E.; Forand, D.; Antonacci, S.; Postovit, A.J.; Mortimer, N.T.; Killian, D.J.; Olesnicky, E.C. Drosophila Shep and C. elegans SUP-26 are RNA-binding proteins that play diverse roles in nervous system development. Dev. Genes Evol. 2015, 225, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Olesnicky, E.C.; Bono, J.M.; Bell, L.; Schachtner, L.T.; Lybecker, M.C. The RNA-binding protein caper is required for sensory neuron development in Drosophila melanogaster. Dev. Dyn. 2017, 246, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Antonacci, S.; Forand, D.; Wolf, M.; Tyus, C.; Barney, J.; Kellogg, L.; Simon, M.A.; Kerr, G.; Wells, K.L.; Younes, S.; et al. Conserved RNA-binding proteins required for dendrite morphogenesis in Caenorhabditis elegans sensory neurons. G3 (Bethesda) 2015, 5, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Misra, M.; Edmund, H.; Ennis, D.; Schlueter, M.A.; Marot, J.E.; Tambasco, J.; Barlow, I.; Sigurbjornsdottir, S.; Mathew, R.; Vallés, A.M.; et al. A Genome-Wide Screen for Dendritically Localized RNAs Identifies Genes Required for Dendrite Morphogenesis. G3 Genes Genomes Genet. 2016, 6, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Taliaferro, J.M.; Vidaki, M.; Oliveira, R.; Olson, S.; Zhan, L.; Saxena, T.; Wang, E.T.; Graveley, B.R.; Gertler, F.B.; Swanson, M.S.; et al. Distal Alternative Last Exons Localize mRNAs to Neural Projections. Mol. Cell 2016, 61, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Bigler, R.L.; Kamande, J.W.; Dumitru, R.; Niedringhaus, M.; Taylor, A.M. Messenger RNAs localized to distal projections of human stem cell derived neurons. Sci. Rep. 2017, 7, 611. [Google Scholar] [CrossRef] [PubMed]
- Mitsumori, K.; Takei, Y.; Hirokawa, N. Components of RNA granules affect their localization and dynamics in neuronal dendrites. Mol. Biol. Cell 2017, 28, 1412–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouwenga, R.; Lake, A.M.; O’Brien, D.; Mogha, A.; Dani, A.; Dougherty, J.D. Transcriptomic Analysis of Ribosome-Bound mRNA in Cortical Neurites In Vivo. J. Neurosci. 2017, 37, 8688–8705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappulo, A.; van den Bruck, D.; Ciolli Mattioli, C.; Franke, V.; Imami, K.; McShane, E.; Moreno-Estelles, M.; Calviello, L.; Filipchyk, A.; Peguero-Sanchez, E.; et al. RNA localization is a key determinant of neurite-enriched proteome. Nat. Commun. 2017, 8, 583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Driesche, S.J.; Martin, K.C. New frontiers in RNA transport and local translation in neurons. Dev. Neurobiol. 2018, 78, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, P.K.; Smith, D.S.; Perrone-Bizzozero, N.; Twiss, J.L. Axonal mRNA transport and translation at a glance. J. Cell Sci. 2018, 131, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tushev, G.; Glock, C.; Heumüller, M.; Biever, A.; Jovanovic, M.; Schuman, E.M. Alternative 3’ UTRs Modify the Localization, Regulatory Potential, Stability, and Plasticity of mRNAs in Neuronal Compartments. Neuron 2018, 98, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Menon, K.P.; Andrews, S.; Murthy, M.; Gavis, E.R.; Zinn, K. The translational repressors Nanos and Pumilio have divergent effects on presynaptic terminal growth and postsynaptic glutamate receptor subunit composition. J. Neurosci. 2009, 29, 5558–5572. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Chen, Y.; Gan, G.; Wang, D.; Ren, J.; Wang, Q.; Xu, Z.; Xie, W.; Zhang, Y.Q. Brain tumor regulates neuromuscular synapse growth and endocytosis in Drosophila by suppressing mad expression. J. Neurosci. 2013, 33, 12352–12363. [Google Scholar] [CrossRef] [PubMed]
- Santana, E.; Casas-Tintó, S. Orb2 as modulator of Brat and their role at the neuromuscular junction. J. Neurogenet. 2017, 31, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Menon, K.P.; Carrillo, R.A.; Zinn, K. The translational regulator Cup controls NMJ presynaptic terminal morphology. Mol. Cell Neurosci. 2015, 67, 126–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Dale, R.K.; Lei, E.P. Shep regulates Drosophila neuronal remodeling by controlling transcription of its chromatin targets. Development 2018, 145, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Gu, T.; Pham, T.N.; Zachary, M.J.; Hewes, R.S. Regulatory Mechanisms of Metamorphic Neuronal Remodeling Revealed Through a Genome-Wide Modifier Screen in Drosophila melanogaster. Genetics 2017, 206, 1429–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, G.; Reichardt, I.; Knoblich, J.A.; Besse, F. The TRIM-NHL protein Brat promotes axon maintenance by repressing src64B expression. J. Neurosci. 2014, 34, 13855–13864. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Manoukian, A.S.; Perrimon, N. Ectopic expression in Drosophila. Methods Cell Biol. 1994, 44, 635–654. [Google Scholar] [PubMed]
- Perrimon, N.; Ni, J.Q.; Perkins, L. In vivo RNAi: Today and tomorrow. Cold Spring Harb. Perspect. Biol. 2010, 2, a003640. [Google Scholar] [CrossRef] [PubMed]
- Duffy, J.B. Genetic Reagents for Making Split-GAL4 Lines in Drosophila. Genesis 2002, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Dionne, H.; Hibbard, K.L.; Cavallaro, A.; Kao, J.C.; Rubin, G.M. Genetic Reagents for Making Split-GAL4 Lines in Drosophila. Genetics 2018, 209, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Yagi, R.; Mayer, F.; Basler, K. Refined LexA transactivators and their use in combination with the Drosophila Gal4 system. Proc. Natl. Acad. Sci. USA 2010, 107, 16166–16171. [Google Scholar] [CrossRef] [PubMed]
- Gnerer, J.P.; Venken, K.J.; Dierick, H.A. Gene-specific cell labeling using MiMIC transposons. Nucleic Acids Res. 2015, 43, e56. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.C.; Rahman, R.; Jin, H.; Shen, J.L.; Fieldsend, A.; Luo, W.; Rosbash, M. TRIBE: Hijacking an RNA-Editing Enzyme to Identify Cell-Specific Targets of RNA-Binding Proteins. Cell 2016, 165, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Jensen, K.; Mele, A.; Darnell, R.B. CLIP: A method for identifying protein-RNA interaction sites in living cells. Methods 2005, 37, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.J.; Yario, T.A.; Steitz, J.A. Association of Argonaute proteins and microRNAs can occur after cell lysis. RNA 2012, 18, 1581–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, E.H.; Vanhoven, M.K.; Bendesky, A.; Wang, G.; Fetter, R.D.; Shen, K.; Bargmann, C.I. GFP Reconstitution Across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron 2008, 57, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, L.J.; Zaharieva, E.E.; Kearney, P.J.; Alpert, M.H.; Lin, T.Y.; Turan, Z.; Lee, C.H.; Gallio, M. Dynamic labelling of neural connections in multiple colours by trans-synaptic fluorescence complementation. Nat. Commun. 2015, 6, 10024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Guo, A.; Li, H. CRASP: CFP reconstitution across synaptic partners. Biochem. Biophys. Res. Commun. 2016, 469, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Owald, D.; Lin, S.; Waddell, S. Light, heat, action: Neural control of fruit fly behaviour. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2015, 370, 20140211. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.E.; Toettcher, J.E. Illuminating developmental biology with cellular optogenetics. Curr. Opin. Biotechnol. 2018, 52, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Morikawa, R.K.; Hasegawa, E.; Emoto, K. Neural Circuitry that Evokes Escape Behavior upon Activation of Nociceptive Sensory Neurons in Drosophila Larvae. Curr. Biol. 2017, 27, 2499–2504. [Google Scholar] [CrossRef] [PubMed]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olesnicky, E.C.; Wright, E.G. Drosophila as a Model for Assessing the Function of RNA-Binding Proteins during Neurogenesis and Neurological Disease. J. Dev. Biol. 2018, 6, 21. https://doi.org/10.3390/jdb6030021
Olesnicky EC, Wright EG. Drosophila as a Model for Assessing the Function of RNA-Binding Proteins during Neurogenesis and Neurological Disease. Journal of Developmental Biology. 2018; 6(3):21. https://doi.org/10.3390/jdb6030021
Chicago/Turabian StyleOlesnicky, Eugenia C., and Ethan G. Wright. 2018. "Drosophila as a Model for Assessing the Function of RNA-Binding Proteins during Neurogenesis and Neurological Disease" Journal of Developmental Biology 6, no. 3: 21. https://doi.org/10.3390/jdb6030021
APA StyleOlesnicky, E. C., & Wright, E. G. (2018). Drosophila as a Model for Assessing the Function of RNA-Binding Proteins during Neurogenesis and Neurological Disease. Journal of Developmental Biology, 6(3), 21. https://doi.org/10.3390/jdb6030021