I Spy in the Developing Fly a Multitude of Ways to Die



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
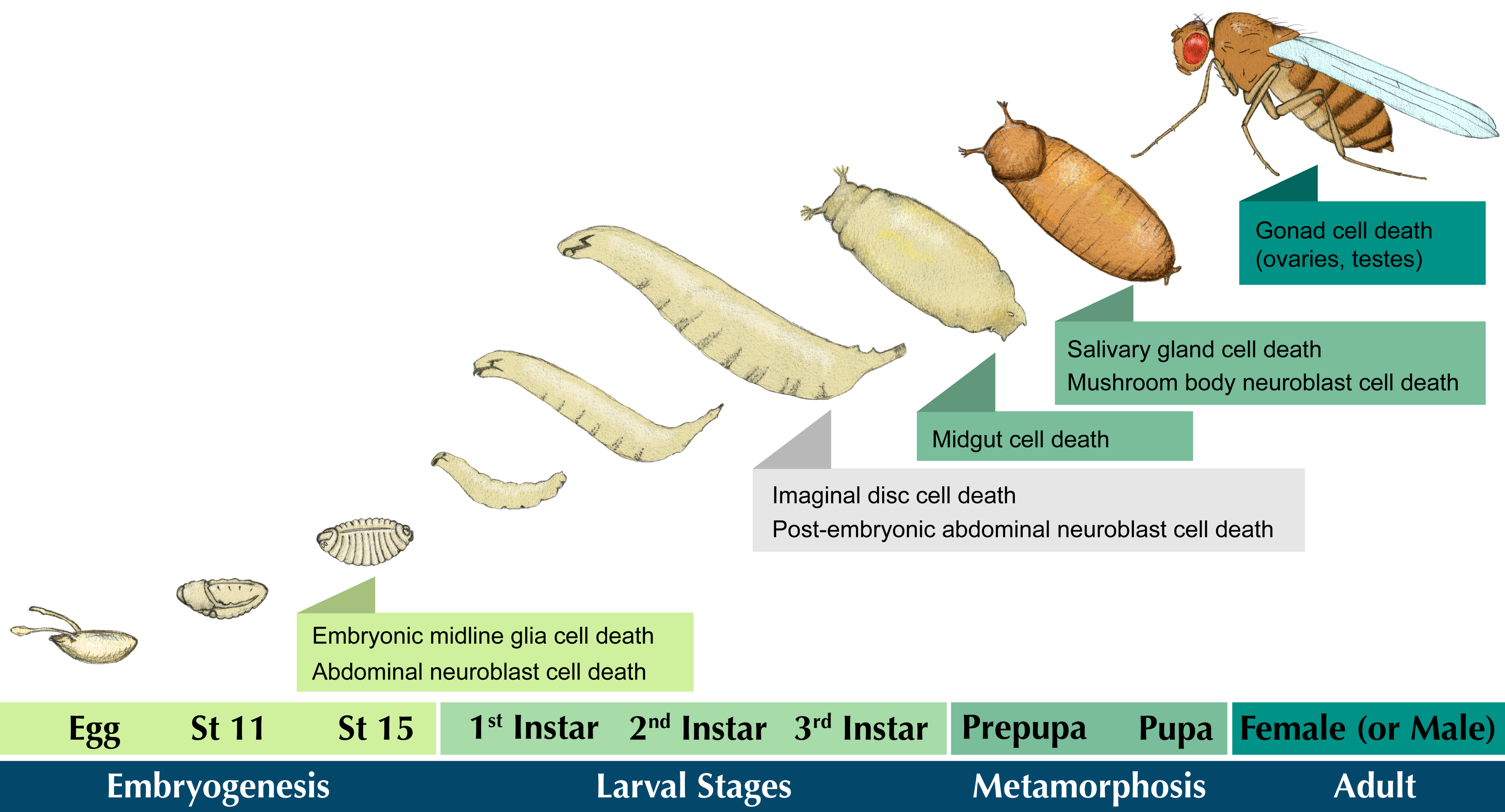
1.1. Cell Death in Development and Disease
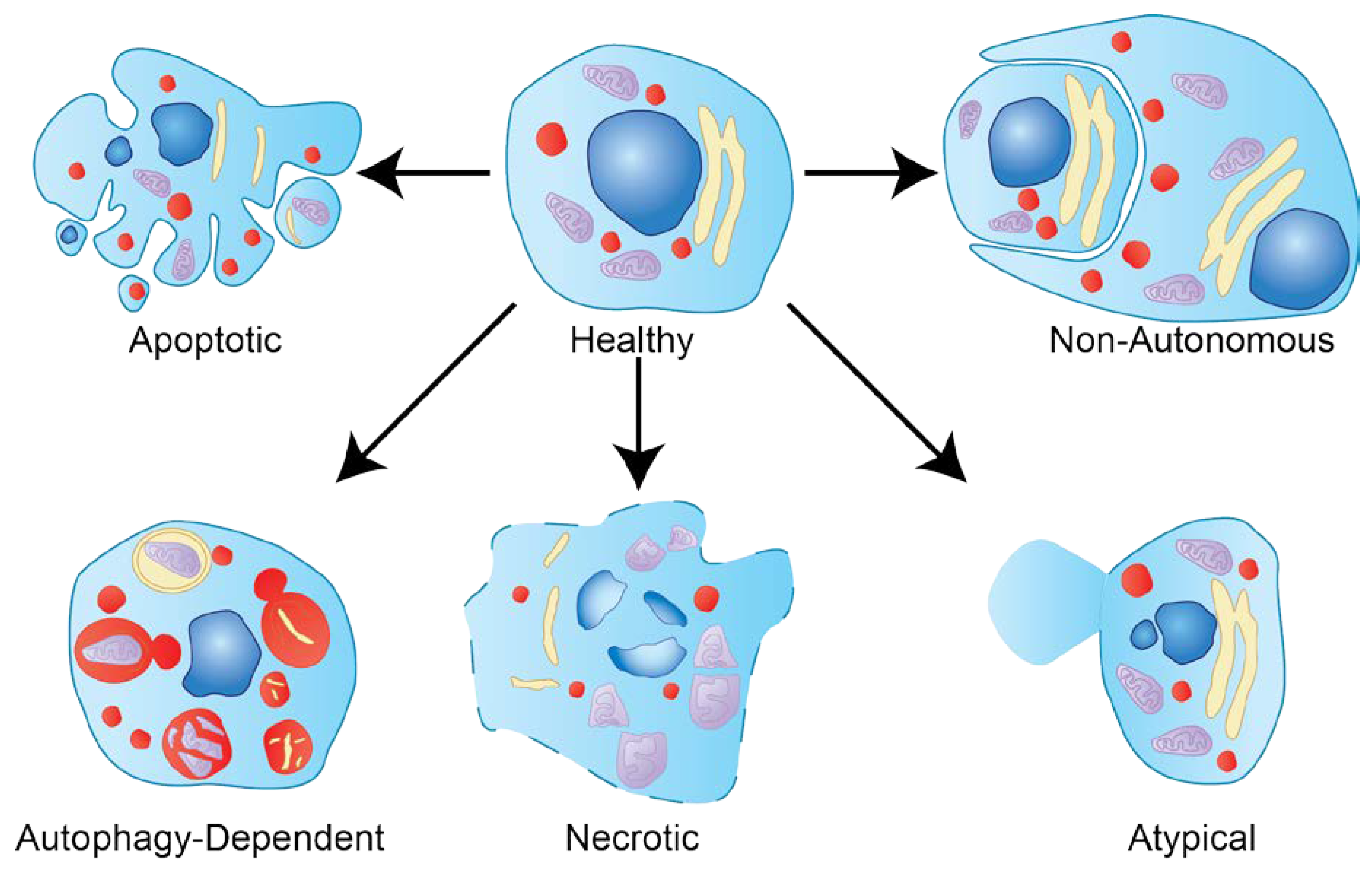
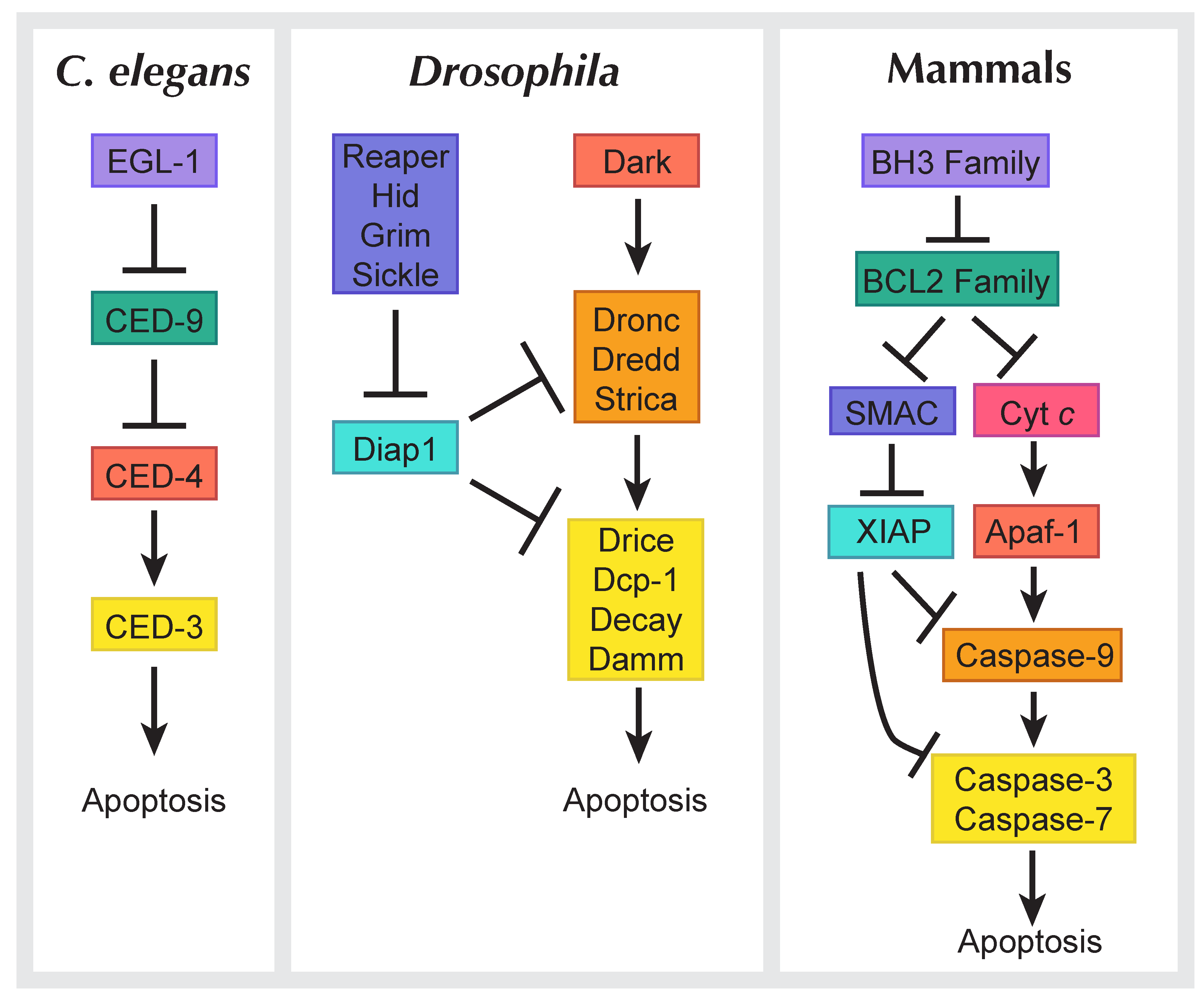
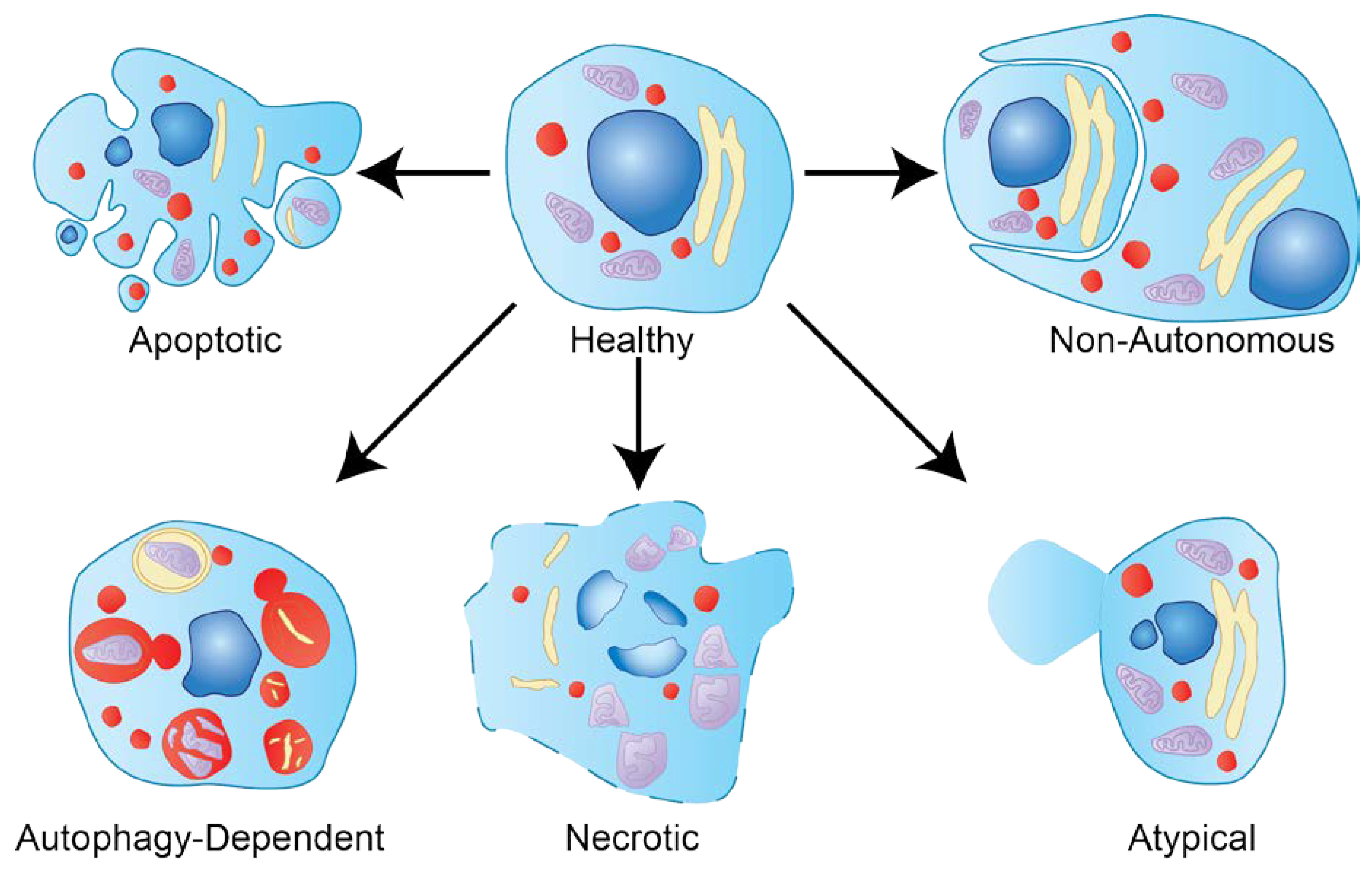
1.2. Types of Cell Death
2. Cell Death in the Developing Drosophila Central Nervous System
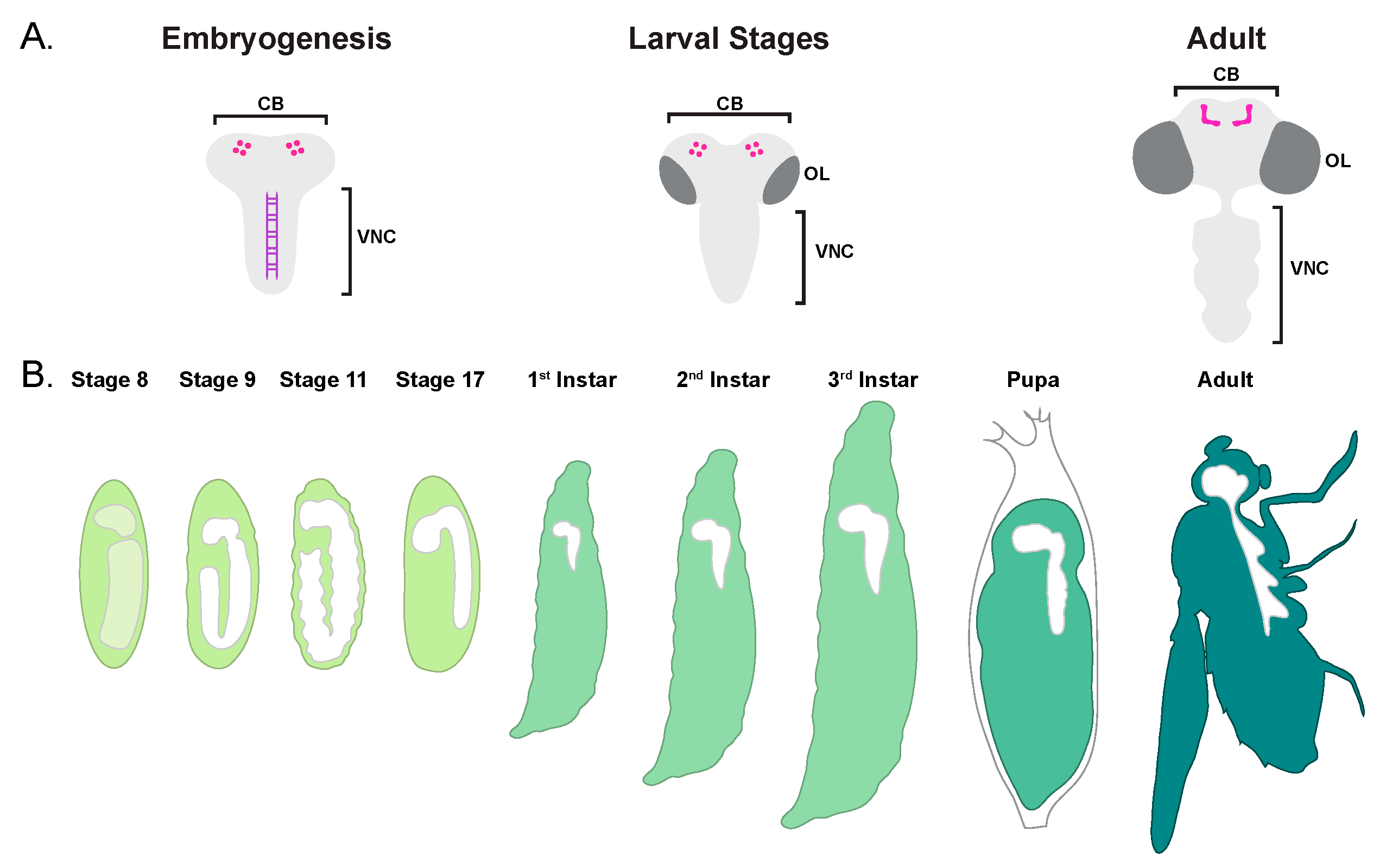
2.1. Brief Overview of CNS Development
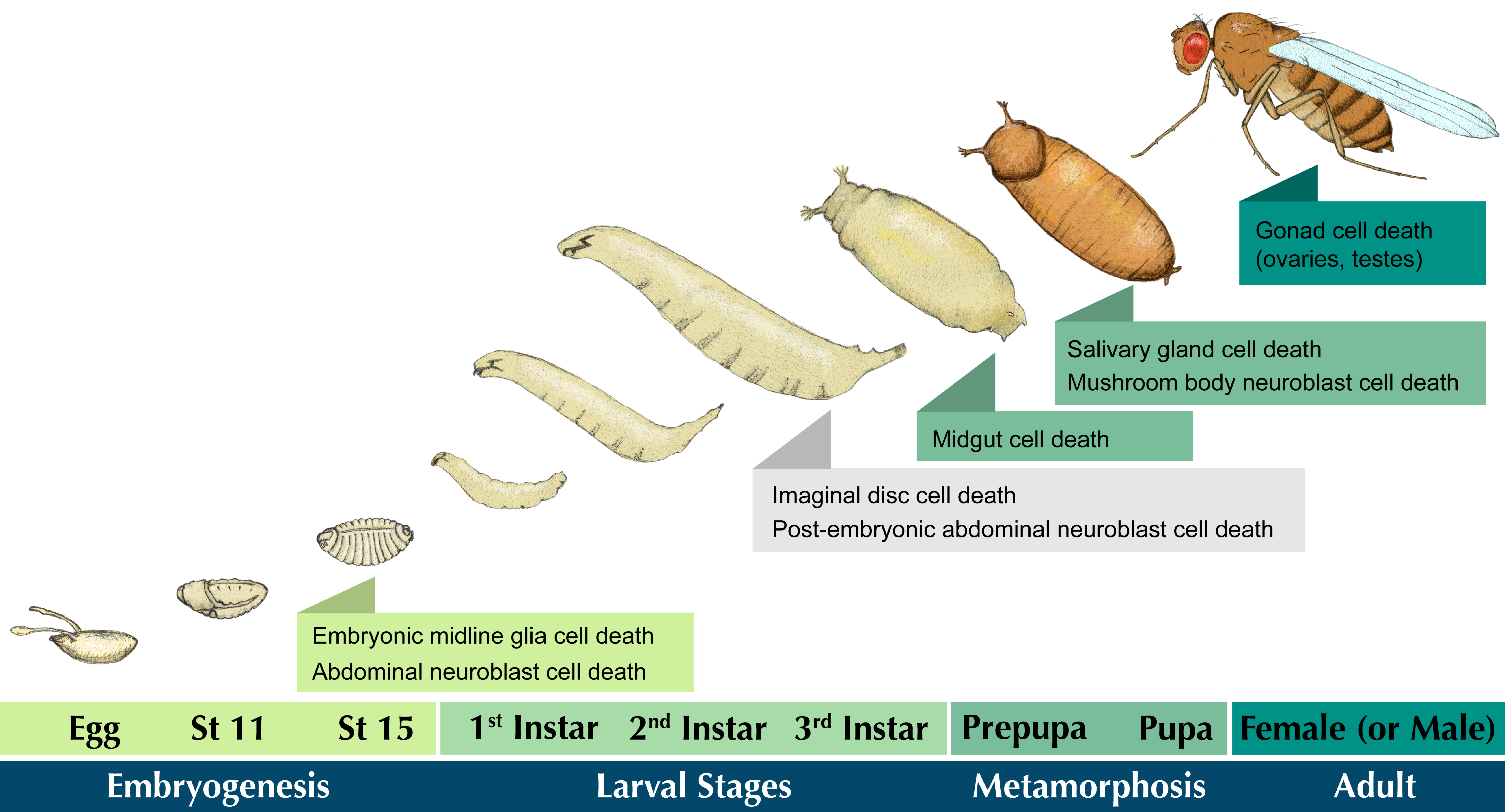
2.2. Embryonic Neuroblast Cell Death
2.3. Midline Glia PCD
2.4. Mushroom Body Neuroblast PCD
3. Death in Reproductive Systems
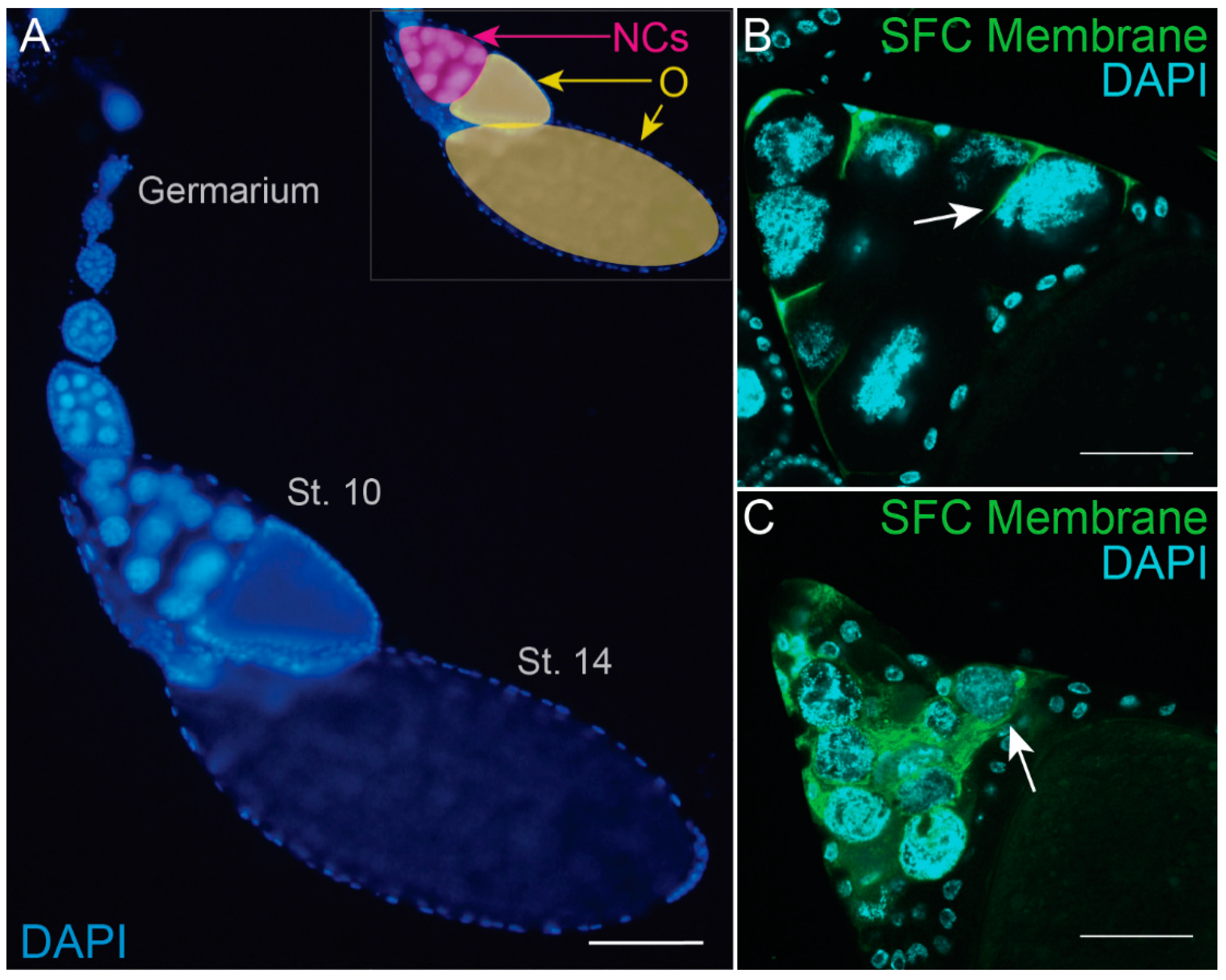
3.1. Developmental Phagoptosis in the Drosophila Ovary
3.2. Non-Apoptotic Cell Death in the Drosophila Testis
4. Steroid Hormone-Induced Cell Death in the Elimination of Larval Tissues
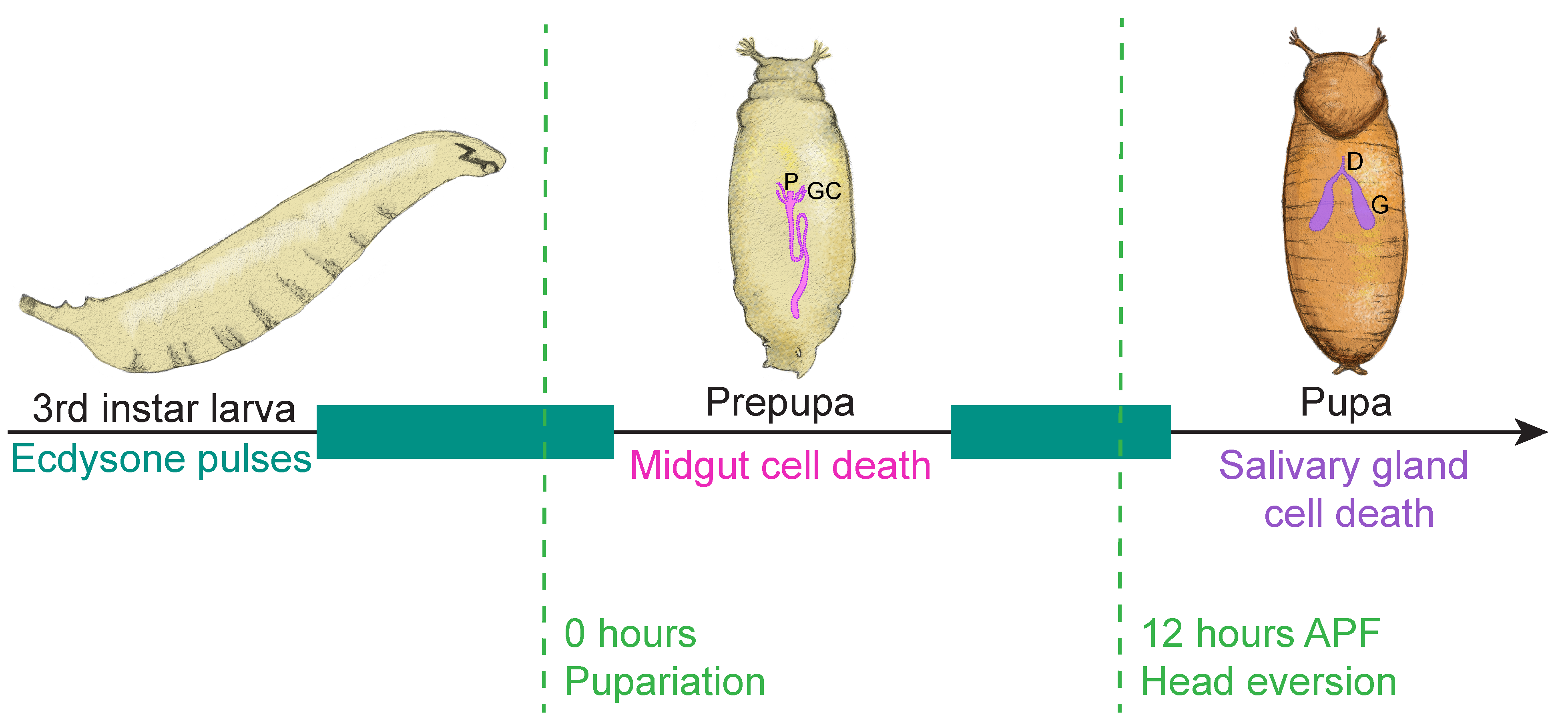
4.1. Removal of the Larval Salivary Glands
4.2. Removal of the Larval Midgut
5. Conclusions
- Why are certain PCD modalities favored in certain tissues? For example, cell death in the nervous system seems to occur through apoptosis, while non-apoptotic forms of cell death are observed in reproductive systems.
- Is there crosstalk between cell death programs, and how does that conversation occur? For example, when one form of cell death is blocked, the cell may still die through an alternative (or secondary) cell death mechanism. In addition, in some instances two different cell death programs contribute to the elimination of cells or tissue, as in larval salivary glands.
- To what extent are non-apoptotic modes of cell death conserved across species? For example, which types of non-apoptotic death are (or are not) conserved among organisms, and how similar are the signaling pathways?
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Milligan, C.E.; Prevette, D.; Yaginuma, H.; Homma, S.; Cardwellt, C.; Fritz, L.C.; Tomaselli, K.J.; Oppenheim, R.W.; Schwartz, L.M. Peptide inhibitors of the ice protease family arrest programmed cell death of motoneurons in vivo and in vitro. Neuron 1995, 15, 385–393. [Google Scholar] [CrossRef]
- Coucouvanis, E.; Martin, G.R. Signals for death and survival: A two-step mechanism for cavitation in the vertebrate embryo. Cell 1995, 83, 279–287. [Google Scholar] [CrossRef]
- Roberts, L.M.; Visser, J.A.; Ingraham, H.A. Involvement of a matrix metalloproteinase in MIS-induced cell death during urogenital development. Development 2002, 129, 1487–1496. [Google Scholar] [PubMed]
- Dekkers, M.P.J.; Nikoletopoulou, V.; Barde, Y.A. Death of developing neurons: New insights and implications for connectivity. J. Cell Biol. 2013, 203, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.; Weil, M.; Raff, M. Programmed cell death in animal development. Cell 1997, 88, 347–354. [Google Scholar] [CrossRef]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.M.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef] [PubMed]
- King, K.L.; Cidlowski, J.A. Cell cycle regulation and apoptosis. Annu. Rev. Physiol. 1998, 60, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Worth, A.; Thrasher, A.J.; Gaspar, H.B. Autoimmune lymphoproliferative syndrome: Molecular basis of disease and clinical phenotype. Br. J. Haematol. 2006, 133, 124–140. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K. Apoptosis in autoimmune diseases. Intern. Med. 2001, 40, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, F.A.; Peréz-Garijo, A.; Morata, G. Apoptosis in Drosophila: Compensatory proliferation and undead cells. Int. J. Dev. Biol. 2009, 53, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Córdoba, S.; Estella, C. The transcription factor Dysfusion promotes fold and joint morphogenesis through regulation of Rho1. PLoS Genet. 2018, 14, e1007584. [Google Scholar] [CrossRef] [PubMed]
- Guarner, A.; Manjón, C.; Edwards, K.; Steller, H.; Suzanne, M.; Sánchez-Herrero, E. The zinc finger homeodomain-2 gene of Drosophila controls Notch targets and regulates apoptosis in the tarsal segments. Dev. Biol. 2014, 385, 350–365. [Google Scholar] [CrossRef] [PubMed]
- Suzanne, M.; Petzoldt, A.G.; Spéder, P.; Coutelis, J.-B.; Steller, H.; Noselli, S. Coupling of apoptosis and L/R patterning controls stepwise organ looping. Curr. Biol. 2010, 20, 1773–1778. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.M.; Levayer, R.; Moreno, E. Survival of the Fittest: Essential Roles of Cell Competition in Development, Aging, and Cancer. Trends Cell Biol. 2016, 26, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Qasim Raza, S.; Voisin, L.; Dakhli, H.; Ed Eric Law, F.; De Jong, D.; Allouch, A.; Thoreau, M.; Brenner, C.; Deutsch, E.; et al. Entosis: The emerging face of non-cell-autonomous type IV programmed death. Biomed. J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Sulston, J.E.; Horvitz, H.R. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 1977, 56, 110–156. [Google Scholar] [CrossRef]
- Sulston, J.E.; Schierenberg, E.; White, J.G.; Thomson, J.N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 1983, 100, 64–119. [Google Scholar] [CrossRef]
- Ellis, H.M.; Horvitz, H.R. Genetic control of programmed cell death in the nematode C. elegans. Cell 1986, 44, 817–829. [Google Scholar] [CrossRef]
- Hedgecock, E.M.; Sulston, J.E.; Thomson, J.N. Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science 1983, 220, 1277–1279. [Google Scholar] [CrossRef] [PubMed]
- Conradt, B.; Horvitz, H.R. The C. elegans Protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 1998, 93, 519–529. [Google Scholar] [CrossRef]
- Del Peso, L.; González, V.M.; Núnez, G. Caenorhabditis elegans EGL-1 disrupts the interaction of CED-9 with CED-4 and promotes CED-3 activation. J. Biol. Chem. 1998, 273, 33495–33500. [Google Scholar] [CrossRef] [PubMed]
- Sandu, C.; Ryoo, H.D.; Steller, H. Drosophila IAP antagonists form multimeric complexes to promote cell death. J. Cell Biol. 2010, 190, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; McCall, K.; Agapite, J.; Hartwieg, E.; Steller, H. Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J. 2000, 19, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Oliver, H.; Zou, H.; Chen, P.; Wang, X.; Abrams, J.M. Dark is a Drosophila homologue of Apaf-1/CED-4 and functions in an evolutionarily conserved death pathway. Nat. Cell Biol. 1999, 1, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, L.; Acehan, D.; Wang, X.; Akey, C.W. Three-dimensional structure of a double apoptosome formed by the Drosophila apaf-1 related killer. J. Mol. Biol. 2006, 355, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; McCall, K.; Steller, H. DCP-1, a Drosophila cell death protease essential for development. Science 1997, 275, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Fraser, A.G.; Evan, G.I. Identification of a Drosophila melanogaster ICE/CED-3-related protease, drICE. EMBO J. 1997, 16, 2805–2813. [Google Scholar] [CrossRef] [PubMed]
- Kulda, K.; Zheng, T.S.; Na, S.; Kuan, C.Y.; Yang, D.; Karasuyama, H.; Rakic, P.; Flavell, R.A. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 1996, 384, 368–372. [Google Scholar] [CrossRef]
- Yoshida, H.; Kong, Y.; Yoshida, R.; Elia, A.J.; Hakem, A.; Hakem, R.; Penninger, J.M.; Mak, T.W. Apaf1 Is Required for Mitochondrial Pathways of Apoptosis and Brain Development. Cell 1998, 94, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Lindsten, T.; Ross, A.J.; King, A.; Zong, W.; Rathmell, J.C.; Shiels, H.A.; Ulrich, E.; Waymire, K.G.; Mahar, P.; Frauwirth, K.; et al. The Combined Functions of Proapoptotic Bcl-2 Family Members Bak and Bax Are Essential for Normal Development of Multiple Tissues. Mol. Cell 2000, 6, 1389–1399. [Google Scholar] [CrossRef]
- Nonomura, K.; Yamaguchi, Y.; Hamachi, M.; Koike, M.; Uchiyama, Y.; Nakazato, K.; Mochizuki, A.; Sakaue-Sawano, A.; Miyawaki, A.; Yoshida, H.; et al. Local apoptosis modulates early mammalian brain development through the elimination of morphogen-producing cells. Dev. Cell 2013, 27, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Kroemer, G. Alternative cell death mechanisms in development and beyond. Genes Dev. 2010, 24, 2592–2602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stjepanovic, G.; Davies, C.W.; Stanley, R.E.; Ragusa, M.J.; Kim, D.J.; Hurley, J.H. Assembly and dynamics of the autophagy-initiating Atg1 complex. Proc. Natl. Acad. Sci. USA 2014, 111, 12793–12798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell Biol. 2010, 12, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Nair, U.; Klionsky, D.J. Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell 2008, 19, 3290–3298. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Nicolson, S.; Denton, D.; Kumar, S. Distinct requirements of Autophagy-related genes in programmed cell death. Cell Death Differ. 2015, 22, 1792–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, D.; Xu, T.; Kumar, S. Autophagy as a pro-death pathway. Immunol. Cell Biol. 2014, 93, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, T.; Kim, J.; Huang, W.P.; Baba, M.; Tokunaga, C.; Ohsumi, Y.; Klionsky, D.J. Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J. Cell Biol. 2000, 148, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debnath, J.; Baehrecke, E.H.; Kroemer, G. Does autophagy contribute to cell death? Autophagy 2005, 1, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Shravage, B.; Simin, R.; Mills, K.; Berry, D.L.; Baehrecke, E.H.; Kumar, S. Autophagy, Not Apoptosis, Is Essential for Midgut Cell Death in Drosophila. Curr. Biol. 2009, 19, 1741–1746. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.L.; Baehrecke, E.H. Growth Arrest and Autophagy Are Required for Salivary Gland Cell Degradation in Drosophila. Cell 2007, 131, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Escobar, M.L.; Echeverría, O.M. Role of Autophagy in the Ovary Cell Death in Mammals. In Autophagy—A Double Edged Sword—Cell Survival or Death? InTech: Munich, Germany, 2013; pp. 423–441. [Google Scholar]
- Kroemer, G.; El-Deiry, W.S.; Golstein, P.; Peter, M.E.; Vaux, D.; Vandenabeele, P.; Zhivotovsky, B.; Blagosklonny, M.V.; Malorni, W.; Knight, R.A.; et al. Nomenclature Committee on Cell Death Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005, 12, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Meng, L.; Xu, T.; Su, Y.; Liu, X.; Zhang, Z.; Wang, X. RIPK1-RIPK3-MLKL-dependent necrosis promotes the aging of mouse male reproductive system. eLife 2017, 6, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Aachoui, Y.; Sagulenko, V.; Miao, E.A.; Stacey, K.J. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr. Opin. Microbiol. 2013, 16, 319–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Akino, K.; Toyota, M.; Suzuki, H.; Imai, T.; Maruyama, R.; Kusano, M.; Nishikawa, N.; Watanabe, Y.; Sasaki, Y.; Abe, T.; et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci. 2007, 98, 88–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overholtzer, M.; Mailleux, A.A.; Mouneimne, G.; Normand, G.; Schnitt, S.J.; King, R.W.; Cibas, E.S.; Brugge, J.S. A Nonapoptotic Cell Death Process, Entosis, that Occurs by Cell-in-Cell Invasion. Cell 2007, 131, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Florey, O.; Kim, S.E.; Sandoval, C.P.; Haynes, C.M.; Overholtzer, M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 2011, 13, 1335–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamann, J.C.; Surcel, A.; Chen, R.; Teragawa, C.; Albeck, J.G.; Robinson, D.N.; Overholtzer, M. Entosis Is Induced by Glucose Starvation. Cell Rep. 2017, 20, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Neher, J.J. Eaten alive! Cell death by primary phagocytosis: “Phagoptosis”. Trends Biochem. Sci. 2012, 37, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Neniskyte, U.; Neher, J.J.; Brown, G.C. Neuronal death induced by nanomolar amyloid β is mediated by primary phagocytosis of neurons by microglia. J. Biol. Chem. 2011, 286, 39904–39913. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Neniskyte, U.; Zhao, J.-W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of Microglial Phagocytosis Is Sufficient To Prevent Inflammatory Neuronal Death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricker, M.; Neher, J.J.; Zhao, J.-W.; Thery, C.; Tolkovsky, A.M.; Brown, G.C. MFG-E8 Mediates Primary Phagocytosis of Viable Neurons during Neuroinflammation. J. Neurosci. 2012, 32, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.I.; Timmons, A.K.; Klein, A.P.; Pritchett, T.L.; Welch, E.; Meehan, T.L.; Li, C.; McCall, K. Draper acts through the JNK pathway to control synchronous engulfment of dying germline cells by follicular epithelial cells. Development 2012, 139, 4029–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homem, C.C.F.; Knoblich, J.A. Drosophila neuroblasts: A model for stem cell biology. Development 2012, 139, 4297–4310. [Google Scholar] [CrossRef] [PubMed]
- Truman, J.W.; Bate, M. Spatial and temporal patterns of neurogenesis in the central nervous system of Drosophila melanogaster. Dev. Biol. 1988, 125, 145–157. [Google Scholar] [CrossRef]
- Campos-Ortega, J. Genetic mechanisms of early neurogenesis in Drosophila melanogaster. Mol. Neurobiol. 1995, 111–122. [Google Scholar] [CrossRef]
- Birkholz, O.; Rickert, C.; Berger, C.; Urbach, R.; Technau, G.M. Neuroblast pattern and identity in the Drosophila tail region and role of doublesex in the survival of sex-specific precursors. Development 2013, 140, 1830–1842. [Google Scholar] [CrossRef] [PubMed]
- Urbach, R. Molecular markers for identified neuroblasts in the developing brain of Drosophila. Development 2003, 130, 3621–3637. [Google Scholar] [CrossRef] [PubMed]
- Nériec, N.; Desplan, C. Chapter Fourteen—From the Eye to the Brain: Development of the Drosophila Visual System. Curr. Top. Dev. Biol. 2016, 116, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hotta, Y. Proliferation pattern of postembryonic neuroblasts in the brain of Drosophila melanogaster. Dev. Biol. 1992, 149, 134–148. [Google Scholar] [CrossRef]
- Prokop, A.; Technau, G.M. The origin of postembryonic neuroblasts in the ventral nerve cord of Drosophila melanogaster. Development 1991, 111, 79–88. [Google Scholar] [PubMed]
- Tan, Y.; Yamada-Mabuchi, M.; Arya, R.; St Pierre, S.; Tang, W.; Tosa, M.; Brachmann, C.; White, K. Coordinated expression of cell death genes regulates neuroblast apoptosis. Development 2011, 138, 2197–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chell, J.M.; Brand, A.H. Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell 2010, 143, 1161–1173. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Nunes, R.; Yee, L.L.; Gould, A.P. Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature 2011, 471, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Maurange, C.; Cheng, L.; Gould, A.P. Temporal Transcription Factors and Their Targets Schedule the End of Neural Proliferation in Drosophila. Cell 2008, 133, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Bello, B.C.; Hirth, F.; Gould, A.P. A pulse of the Drosophila Hox protein Abdominal-A schedules the end of neural proliferation via neuroblast apoptosis. Neuron 2003, 37, 209–219. [Google Scholar] [CrossRef]
- Pinto-Teixeira, F.; Konstantinides, N.; Desplan, C. Programmed cell death acts at different stages of Drosophila neurodevelopment to shape the central nervous system. FEBS Lett. 2016, 590, 2435–2453. [Google Scholar] [CrossRef] [PubMed]
- Page, D.T.; Olofsson, B. Multiple roles for apoptosis facilitating condensation of the Drosophila ventral nerve cord. Genesis 2008, 46, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Schnitzler, A.; Agapite, J.; Schwartz, L.M.; Steller, H.; Nambu, J.R. Cooperative functions of the reaper and head involution defective genes in the programmed cell death of Drosophila central nervous system midline cells. Proc. Natl. Acad. Sci. USA 1997, 94, 5131–5136. [Google Scholar] [CrossRef] [PubMed]
- Tissot, M.; Stocker, R.F. Metamorphosis in Drosophila and other insects: The fate of neurons throughout the stages. Prog. Neurobiol. 2000, 62, 89–111. [Google Scholar] [CrossRef]
- Kimura, K.I.; Truman, J.W. Postmetamorphic cell death in the nervous and muscular systems of Drosophila melanogaster. J. Neurosci. 1990, 10, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Hirai, K.; Togane, Y.; Akagawa, H.; Iwabuchi, K.; Tsujimura, H. Ecdysone-dependent and ecdysone-independent programmed cell death in the developing optic lobe of Drosophila. Dev. Biol. 2013, 374, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.; White, K. Cell death in development: Signaling pathways and core mechanisms. Semin. Cell Dev. Biol. 2015, 39, 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auld, V. Glia as mediators of growth cone guidance: Studies from insect nervous systems. Cell. Mol. Life Sci. 1999, 55, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.R. The Midline Glia of Drosophila: A molecular genetic model for the developmental functions of Glia. Prog. Neurobiol. 2000, 62, 475–508. [Google Scholar] [CrossRef]
- Sonnenfeld, M.J.; Jacobs, J.R. Apoptosis of the midline glia during Drosophila embryogenesis: A correlation with axon contact. Development 1995, 121, 569–578. [Google Scholar] [PubMed]
- Seeger, M.; Tear, G.; Ferres-Marco, D.; Goodman, C.S. Mutations affecting growth cone guidance in Drosophila: Genes necessary for guidance toward or away from the midline. Neuron 1993, 10, 409–426. [Google Scholar] [CrossRef]
- Bergmann, A.; Tugentman, M.; Shilo, B.Z.; Steller, H. Regulation of cell number by MAPK-dependent control of apoptosis: A mechanism for trophic survival signaling. Dev. Cell 2002, 2, 159–170. [Google Scholar] [CrossRef]
- Stork, T.; Thomas, S.; Rodrigues, F.; Silies, M.; Naffin, E.; Wenderdel, S.; Klambt, C. Drosophila Neurexin IV stabilizes neuron-glia interactions at the CNS midline by binding to Wrapper. Development 2009, 136, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.R.; Banerjee, S.; Blauth, K.; Rogers, S.L.; Bhat, M.A.; Crews, S.T. Neurexin IV and Wrapper interactions mediate Drosophila midline glial migration and axonal ensheathment. Development 2009, 136, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.R.; Pearson, J.C.; Crews, S.T. Time-lapse imaging reveals stereotypical patterns of Drosophila midline glial migration. Dev. Biol. 2012, 361, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Aso, Y.; Grübel, K.; Busch, S.; Friedrich, A.B.; Siwanowicz, I.; Tanimoto, H. The mushroom body of adult Drosophila characterized by GAL4 drivers. J. Neurogenet. 2009, 23, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Kunz, T.; Kraft, K.F.; Technau, G.M.; Urbach, R. Origin of Drosophila mushroom body neuroblasts and generation of divergent embryonic lineages. Development 2012, 139, 2510–2522. [Google Scholar] [CrossRef] [PubMed]
- Siegrist, S.E.; Haque, N.S.; Chen, C.H.; Hay, B.A.; Hariharan, I.K. Inactivation of Both foxo and reaper Promotes Long-Term Adult Neurogenesis in Drosophila. Curr. Biol. 2010, 20, 643–648. [Google Scholar] [CrossRef] [PubMed]
- King, R.C. Ovarian Development in Drosophila Melanogaster; Academic Press: New York, NY, USA, 1970. [Google Scholar]
- Spradling, A.C. Developmental genetics of oogenesis. In The Development of Drosophila Melanogaster; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1993; pp. 1–70. ISBN 978-087969899-7. [Google Scholar]
- Cooley, L.; Verheyen, E.; Ayers, K. chickadee encodes a profilin required for intercellular cytoplasm transport during Drosophila oogenesis. Cell 1992, 69, 173–184. [Google Scholar] [CrossRef]
- Guild, G.M.; Connelly, P.S.; Shaw, M.K.; Tilney, L.G. Actin filament cables in Drosophila nurse cells are composed of modules that slide passively past one another during dumping. J. Cell Biol. 1997, 138, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Hudson, A.M.; Cooley, L. Understanding the Function of Actin-Binding Proteins through Genetic Analysis of Drosophila Oogenesis. Annu. Rev. Genet. 2002, 36, 455–488. [Google Scholar] [CrossRef] [PubMed]
- McCall, K.; Steller, H. Requirement for DCP-1 caspase during Drosophila oogenesis. Science 1998, 279, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Nezis, I.P.; Stravopodis, D.J.; Papassideri, I.; Robert-Nicoud, M.; Margaritis, L.H. Stage-specific apoptotic patterns during Drosophila oogenesis. Eur. J. Cell Biol. 2000, 79, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Foley, K.; Cooley, L. Apoptosis in late stage Drosophila nurse cells does not require genes within the H99 deficiency. Development 1998, 125, 1075–1082. [Google Scholar] [PubMed]
- Peterson, J.S.; Barkett, M.; McCall, K. Stage-specific regulation of caspase activity in Drosophila oogenesis. Dev. Biol. 2003, 260, 113–123. [Google Scholar] [CrossRef]
- Baum, J.S.; Arama, E.; Steller, H.; McCall, K. The Drosophila caspases Strica and Dronc function redundantly in programmed cell death during oogenesis. Cell Death Differ. 2007, 14, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.S.; McCall, K. Combined inhibition of autophagy and caspases fails to prevent developmental nurse cell death in the Drosophila melanogaster ovary. PLoS ONE 2013, 8, e76046. [Google Scholar] [CrossRef] [PubMed]
- Horne-Badovinac, S.; Bilder, D. Mass transit: Epithelial morphogenesis in the Drosophila egg chamber. Dev. Dyn. 2005, 232, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Timmons, A.K.; Mondragon, A.A.; Schenkel, C.E.; Yalonetskaya, A.; Taylor, J.D.; Moynihan, K.E.; Etchegaray, J.I.; Meehan, T.L.; McCall, K. Phagocytosis genes nonautonomously promote developmental cell death in the Drosophila ovary. Proc. Natl. Acad. Sci. USA 2016, 113, E1246–E1255. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, V.; Taddei, C.; Gargiulo, G. Apoptosis of nurse cells at the late stages of oogenesis of Drosophila melanogaster. Dev. Genes Evol. 1998, 208, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Timmons, A.K.; Mondragon, A.A.; Meehan, T.L.; McCall, K. Control of non-apoptotic nurse cell death by engulfment genes in Drosophila. Fly (Austin) 2017, 11, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Santoso, C.S.; Meehan, T.L.; Peterson, J.S.; Cedano, T.M.; Turlo, C.V.; McCall, K. The ABC Transporter Eato Promotes Cell Clearance in the Drosophila melanogaster Ovary. G3 (Bethesda) 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Bass, B.P.; Tanner, E.A.; Mateos San Martín, D.; Blute, T.; Kinser, R.D.; Dolph, P.J.; McCall, K. Cell-autonomous requirement for DNaseII in nonapoptotic cell death. Cell Death Differ. 2009, 16, 1362–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, Y.; Fujitani, K.; Kurihara, J.; Usui-aoki, K.; Shimoda, L.; Suzuki, K.; Sezaki, M.; Sano, Y.; Ueda, R.; Awano, W.; et al. Mutations in the Novel Membrane Protein Spinster Interfere with Programmed Cell Death and Cause Neural Degeneration in Drosophila melanogaster. Mol. Cell. Biol. 2001, 21, 3775–3788. [Google Scholar] [CrossRef] [PubMed]
- Mondragon, A.; Yalonetskaya, A.; Oretga, A.; Zhang, Y.; Naranjo, O.; Elguero, J.; Chung, W.-S.; McCall, K. Lysosomal machinery drives extracellular acidification to direct non-apoptotic cell death. Under Review.
- Fuller, M.T. Spermatogenesis in Drosophila. In The Development of Drosophila melanogaster; Bate, M., Arias, A.M., Eds.; Cold Spring Harbor Lab Press: Cold Spring Harbor, NY, USA, 1993. [Google Scholar]
- Yacobi-Sharon, K.; Namdar, Y.; Arama, E. Alternative germ cell death pathway in Drosophila involves HtrA2/Omi, lysosomes, and a caspase-9 counterpart. Dev. Cell 2013, 25, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Arama, E.; Agapite, J.; Steller, H. Caspase activity and a specific cytochrome C are required for sperm differentiation in Drosophila. Dev. Cell 2003, 4, 687–697. [Google Scholar] [CrossRef]
- Napoletano, F.; Gibert, B.; Yacobi-Sharon, K.; Vincent, S.; Favrot, C.; Mehlen, P.; Girard, V.; Teil, M.; Chatelain, G.; Walter, L.; et al. P53-Dependent Programmed Necrosis Controls Germ Cell Homeostasis During Spermatogenesis. PLoS Genet. 2017, 13, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Drummond-Barbosa, D.; Spradling, A.C. Stem cells and their progeny respond to nutritional changes during Drosophila oogenesis. Dev. Biol. 2001, 231, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Kacsoh, B.Z.; Bozler, J.; Ramaswami, M.; Bosco, G. Social communication of predator-induced changes in Drosophila behavior and germ line physiology. eLife 2015. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, V.K.; Timmons, A.K.; McCall, K. Diversity of cell death pathways: Insight from the fly ovary. Trends Cell Biol. 2013, 23, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Baehrecke, E.H. Steroid regulation of programmed cell death during Drosophila development. Cell Death Differ. 2000, 7, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.A.; Kuo, Y.M.; Sun, Y.H.; Beckendorf, S.K. The Drosophila Pax gene eye gone is required for embryonic salivary duct development. Development 1998, 125, 4163–4174. [Google Scholar] [PubMed]
- Andrew, D.J.; Henderson, K.D.; Seshaiah, P. Salivary gland development in Drosophila melanogaster. Mech. Dev. 2000, 92, 5–17. [Google Scholar] [CrossRef]
- Fraenkel, G.; Brookes, V.J. The process by which the puparia of many species of flies become fixed to a substrate. Biol. Bull. 1953, 105, 442–449. [Google Scholar] [CrossRef]
- Richards, G. The radioimmune assay of ecdysteroid titres in Drosophila melanogaster. Mol. Cell. Endocrinol. 1981, 21, 181–197. [Google Scholar] [CrossRef]
- Yamanaka, N.; Rewitz, K.F.; O’Connor, M.B. Ecdysone Control of Developmental Transitions: Lessons from Drosophila Research. Annu. Rev. Entomol. 2013, 58, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Koelle, M.R.; Talbot, W.S.; Segraves, W.A.; Bender, M.T.; Cherbas, P.; Hogness, D.S. The Drosophila EcR gene encodes an ecdysone receptor, a new member of the steroid receptor superfamily. Cell 1991, 67, 59–77. [Google Scholar] [CrossRef]
- Thomas, H.E.; Stunnenberg, H.G.; Steward, A.F. Heterodimerization of the Drosophila ecdysone receptor with retinoid X receptor and ultraspiracle. Nature 1993, 362, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.-P.; Segraves, W.A.; Oro, A.E.; McKeown, M.; Evans, R.M. Drosophila ultraspiracle modulates ecdysone receptor function via heterodimer formation. Cell 1992, 71, 63–72. [Google Scholar] [CrossRef]
- Oro, A.E.; McKeown, M.; Evans, R.M. Relationship between the product of the Drosophila ultraspiracle locus and the vertebrate retinoid X receptor. Nature 1990, 347, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Baehrecke, E.H.; Thummel, C.S. The Drosophila E93 gene from the 93F early puff displays stage- and tissue-specific regulation by 20-hyroxyecdysone. Dev. Biol. 1995, 171, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Broadus, J.; McCabe, J.R.; Endrizzi, B.; Thummel, C.S.; Woodard, C.T. The Drosophila FTZ-F1 Orphan Nuclear Receptor Provides Competence for Stage-Specific Responses to the Steroid Hormone Ecdysone. Mol. Cell 1999, 3, 143–149. [Google Scholar] [CrossRef]
- Woodard, C.T.; Baehrecke, E.H.; Thummel, C.S. A molecular mechanism for the stage specificity of the Drosophila prepupal genetic response to ecdysone. Cell 1994, 79, 607–615. [Google Scholar] [CrossRef]
- Jiang, C.; Baehrecke, E.H.; Thummel, C.S. Steroid regulated programmed cell death during Drosophila metamorphosis. Development 1997, 124, 4673–4683. [Google Scholar] [PubMed]
- Martin, D.N.; Baehrecke, E.H. Caspases function in autophagic programmed cell death in Drosophila. Development 2004, 131, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Von Gaudecker, B.; Schmale, E.-M. Substrate-Histochemical Investigations and Ultrahistochemical Demonstrations of Acid Phosphatase in Larval and Prepupal Salivary Glands of Drosophila melanogaster; Springer: New York, NY, USA, 1974; Volume 155. [Google Scholar]
- Lee, C.-Y.; Baehrecke, E.H. Steroid regulation of autophagic programmed cell death during development. Development 2001, 1443–1455. [Google Scholar] [CrossRef]
- Jiang, C.; Lamblin, A.-F.J.; Steller, H.; Thummel, C.S. A steroid-triggered transcriptional hierarchy controls salivary gland cell death during Drosophila metamorphosis. Mol. Cell 2000, 5, 445–455. [Google Scholar] [CrossRef]
- Lee, C.-Y.; Wendel, D.P.; Reid, P.; Lam, G.; Thummel, C.S.; Baehrecke, E.H. E93 Directs Steroid-Triggered Programmed Cell Death in Drosophila. Mol. Cell 2000, 6, 433–443. [Google Scholar] [CrossRef]
- Gorski, S.M.; Chittaranjan, S.; Pleasance, E.D.; Freeman, J.D.; Anderson, C.L.; Varhol, R.J.; Coughlin, S.M.; Zuyderduyn, S.D.; Jones, S.J.M.; Marra, M.A. A SAGE approach to discovery of genes involved in autophagic cell death. Curr. Biol. 2003, 13, 358–363. [Google Scholar] [CrossRef]
- McPhee, C.K.; Balgley, B.M.; Nelson, C.; Hill, J.H.; Batlevi, Y.; Fang, X.; Lee, C.S.; Baehrecke, E.H. Identification of factors that function in Drosophila salivary gland cell death during development using proteomics. Cell Death Differ. 2013, 20, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-Y.; Clough, E.A.; Yellon, P.; Teslovich, T.M.; Stephan, D.A.; Baehrecke, E.H. Genome-wide analyses of steroid-and radiation-triggered programmed cell death in Drosophila. Curr. Biol. 2003, 13, 350–357. [Google Scholar] [CrossRef]
- Denton, D.; Nicolson, S.; Kumar, S. Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ. 2012, 19, 87–95. [Google Scholar] [CrossRef] [PubMed]
- McPhee, C.K.; Logan, M.A.; Freeman, M.R.; Baehrecke, E.H. Activation of autophagy during cell death requires the engulfment receptor Draper. Nature 2010, 465, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Rodrigues, F.S.L.M.; Kary, C.; Contet, A.; Logan, M.; Baxter, R.H.G.; Wood, W.; Baehrecke, E.H. Complement-Related Regulates Autophagy in Neighboring Cells. Cell 2017, 170, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Frawley, L.E.; Orr-Weaver, T.L. Ploidy. Curr. Biol. 2015, 25, R353–R358. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, B.; Miguel-Aliaga, I. The Digestive Tract of Drosophila melanogaster. Annu. Rev. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-Y.; Cooksey, B.A.K.; Baehrecke, E.H. Steroid Regulation of Midgut Cell Death during Drosophila Development. Dev. Biol. 2002, 250, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Chang, T.-K.; Nicolson, S.; Shravage, B.; Simin, R.; Baehrecke, E.H.; Kumar, S. Relationship between growth arrest and autophagy in midgut programmed cell death in Drosophila. Cell Death Differ. 2012, 19, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Shravage, B.; Simin, R.; Baehrecke, E.H.; Kumar, S. Larval midgut destruction in Drosophila: Not dependent on caspases but suppressed by the loss of autophagy. Autophagy 2010, 6, 163. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-K.; Shravage, B.V.; Hayes, S.D.; Powers, C.M.; Simin, R.T.; Harper, J.W.; Baehrecke, E.H. Uba1 functions in Atg7- and Atg3-independent autophagy. Nature 2013, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, D.; Xu, T.; Dayan, S.; Nicolson, S.; Kumar, S. Dpp regulates autophagy-dependent midgut removal and signals to block ecdysone production. Cell Death Differ. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Neuman, S.D.; Bashirullah, A. Tango7 regulates cortical activity of caspases during reaper-triggered changes in tissue elasticity. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Zirin, J.; Cheng, D.; Dhanyasi, N.; Cho, J.; Dura, J.-M.; Vijayraghavan, K.; Perrimon, N. Ecdysone signaling at metamorphosis triggers apoptosis of Drosophila abdominal muscles. Dev. Biol. 2013, 383, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.I.; Kuranaga, E. Caspase-dependent non-apoptotic processes in development. Cell Death Differ. 2017, 24, 1422–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, J.; Broemer, M. Nerve-racking—Apoptotic and non-apoptotic roles of caspases in the nervous system of Drosophila. Eur. J. Neurosci. 2016, 44, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.C.; Lu, Y.; Shaham, S. A Morphologically Conserved Nonapoptotic Program Promotes Linker Cell Death in Caenorhabditis elegans. Dev. Cell 2007, 12, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Kutscher, L.M.; Shaham, S. Non-apoptotic cell death in animal development. Cell Death Differ. 2017, 24, 1326–1336. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yalonetskaya, A.; Mondragon, A.A.; Elguero, J.; McCall, K. I Spy in the Developing Fly a Multitude of Ways to Die. J. Dev. Biol. 2018, 6, 26. https://doi.org/10.3390/jdb6040026
Yalonetskaya A, Mondragon AA, Elguero J, McCall K. I Spy in the Developing Fly a Multitude of Ways to Die. Journal of Developmental Biology. 2018; 6(4):26. https://doi.org/10.3390/jdb6040026
Chicago/Turabian StyleYalonetskaya, Alla, Albert A. Mondragon, Johnny Elguero, and Kimberly McCall. 2018. "I Spy in the Developing Fly a Multitude of Ways to Die" Journal of Developmental Biology 6, no. 4: 26. https://doi.org/10.3390/jdb6040026
APA StyleYalonetskaya, A., Mondragon, A. A., Elguero, J., & McCall, K. (2018). I Spy in the Developing Fly a Multitude of Ways to Die. Journal of Developmental Biology, 6(4), 26. https://doi.org/10.3390/jdb6040026