1. Introduction
Electroporation is an effective non-viral technique that breaches the cell membrane [
1] and continues to be accepted as a delivery system at the cellular level for many molecules like DNA, dsRNA, siRNA, mRNA, proteins, peptides, antibodies and drugs [
2,
3,
4,
5,
6,
7,
8,
9,
10,
11]. Since its inception, various techniques have been attempted to improve the transfection efficiencies in various cell types by optimizing the transfection parameters, including voltage, pulse duration, number of pulses and pulse interval. In the present study, the fluorescence expression property of green fluorescent protein (GFP) was explored for evaluation and assessment of transfection efficiency. GFP acts as an energy-transfer acceptor and transduces the blue chemiluminescence of the protein aequorin into green fluorescent light by energy transfer. For the first time, we optimized electroporation parameters and demonstrated delivery of DNA/RNA across the zona pellucida (ZP), along with increased molecular uptake, while maintaining cell viability.
Reproduction is an essential progressive process for the survival and conservation of all living organisms. In females, it requires a developmentally competent oocyte that develops within the follicle inside the ovary. Ovarian follicle development commences with the formation of primordial follicles, which further advances to subsequent stages by changing the shape and expanding the granulosa cell network around the oocyte. During this development, a thick layer of glycoprotein, namely the zona pellicuda (ZP), takes up space between the oocyte and granulosa cells. The ZP surrounds oocytes and embryos until the early blastocyst stage, after which its hatching occurs to allow the embryo to implant [
12]. Thus, the ZP attributes oogenesis, fertilization and preimplantation development [
13,
14]. Apart from this, the ZP acts as a natural barrier against untoward chemical and biological agents such as viruses [
6]. However, this barrier is capable of reducing the efficacy of available somatic cell transfection methods that aid in deciphering the function of genes and proteins and hence precluding scientific interventions.
In order to gain insights into biological processes and molecular functions during folliculogenesis, oocyte maturation as well as early embryonic development, it is important to alter gene expression either at the transcriptional or translational level in target cells. For this, a microinjection technique has been utilized to deliver foreign genes into oocytes, zygotes and embryos by traversing the ZP layer. Even though microinjection is useful, it is difficult, tedious, low in throughput, and requires expensive equipment. Other gene delivery methods like liposomal, cationic polymer-based, virus-mediated and electroporation have also been attempted but with limited success [
15]. Some studies have successfully transfected exogenous vector DNAs via electroporation in oocytes and zygotes but this required removal or weakening of the ZP with harsh acid Tyrode’s solution [
16,
17]. In view of the points mentioned above, this study was carried out to find out whether in vitro electroporation alone can be sufficient to deliver foreign genes to intact follicles, oocytes, and embryos without the ZP weakening treatments.
2. Materials and Methods
2.1. Follicle Collection
Follicles were collected from ovaries of 8-week female Swiss mice aseptically followed by digestion in collagenase (1 mg/mL) and DNase-I (0.2 mg/mL) in Leibovitz-15 (L-15) medium at 37 °C for 20 min. Repeat pipetting was utilized every 8–10 min to facilitate mechanical dissociation of each follicle. The criteria adopted for selection of preantral follicles was determined by their morphology, i.e., having centralized round oocytes surrounded by 2–3 layers of granulosa cells. Overall, it should be a structurally undamaged round follicular structure with a diameter of 100–120 μm [
18]. Further, these designated follicles were mixed and distributed into randomized groups for the in vitro transfection experiments.
2.2. In Vitro Culture of Mice Preantral Follicles and Their Ovulation Induction
Following transfection via electroporation, follicles were grown in α-MEM media supplemented with 5% FBS, 8 μg/mL insulin,10 mIU/mL LH and 100 mIU/mL FSH, 100 μg/mL penicillin and 50 μg/mL streptomycin as drop culture (10 follicles/30 µL medium drop) in 35-mm culture dishes under mineral oil. After 48 h of culture, these follicles were washed three times in phosphate buffer saline (PBS) and then fixed for the assessment of transfection efficiency experiment. For the in vitro oocyte maturation experiment, follicles were maintained at 37 °C with 5% CO
2 for 12 days. Every second day, half of the medium was replenished with the new medium. Following 12 days of culture, the follicles were further allowed to mature in the medium above containing 1.5 IU/mL human chorionic gonadotrophin (hCG) and 5 ng/mL epidermal growth factor (EGF) for 16 h [
19]. The cumulus-oocyte complexes (COC) were denuded using hyaluronidase enzyme and the oocytes were monitored under an inverted microscope (Zeiss, Goettingen, Germany). These oocytes were further utilized for immuno-localization of maturation markers.
2.3. Oocyte and Embryo Collection
Mature (MII) oocytes and embryos were collected from 8–9-week-old super-ovulated swiss mice. The super-ovulation was induced by an injection of 5 IU pregnant mare serum gonadotropin (PMSG, Intervet, Milton Keynes, UK) and, after 48 h of PMSG injection, 5 IU human chorionic gonadotrophin (hCG, Intervet, UK) injection. COCs were collected from oviducts in mineral oil at 12–16 h after hCG injection and surrounding cumulus cells were digested by hyaluronidase. Cumulus cell free oocytes were collected and washed thrice with PBS with 1% bovine serum albumin (BSA) and used in the further experiment.
For embryo collection, super-ovulated female mice were caged with fertile male post hCG injection. Mating was secured by the presence or absence of vaginal plug and day zero of pregnancy was noted. Then, embryos were collected by flushing them out from the oviducts and uterine horn periodically at day two, three and four of gestation. The collected embryos were selected randomly for electroporation and apoptosis experiments. Mouse embryos collected on day 2 and 3 post coitum were classified as early (2–4 cell stage) and late (morula stage) embryos, respectively. The embryos collected on day 4 (blastocyst stage) were used as the control group in the TUNEL assay experiment.
2.4. Optimization of Electroporation Parameters in Follicle, Oocyte and Various Embryonic Stages
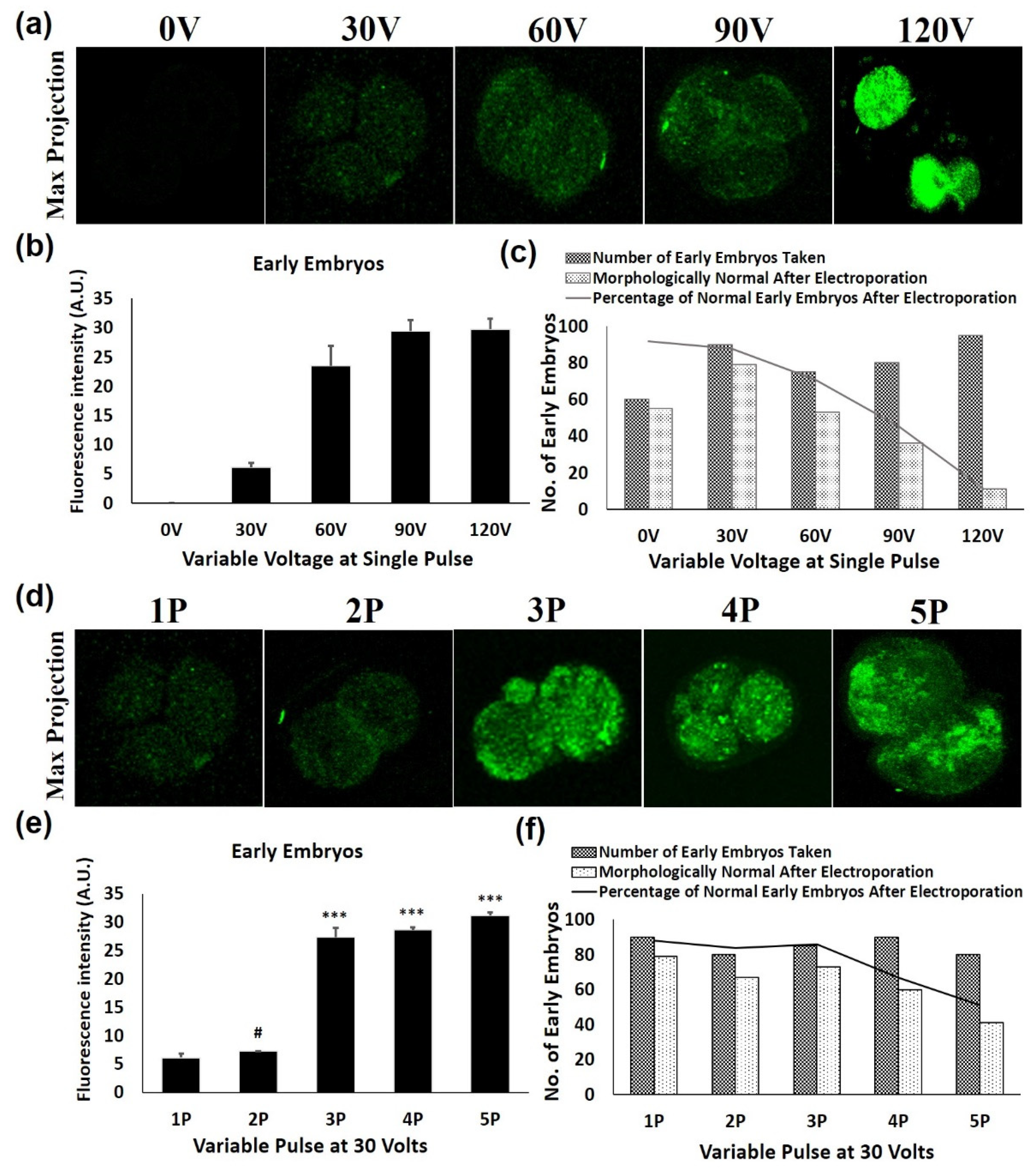
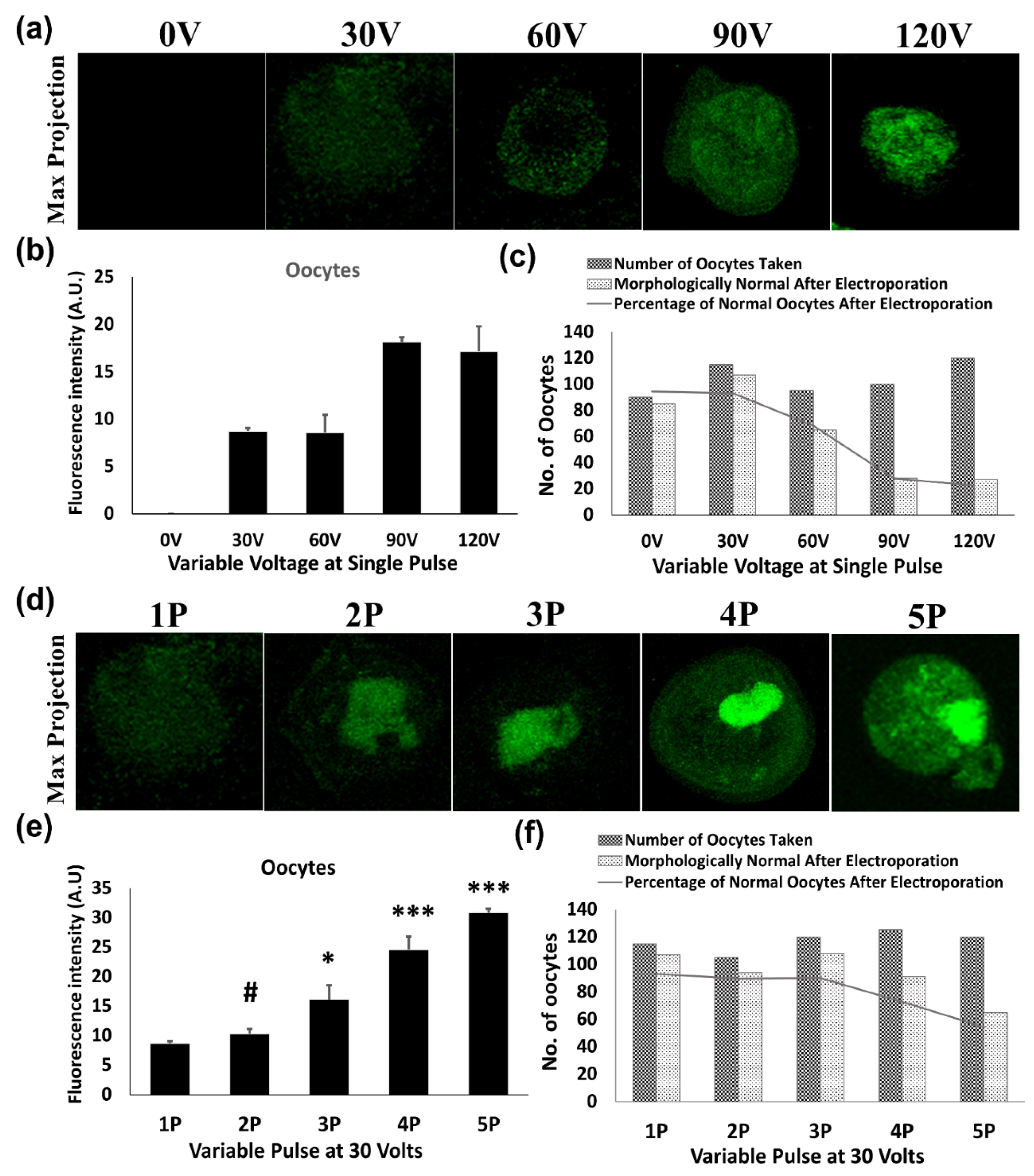
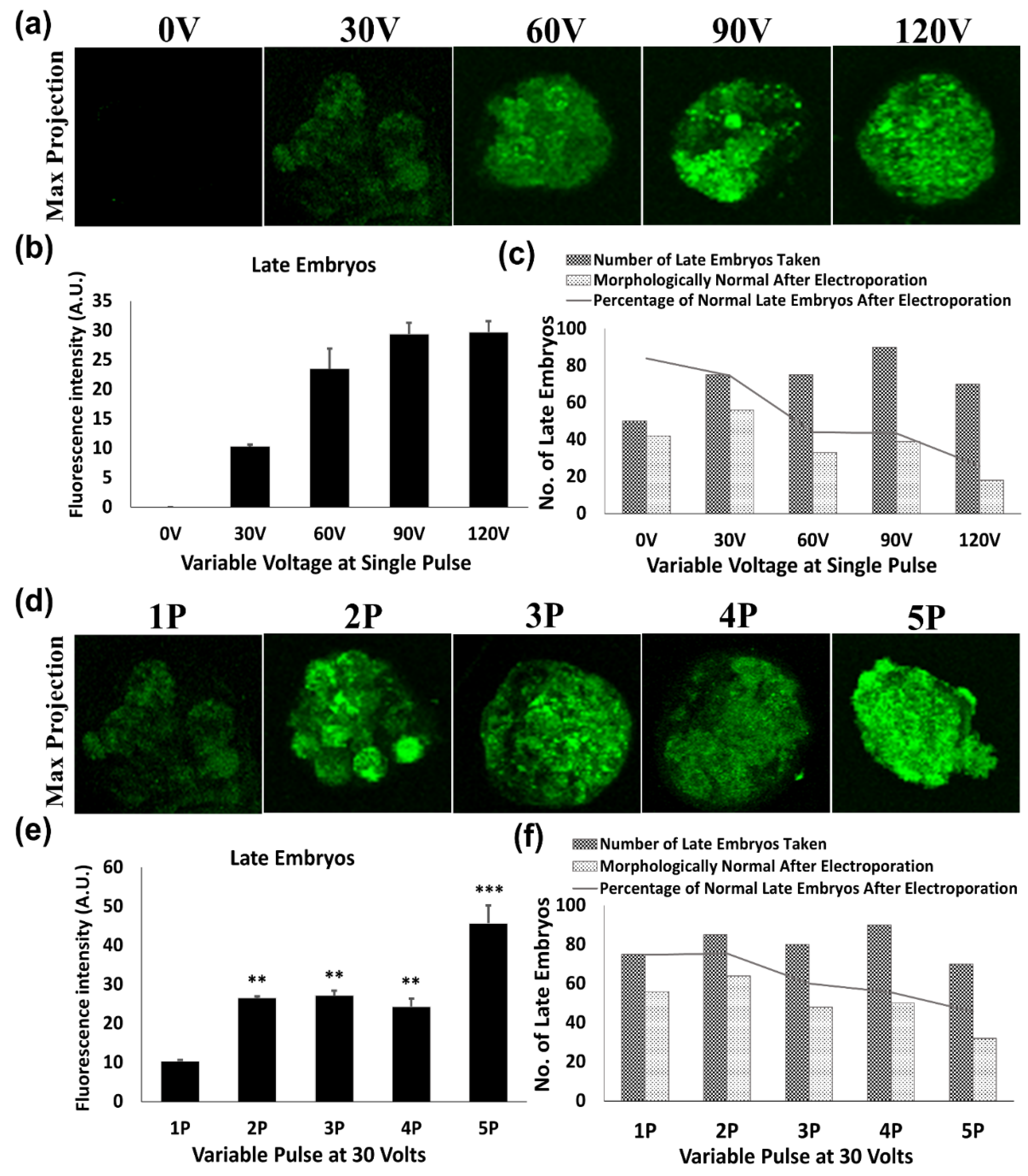
To measure the impact of voltage and pulse number on the transfection of green fluorescent protein-expressing construct (pEGFP-C1 Vector), 0.5 μg of purified plasmid was mixed into 20 μL of HEPES- buffered saline (150 mM NaCl and 20 mM Hepes) containing 30–80 cells from each cell type. The mixture was electroporated in the 1mm gap cuvettes with a single 1 millisecond (ms) pulse of 30, 60, 90 and 120 Volts (V) by Gene Pulser Mxcell System (Bio-rad Laboratories, Hercules, CA, USA). As per the results of a single pulse, 30 V was found to be optimal voltage with minimal effect on cellular morphology as well as on its development. Consequently, multiple pulses (2, 3, 4 and 5) of 1 ms at a gap of 10 s were evaluated in order to increase the transfection efficiency regardless of an uncompromised cellular developmental potential. Then, these follicles, oocytes and embryos were allowed to grow for 48 h in their respective culture media.
2.5. Assessment of Transfection Efficiency
Electroporated follicles, oocytes and embryos were cultured for 48 h, then fixed using 3.7% paraformaldehyde for 20 min at room temperature. Imaging of GFP expression was done using a confocal fluorescence microscope (Olympus, Southborough, MA, USA). Furthermore, GFP intensities of these transfects were measured by using ImageJ software (NIH, Bethesda, MD, USA). The experiments were done in triplicate and a minimum of four samples from each transfected group of the follicles, oocytes and embryos were observed for fluorescent intensities after transfection with their fluorescence intensities plotted as average intensity fluorescence intensities ± SEM.
2.6. Immunofluorescence of Oocyte Maturation Marker
Ovastacin (also known as sperm acrosomal SLIP1 binding protein, SAS1B), an oocyte maturation marker, was localized on in vitro matured oocytes grown from preantral follicles following electroporation [
19,
20]. The oocytes were collected at day 11/13 from culture, as well as after superovulation. Further, these oocytes were fixed with 3.7% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min and permeabilized by 0.1% Triton-X in PBS for 30 min at 37 °C. Subsequently, the permeabilized oocytes were blocked in 3% bovine serum albumin (BSA) in PBS for 30 min at 37 °C and then incubated with the anti-SAS1B primary antibody (1:200; received as a gift from UVA, Charlottesville) in PBS with 1.5% BSA overnight at 4 °C. Following incubation, oocytes were washed thrice in washing buffer (PBS containing 1% BSA) and incubated with anti-guinea pig IgG Cy3 labeled secondary antibody (1:500; Jackson Immuno Research, West Grove, PA, USA) in the dark for 1 h at 37 °C. The oocytes were further co-stained with DNA binding dye Hoechst 33342 (Invitrogen, Carlsbad, CA, USA) for 15 min at 37 °C. Then the oocytes were washed again three times in washing buffer before mounting them on a glass slide. The samples were observed under a confocal laser-scanning microscope (Zeiss, Goettingen, Germany).
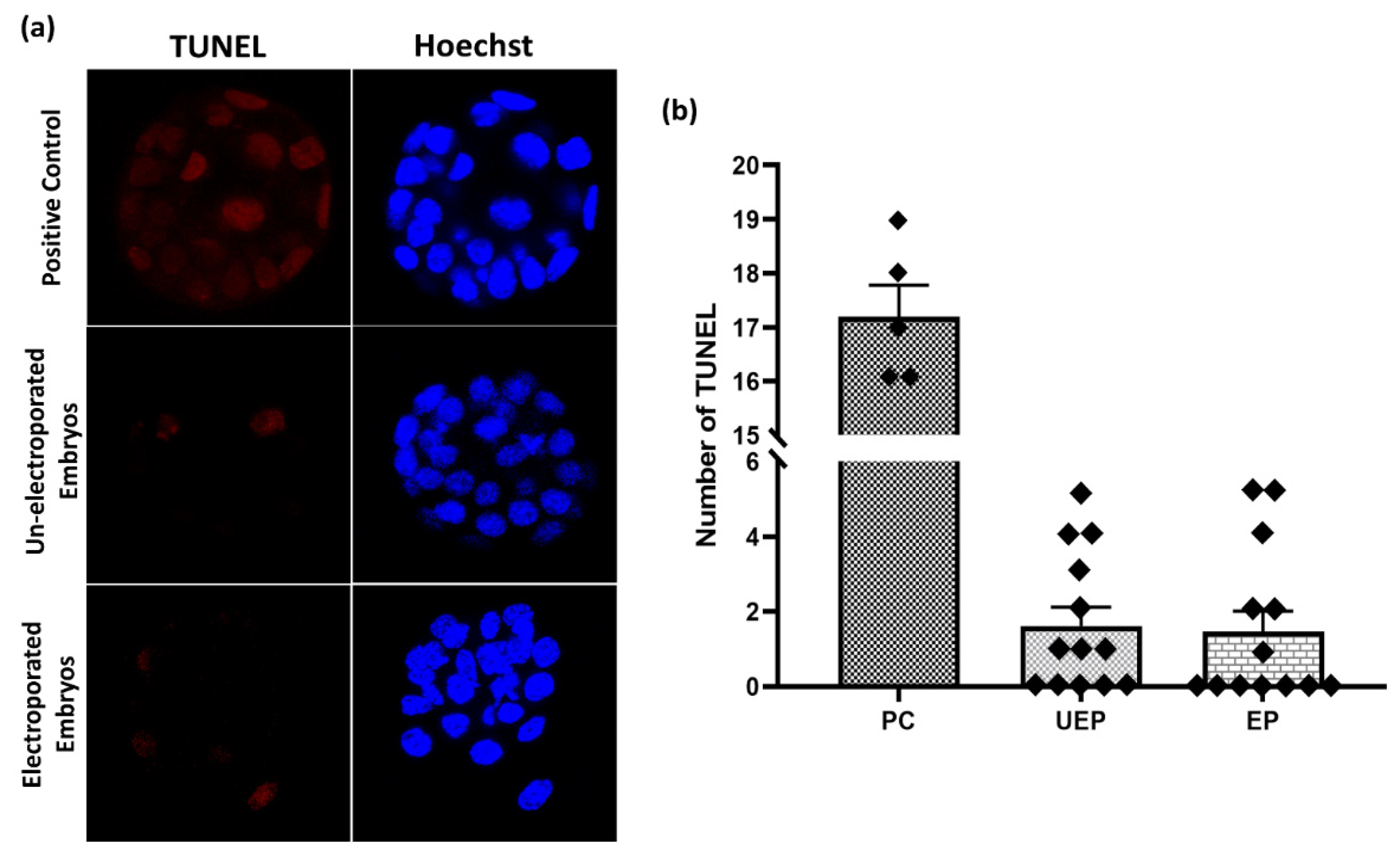
2.7. Assessment of Embryo Damage after Electroporation
A TUNEL (terminal deoxyuridine triphosphate nick end labelling) assay was performed to detect the embryo damage after electroporation by the Click-iTTM Plus TUNEL Assay for in situ Apoptosis Detection Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Briefly, fixed embryos were permeabilized by 0.1% Triton-X in PBS for 30 min at 37 °C. Before the terminal deoxynucleotidyl transferase (TdT) reaction initiation, positive control embryos were incubated with 1 unit of DNase I to induce DNA strand breaks for 30 min at 37 °C and washed with deionized water. Now these embryos were equilibrated with TdT reaction buffer for 10 min at 37 °C and further incubated in TdT reaction mixture for 1 h at 37 °C. Immediately after the TdT reaction, embryos were washed and incubated in the Click-iT™ Plus TUNEL reaction cocktail for 30 min in the dark at 37 °C. Hoechst 33342 solution was used to label the embryo nuclei for 15 min at 37 °C. The standard washing was done three times before each step with PBS containing 3% BSA, otherwise stated. Then the whole mount embryos were analyzed with confocal microscopy.
2.8. miRNA Transfection and Real-Time RT-PCR
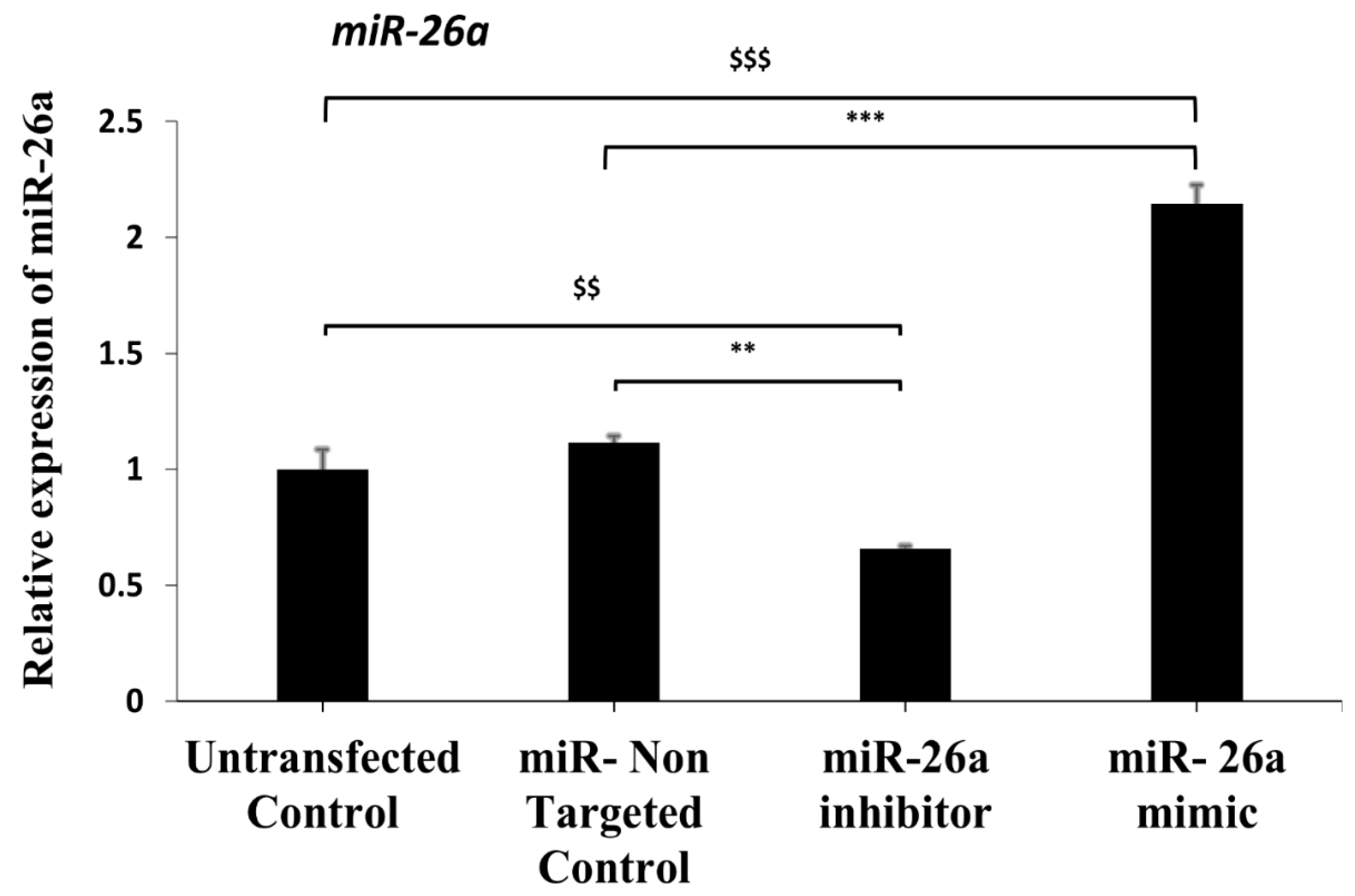
The miR-26a mimic, an inhibitor as well as non-targeted control (Qiagen, Valencia, CA, USA), was transfected in approximately 90–100 preantral follicles in each group using best-optimized electroporation conditions, i.e., 3 pulses at 30 V, and the experiment was repeated three times. For miRNA expression, total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA) from the pooled follicles after 48 h of culture. RNA samples were subjected to reverse transcription using the miScript II RT Kit (Qiagen;#218161), followed by quantitative real-time PCR with miScript SYBR Green PCR Kit (Qiagen; #218073) with miScript Primer assays (Qiagen; #218200) for miR-26a and RNU6-6P on the LightCycler 480 real-time PCR (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instruction. The reference miRNA RNU6-6P values were used for the normalization of miR-26a expression.
2.9. Statistical Analysis
Survival percentage of preantral follicles, oocytes, early and late embryos was calculated using pooled data from at least five independent experiments. These experimental results were analyzed through the χ2 test to compare the survival rate. One-way ANOVA followed by Newman–Keuls post-test was also performed for other comparative analysis. Overall data was expressed as mean ± SEM (standard error mean) and a p value less than 0.05 was considered statistically significant. GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA, USA) was employed for statistical analyses.
4. Discussion
An in vitro follicle culture system has become a vital tool to study folliculogenesis, oogenesis and early embryonic development. It serves as a perfect model to explore the function of genes and thus creates an opportunity to understand the regulatory mechanism of the development of a follicle to a growing embryo. In the course of folliculogenesis, the oocyte attains maturation and is surrounded by the zona pellicuda (ZP). The ZP is a protective glycoprotein coating that encircles the plasma membrane of oocytes and early embryos before implantation [
21]. This covering triggers the sperm to undergo acrosomal reaction and acts as a selective filter for them, only allowing sperm that have completed the acrosomal reaction. However, the ZP acts as a potential hindrance for transfection related studies in these cells [
22]. Regardless, the usage of electroporation for the alteration of genetic material in the cells is being adopted by various laboratories as it is a relatively economical, safe and faster technique for the delivery of exogenous DNA plasmids and RNA. However, fine-tuning of parameters like voltage, pulse amplitude and pulse number needs to be standardized every time to achieve better transfection efficiency [
23]. The transfer of foreign nucleic acid in oocytes and preimplantation embryos using electroporation was demonstrated successfully for the first time in 2002 [
3]. However this initial study demonstrated the electroporation of rhodamine-conjugated dextran in ZP-removed oocytes and zygotes, but the removal of ZP usually hampers in vitro growth in these cells and also reduces their in vivo survival if transferred in pseudo-pregnant foster mothers due to their adhesive property [
24,
25]. A diffuse signal pattern of GFP was observed throughout the cytoplasm in the case of weak transfection with a single pulse, whereas increased transfection efficiency with multiple pulses (
Figure 2b) promoted accumulation of GFP localization. Alternatively, acidic Tyrode’s solution was used to loosen the ZP for the transfection of these cells, which reasonably prevented the damage caused by electroporation. Subsequently, many studies achieved delivery of foreign genes to early embryos via electroporation by this zona loosening strategy [
16,
17]. However, application of caustic treatments is harmful and gradually compromises the developmental potential of embryos in vitro [
17]. Contrarily, techniques involving transfection in intact zygotes require specialized devices that are not widely available [
26,
27]. To overcome these concerns, the present study established an easy transfection method for ZP intact oocytes and embryos, which can be done with any ordinary electroporation equipment.
A series of conditions were optimized for highly efficacious transfection in intact mice follicles, oocytes and various embryonic stages through electroporation while maintaining a high survival rate as well as uncompromised developmental potential. Firstly, a higher voltage for the electroporation of these cells was applied, but it triggered a fall in the survival rate and also changed the overall morphology. Hence the idea of implementing a higher voltage in these cells was dropped and an increased number of pulses at a lower voltage was determined. On applying 30 V of varying pulses, a gradual increase in the electroporation was achieved with high survival rates. However, more than three consecutive pulses of 30 V compromised the survival rate in the follicles, oocytes, and early embryos. Accordingly, 3 pulses of 30 V of 1 ms each at an interval of 10 s were observed to be the best electroporation conditions for highly efficacious transfection.
In the case of late embryos (morulae stage), a non-significant change in electroporation efficiency was observed by increasing the pulses from 2 to 3. Furthermore, two pulses of 30 V increased survival up to 75%, whereas beyond 2 pulses of 30 V decreased the percentage of embryo survival, as well as further development to the subsequent blastocyst stage. This might be due to the zona thinning and the blastocyst expansion process during late embryonic stages prior to implantation [
28]. Moreover, the cell apoptosis assay was performed in order to validate the healthy embryonic development following electroporation. Apoptosis is a process by which defective cells are usually eliminated but it is not generally observed throughout normal embryonic development [
29]. The initiation of apoptosis by chemical or physical injury during preimplantation embryonic development could lead to deletion or damage of important cell lineages and hence affect the normal embryonic development that may lead to embryonic malformation or abortion [
17,
30]. The results of the TUNEL apoptosis assay showed no significant difference in the apoptotic nuclei number of blastocysts developed in vitro from electroporated embryos in contrast with normal un-electroporated blastocysts grown in vitro. To authenticate the TUNEL apoptosis assay, a positive control experiment was also designed simultaneously, which showed ample TUNEL-positive nuclei. Thus, suggesting that these parameters are safe for intact embryo electroporation and do not induce any embryo impairment after electroporation.
In addition, proper maturation of the preantral follicles after electroporation with the selected conditions was successfully evaluated. Previous reports have already documented the change in the staining pattern of ovastacin during oocyte maturation [
20]. As per the previously published results, in ovulated immature oocytes (germinal vesicle stage), ovastacin was observed throughout the ooplasm with enrichment at the oocyte periphery, whereas in the mature MII stage of oocytes, ovastacin concentrates in the microvillar domain of the oolemma antipodal to the nucleus. Hence, this marker was explored in our sequential experiments of in vitro oocyte maturation, and the observations were found to be similar during the in vitro transition of the germinal vesicle to mature MII oocyte, where ovastacin also re-oriented in the oolemma. Our results also showed successful manipulation of miRNA expression by transfection of the miR inhibitor and mimic in preantral follicles through electroporation. The expression of mmu-mir-26a showed a significant decrease in the miR-26a inhibitor-transfected follicles, while a considerable increase in the expression of mmu-mir-26a was observed in the miR-26a mimic-transfected follicles as compared to both vehicle control and non-targeted control.
5. Conclusions
In conclusion, this study offers an efficient transfection method using electroporation for intact mouse follicles that would help to provide a greater understanding during in vitro analysis of folliculogenesis. Concurrently, this transfection procedure is equally applicable for ZP enclosed oocytes and embryos and does not require the corrosive treatments for the weakening of ZP. Other advantages of this technique over other classical transfection methods are that it is simple, efficient, affordable, non-laborious and less toxic; at the same time, instantaneous co-delivery of multiple plasmids and molecular drugs is possible. Thus, these optimized conditions for electroporation can provide insights about the molecular mechanisms modulating folliculogenesis, oogenesis and embryogenesis.
Thus, these results validate the standardization of the electroporation technique for effective transfection and also opens a new vista for its potential application in assisted reproductive technologies (ART) in clinical settings.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}