
A Two-Temperature Open-Source CFD Model for Hypersonic Reacting Flows, Part One: Zero-Dimensional Analysis †

Abstract
:
1. Introduction
2. Methodology
2.1. Governing Equations
2.1.1. Two-Temperature Model
2.1.2. Non-Equilibrium Navier-Stokes-Fourier Equations
2.2. Chemistry Source Terms
2.2.1. Generalities
2.2.2. Chemistry-Vibration Coupling: The Park TTv Model
2.2.3. Chemistry-Vibration Coupling: The Coupled Vibration-Dissociation-Vibration Model
2.3. Implementation in OpenFOAM 2.3.0
3. Results and Discussion
3.1. Vibrational-Translational Relaxation of a Single-Species Gas
3.1.1. Case without Electronic Energy
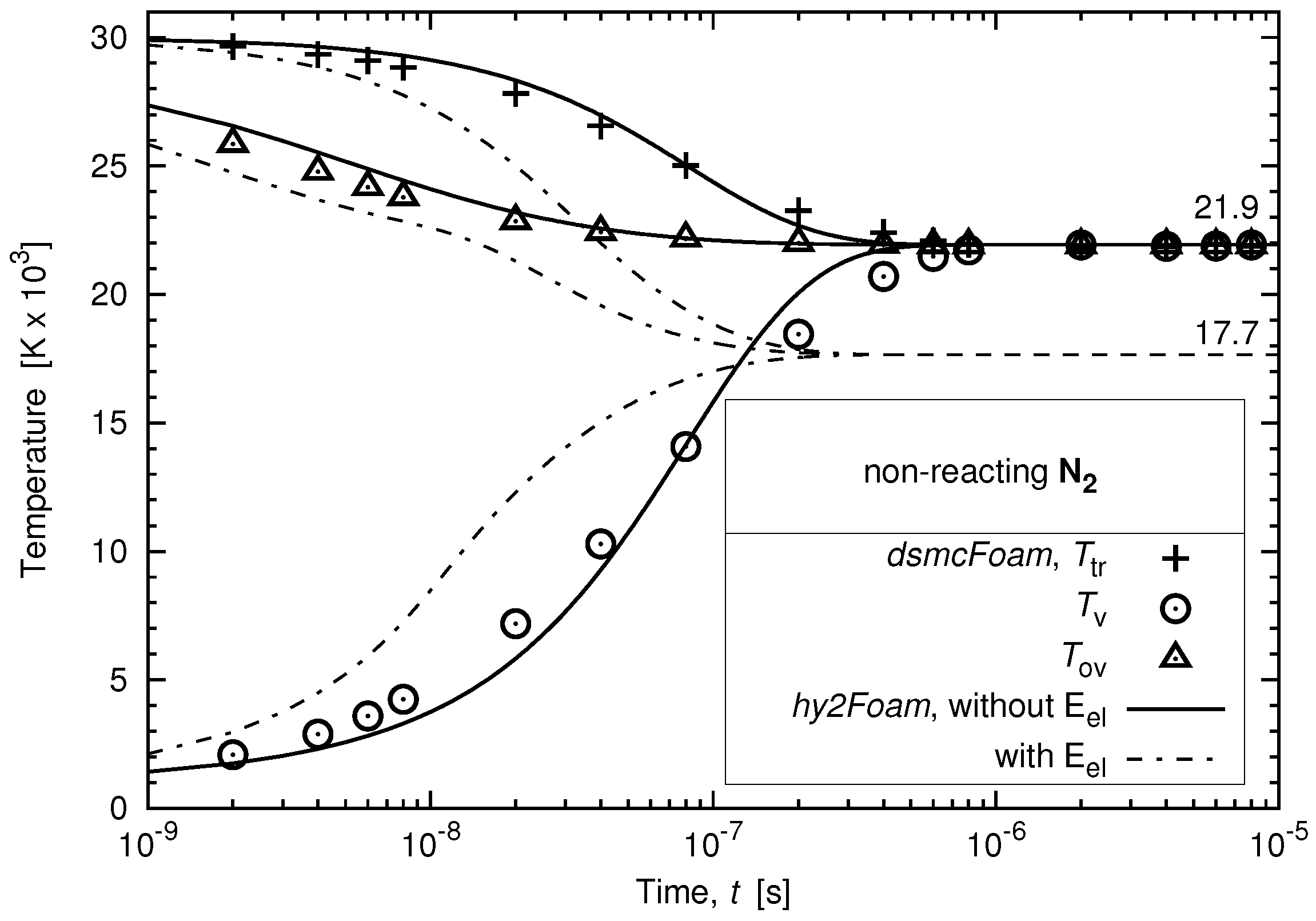
3.1.2. Case with Electronic Energy
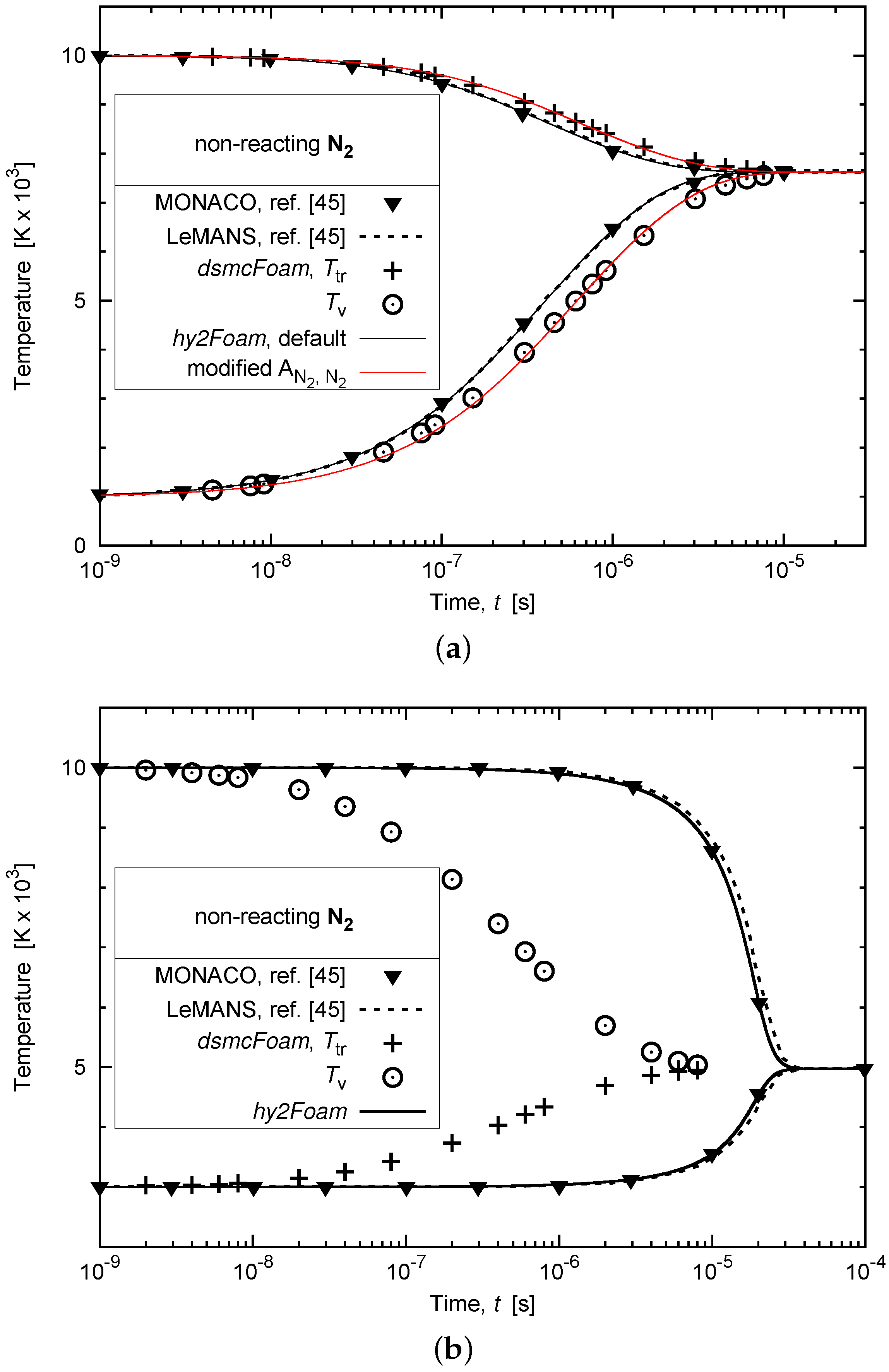
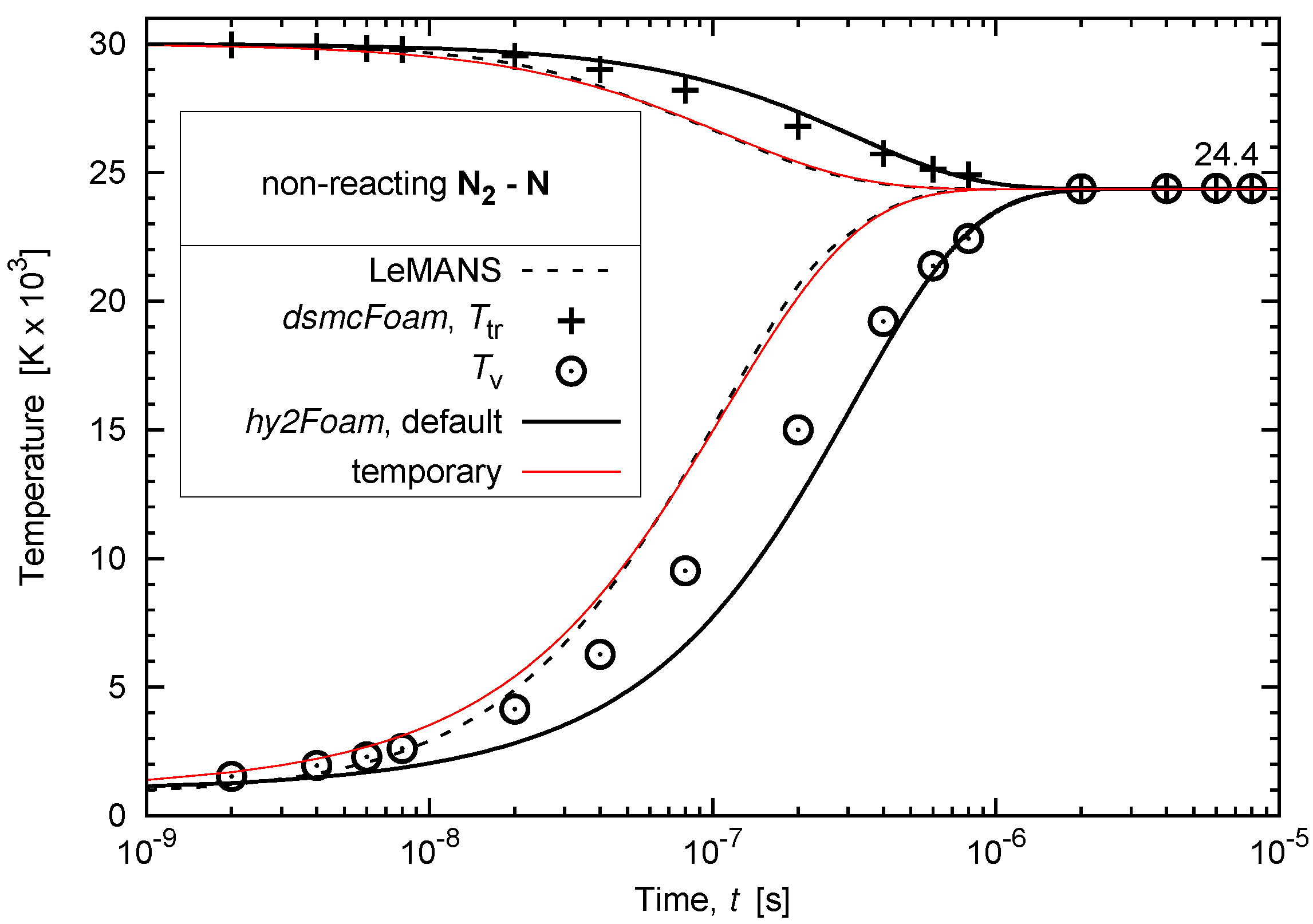
3.2. Vibrational-Translational Relaxation of a Non-Reacting Multi-Species Gas
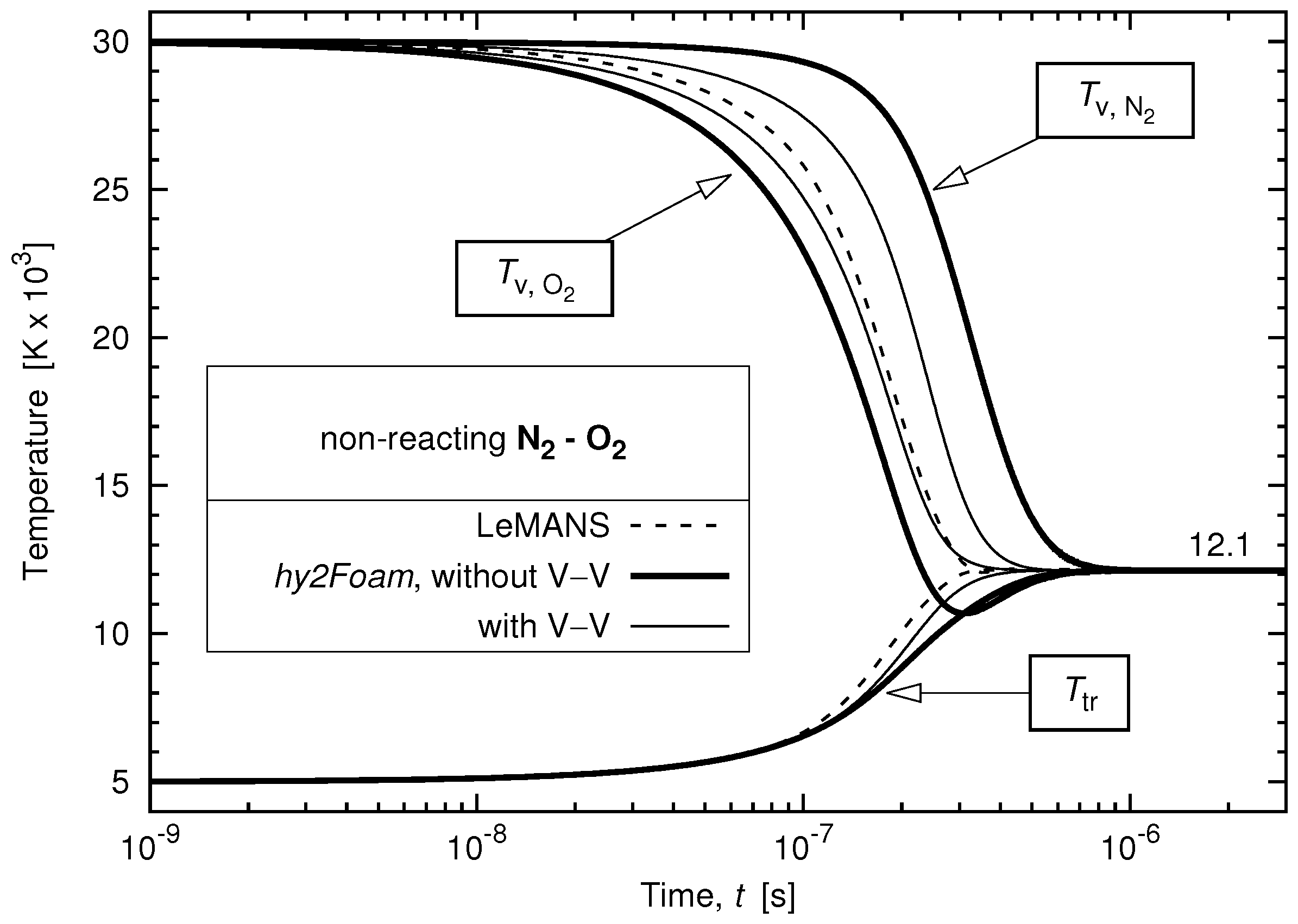
3.3. Vibrational-Translational and Vibrational-Vibrational Relaxations
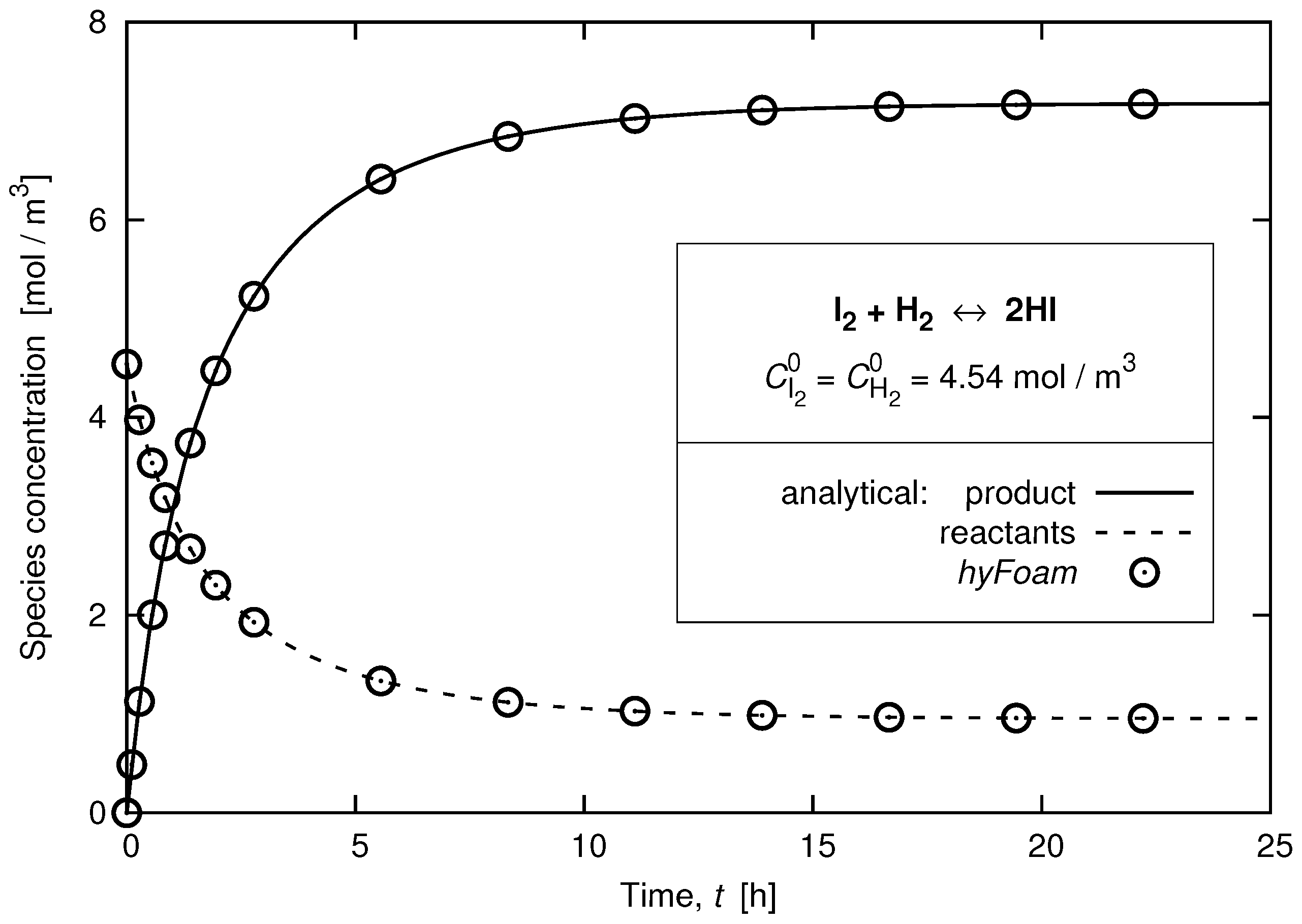
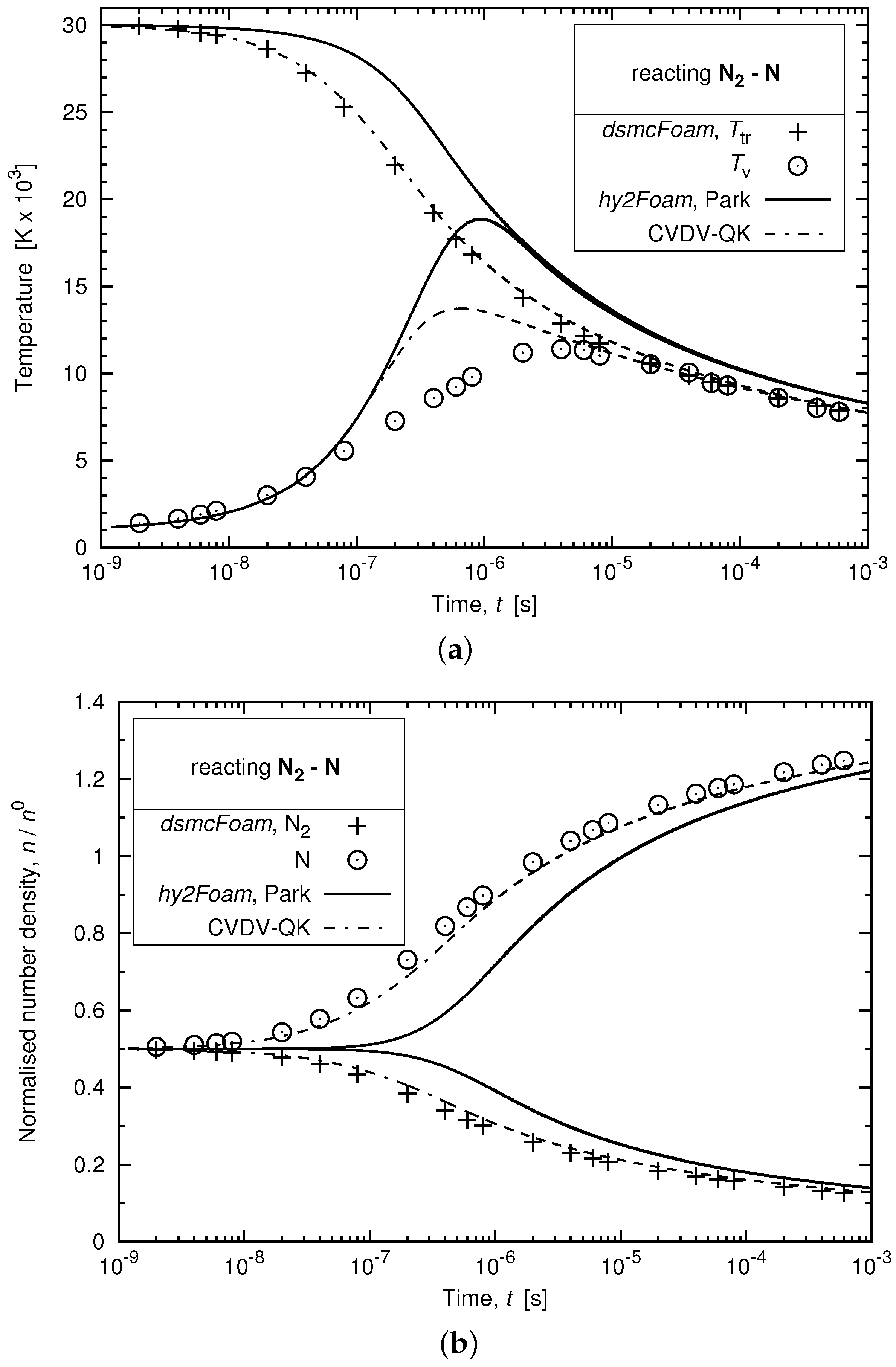
3.4. Relaxation of a Chemically-Reacting Mixture
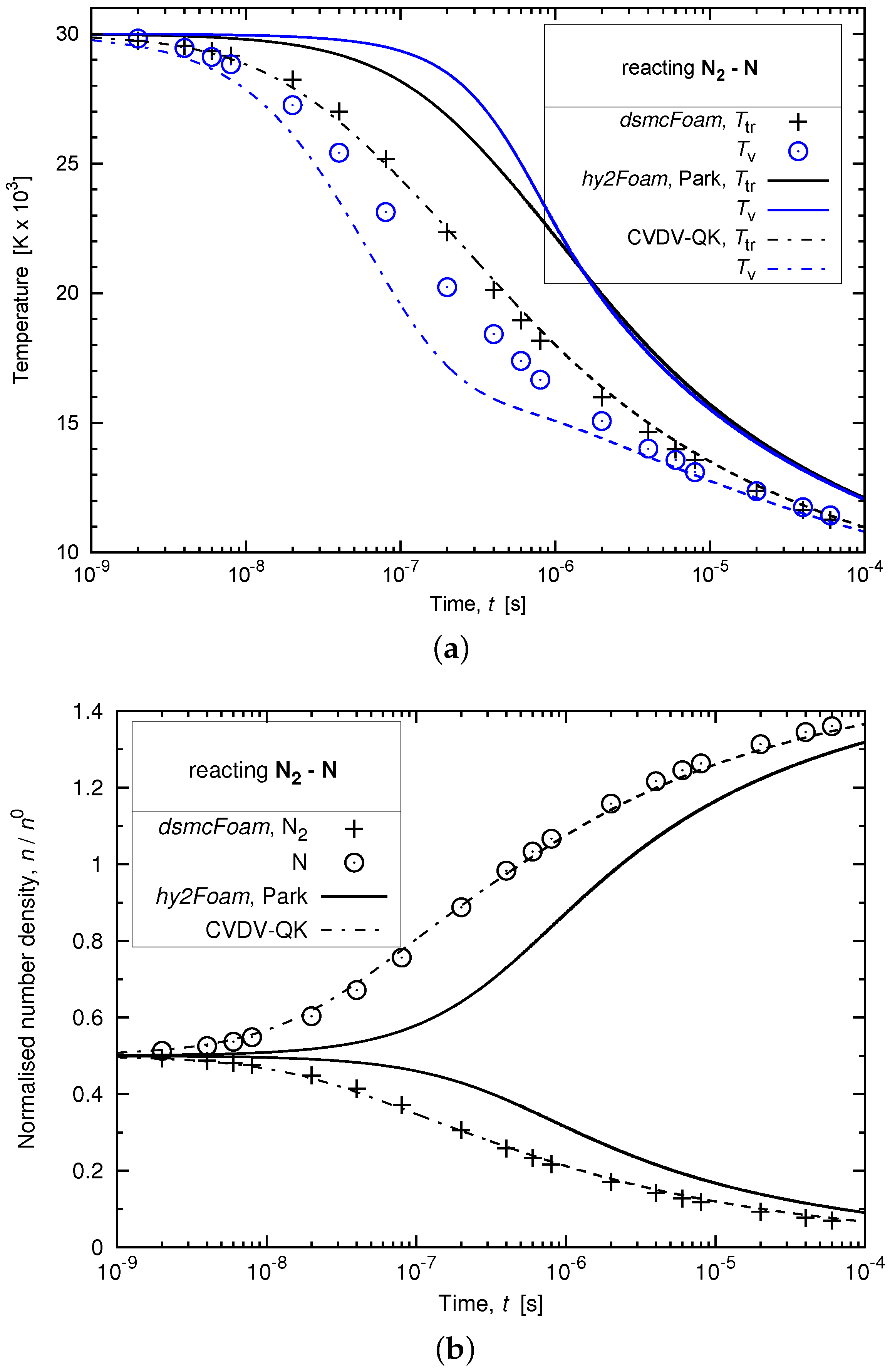
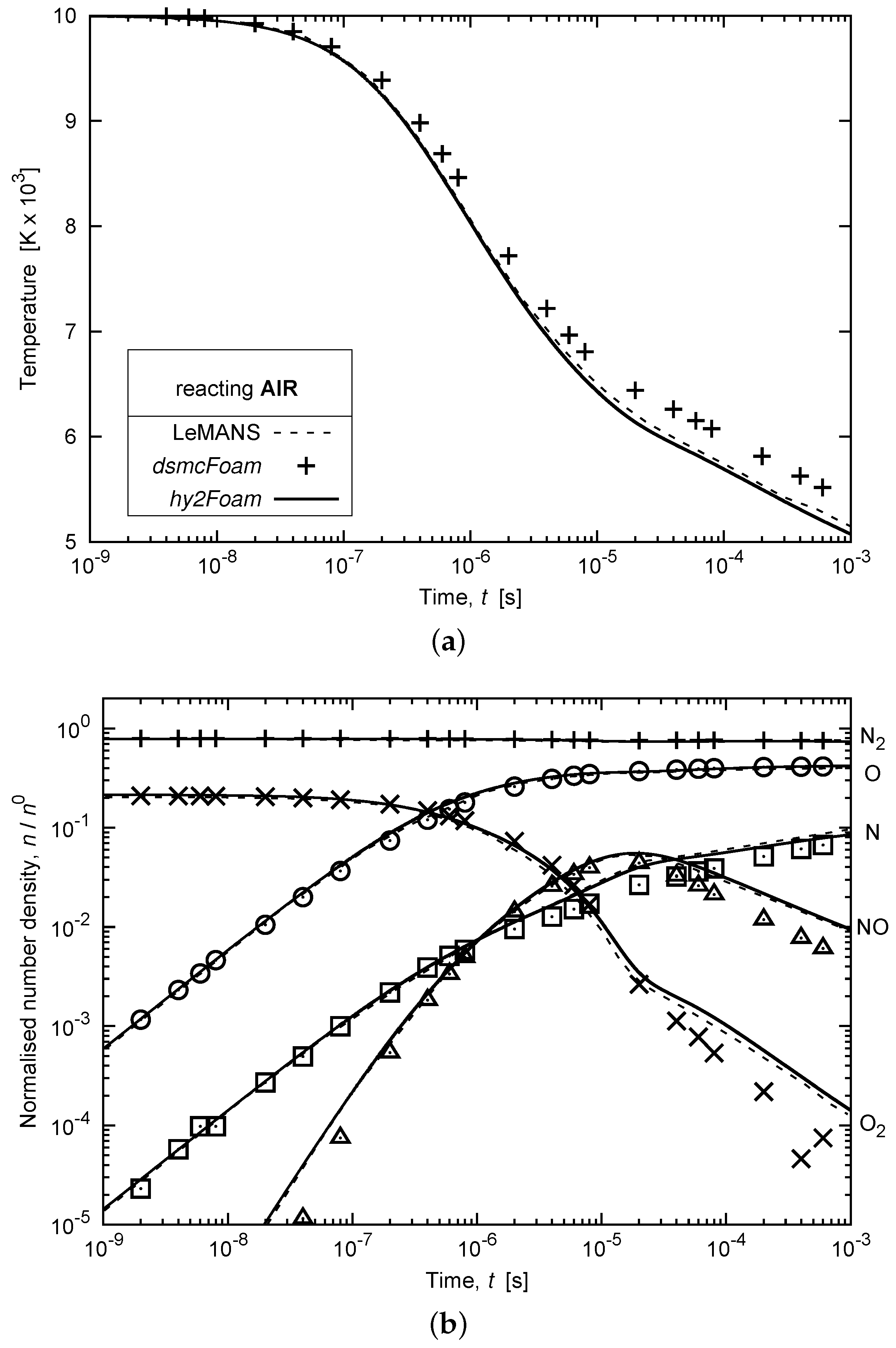
3.5. Chemically-Reacting Air
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species s | (g·m) | (J·kg) | (K) | (J·kg) |
|---|---|---|---|---|
| N2 | 28.0134 | 0 | 3,371 | 3.36 × 10 |
| O2 | 31.9988 | 0 | 2,256 | 1.54 × 10 |
| NO | 30.0061 | 3.04 × 10 | 2,719 | 2.09 × 10 |
| N | 14.0067 | 3.37 × 10 | - | - |
| O | 15.9994 | 1.56 × 10 | - | - |
| Level i | (K) | |
|---|---|---|
| N2 | ||
| ground | 1 | 0 |
| 1 | 3 | 7.223157 × 10 |
| 2 | 6 | 8.577863 × 10 |
| 3 | 6 | 8.605027 × 10 |
| 4 | 3 | 9.535119 × 10 |
| 5 | 1 | 9.805636 × 10 |
| 6 | 2 | 9.968268 × 10 |
| 7 | 2 | 1.048976 × 10 |
| 8 | 5 | 1.116490 × 10 |
| 9 | 1 | 1.225836 × 10 |
| 10 | 6 | 1.248857 × 10 |
| 11 | 6 | 1.282476 × 10 |
| 12 | 10 | 1.338061 × 10 |
| 13 | 6 | 1.404296 × 10 |
| 14 | 6 | 1.504959 × 10 |
| O2 | ||
| ground | 3 | 0 |
| 1 | 2 | 1.139156 × 10 |
| 2 | 1 | 1.898474 × 10 |
| 3 | 1 | 4.755974 × 10 |
| 4 | 6 | 4.991242 × 10 |
| 5 | 3 | 5.092269 × 10 |
| 6 | 3 | 7.189863 × 10 |
| NO | ||
| ground | 4 | 0 |
| 1 | 8 | 5.467346 × 10 |
| 2 | 2 | 6.317140 × 10 |
| 3 | 4 | 6.599450 × 10 |
| 4 | 4 | 6.906121 × 10 |
| 5 | 4 | 7.049998 × 10 |
| 6 | 4 | 7.491055 × 10 |
| 7 | 2 | 7.628875 × 10 |
| 8 | 4 | 8.676189 × 10 |
| 9 | 2 | 8.714431 × 10 |
| 10 | 4 | 8.886077 × 10 |
| 11 | 4 | 8.981756 × 10 |
| 12 | 2 | 8.988446 × 10 |
| 13 | 2 | 9.042702 × 10 |
| 14 | 2 | 9.064284 × 10 |
| 15 | 4 | 9.111763 × 10 |
| N | ||
| ground | 4 | 0 |
| 1 | 10 | 2.766470 × 10 |
| 2 | 6 | 4.149309 × 10 |
| O | ||
| ground | 9 | 0 |
| 3 | 5 | 2.283029 × 10 |
| 4 | 1 | 4.861993 × 10 |
References
- Feldick, A.M.; Modest, M.F.; Levin, D.A.; Gnoffo, P.; Johnston, C.O. Examination of coupled continuum fluid dynamics and radiation in hypersonic simulations. In Proceedings of the AIAA Aerospace Sciences Meeting, Orlando, FL, USA, 5–8 January 2009.
- NASA’s Orion Spacecraft Official Website. Available online: https://www.nasa.gov/exploration/systems/orion/index.html (accessed on 4 September 2016).
- Wuilbercq, R.; Pescetelli, F.; Mogavero, A.; Minisci, E.; Brown, R.E. Robust multidisciplinary design and optimisation of a reusable launch vehicle. In Proceedings of the 19th AIAA International Space Planes and Hypersonic Systems and Technologies Conference, Atlanta, GA, USA, 16 June 2014.
- Brown, R.E. The future of air travel: Dinner in Sydney, London in time for ’The X-Factor’? The Washington Post, 13 October 2014. [Google Scholar]
- Park, C. Review of chemical-kinetic problems of future NASA missions. I-Earth entries. J. Thermophys. Heat Transf. 1993, 7, 385–398. [Google Scholar] [CrossRef]
- Candler, G.V.; Nompelis, I. Computational fluid dynamics for atmospheric entry. In Non-Equilibrium Dynamics: From Physical Models to Hypersonic Flights; The von Karman Institute for Fluid Dynamics: Rhode-Saint-Genèse, Belgium, 2009. [Google Scholar]
- Bird, G.A. Molecular Gas Dynamics and the Direct Simulation of Gas Flows; Clarendon: Oxford, UK, 1994. [Google Scholar]
- Park, C. The limits of two-temperature kinetic model in air. In Proceedings of the 48th AIAA Aerospace Sciences Meeting Including the New Horizons Forum and Aerospace Exposition, Orlando, FL, USA, 4–7 January 2010.
- Park, C. Nonequilibrium Hypersonic Aerothermodynamics; Wiley International: New York, NY, USA, 1990. [Google Scholar]
- Candler, G.V.; Write, M.J.; McDonald, J.D. Data-parallel lower-upper relaxation method for reacting flows. AIAA J. 1994, 32, 2380–2386. [Google Scholar] [CrossRef]
- Cheatwood, F.M.; Gnoffo, P.A. User’s Manual for the Langley Aerothermodynamic Upwind Algorithm (LAURA); Technical Report; NASA Langley Research Center: Hampton, VA, USA, 1996. [Google Scholar]
- VULCAN-CFD Official Website. Available online: http://vulcan-cfd.larc.nasa.gov/ (accessed on 4 September 2016).
- Scalabrin, L.C.; Boyd, I.D. Development of an unstructured navier-stokes solver for hypersonic nonequilibrium aerothermodynamics. In Proceedings of the 38th AIAA Thermophysics Conference, Toronto, ON, Canada, 6–9 June 2005.
- Scalabrin, L.C.; Boyd, I.D. Numerical simulation of weakly ionized hypersonic flow for reentry configurations. In Proceedings of the 9th AIAA/ASME Joint Thermophysics and Heat Transfer Conference, San Francisco, CA, USA, 5–8 June 2006.
- Nompelis, I.; Drayna, T.W.; Candler, G.V. Development of a hybrid unstructured implicit solver for the simulation of reacting flows over complex geometries. In Proceedings of the 34th AIAA Fluid Dynamics Conference and Exhibit, Portland, OR, USA, 28 June–1 July 2004.
- Nompelis, I.; Drayna, T.W.; Candler, G.V. A parallel unstructured implicit solver for hypersonic reacting flow simulation. In Proceedings of the 17th AIAA Computational Fluid Dynamics Conference, Toronto, ON, Canada, 6–9 June 2005.
- OpenFOAM Official Website. Available online: http://www.openfoam.com/ (accessed on 4 September 2016).
- Scanlon, T.J.; White, C.; Borg, M.K.; Palharini, R.C.; Farbar, E.; Boyd, I.D.; Reese, J.M.; Brown, R.E. Open source Direct Simulation Monte Carlo (DSMC) chemistry modelling for hypersonic flows. AIAA J. 2015, 53, 1670–1680. [Google Scholar] [CrossRef]
- Palharini, R.C. Atmospheric Reentry Modelling Using an Open-Source DSMC Code. Ph.D. Thesis, University of Strathclyde, Glasgow, UK, 2014. [Google Scholar]
- Bird, G.A. The DSMC Method, 2nd ed.; CreateSpace Independent Publishing Platform: Sydney, Australia, 2013. [Google Scholar]
- Borgnakke, C.; Larsen, P.S. Statistical collision model for simulating polyatomic gas with restricted energy exchange. In Rarefied Gas Dynamics; DFVLR Press: Porz-Wahn, Germany, 1974; Volume 1, p. A7. [Google Scholar]
- Landau, L.; Teller, E. On the theory of sound dispersion. Physikalische Z. Sowjetunion 1936, 10, 34–43. [Google Scholar]
- Millikan, R.C.; White, D.R. Systematics of vibrational relaxation. J. Chem. Phys. 1963, 39, 3209–3213. [Google Scholar] [CrossRef]
- Andrienko, D.A. Non-Equilibrium Models for High Temperature Gas Flows. Ph.D. Thesis, Moscow Institute of Physics and Technology, Dolgoprudny, Russia, 2014. [Google Scholar]
- Knab, O.; Frühauf, H.H.; Jonas, S. Multiple temperature descriptions of reaction rate constants with regard to consistent chemical-vibrational coupling. In Proceedings of the 27th Thermophysics Conference, Nashville, TN, USA, 6–8 July 1992.
- Knab, O.; Frühauf, H.H.; Messerschmid, E.W. Theory and validation of the physically consistent coupled vibration-chemistry-vibration model. J. Thermophys. Heat Transf. 1995, 9, 219–226. [Google Scholar] [CrossRef]
- Candler, G.V.; MacCormack, R.W. Computation of weakly ionized hypersonic flows in thermochemical nonequilibrium. J. Thermophys. Heat Transf. 1991, 5, 266–273. [Google Scholar] [CrossRef]
- Jacobs, P.A.; Gollan, R.J.; Potter, D.F.; Gildfind, D.E.; Eichmann, T.N.; O’Flaherty, B.T.; Buttsworth, D.R. CFD tools for design and simulation of transient flows in hypersonic facilities. In Proceedings of the RTO-AVT-VKI Lecture Series 2010-AVT-186-Aerothermodynamic Design, Review on Ground Testing and CFD, Rhode-Saint-Genèse, Belgium, 29 March–1 April 2010.
- Brun, R. Introduction to Reactive Gas Dynamics; Oxford University Press: New York, NY, USA, 2009; p. 429. [Google Scholar]
- Scalabrin, L. Numerical Simulation of Weakly Ionized Hypersonic Flow over Reentry Capsules. Ph.D. Thesis, The University of Michigan, Ann Arbor, MI, USA, 2007. [Google Scholar]
- Gnoffo, P.A.; Gupta, R.; Shinn, J.L. Conservation Equations and Physical Models for Hypersonic Air Flows in Thermal and Chemical Nonequilibrium; Technical Report NASA-2867; NASA Langley Research Center: Hampton, VA, USA, 1989. [Google Scholar]
- Marrone, P.V.; Treanor, C.E. Chemical relaxation with preferential dissociation from excited vibrational levels. Phys. Fluids 1963, 6, 1215–1221. [Google Scholar] [CrossRef]
- Holman, T.D.; Boyd, I. Effects of continuum breakdown on hypersonic aerothermodynamics for reacting flow. Phys. Fluids 2011, 23, 027101. [Google Scholar] [CrossRef]
- Treanor, C.E.; Marrone, P.V. Effect of dissociation on the rate of vibrational relaxation. Phys. Fluids 1962, 9, 1022–1026. [Google Scholar] [CrossRef]
- OpenFOAM Version Used. Available online: http://openfoam.org/version/2-3-0/ (accessed on 4 September 2016).
- Greenshields, C.J.; Weller, H.G.; Gasparini, L.; Reese, J.M. Implementation of semi-discrete, non-staggered central schemes in a collocated, polyhedral, finite volume framework, for high-speed viscous flows. Int. J. Numer. Methods Fluids 2010, 63, 1–21. [Google Scholar]
- Kurganov, A.; Noelle, S.; Petrova, G. Semi-discrete central-upwind schemes for hypersonic conservation laws and Hamilton-Jacobi equations. SIAM J. Sci. Comput. 2001, 23, 707–740. [Google Scholar] [CrossRef]
- Vos, J.B.; Rizzi, A.; Darracq, D.; Hirschel, E.H. Navier-Stokes solvers in European aircraft design. Prog. Aerosp. Sci. 2002, 38, 601–697. [Google Scholar] [CrossRef]
- Mack, A.; Hannemann, V. Validation of the unstructured DLR TAU-Code for hypersonic flows. In Proceedings of the 32nd AIAA Fluid Dynamics Conference and Exhibit, St. Louis, MO, USA, 24–26 June 2002.
- Scanlon, T.J.; Cassineli Palharini, R.; White, C.; Espinoza, D.; Casseau, V. Simulations of rarefied and continuum hypersonic flow over re-entry objects. In Proceedings of the 8th European Symposium on Aerothermodynamics for Space Vehicles, Lisbon, Portugal, 2–6 March 2015.
- Williams, F.A. Combustion Theory, 2nd ed.; Addison-Wesley: Redwood City, CA, USA, 1985. [Google Scholar]
- Dietrich, S.; Boyd, I.D. Scalar and parallel optimized implementation of the direct simulation Monte Carlo method. J. Comput. Phys. 1996, 126, 328–342. [Google Scholar] [CrossRef]
- Haas, B.L.; McDonald, J.D. Validation of chemistry models employed in a particle simulation method. J. Thermophys. Heat Transf. 1993, 7, 42–48. [Google Scholar] [CrossRef]
- Casseau, V.; Scanlon, T.J.; Brown, R.E. Development of a two-temperature open-source CFD model for hypersonic reacting flows. In Proceedings of the 20th International Space Planes and Hypersonic Systems and Technologies Conference, Glasgow, UK, 6–9 July 2015.
- Boyd, I.D.; Josyula, E. State resolved vibrational relaxation modeling for strongly nonequilibrium flows. Phys. Fluids 2011, 23, 057101. [Google Scholar] [CrossRef]
- Boyd, I.D. Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen. Phys. Fluids 1992, 4, 178–185. [Google Scholar] [CrossRef]
- Magin, T.E.; Panesi, M.; Bourdon, A.; Jaffe, R.; Schwenke, D. Internal energy excitation and dissociation of molecular nitrogen in a compressing flow. In Proceedings of the 41st AIAA Thermophysics Conference, San Antonio, TX, USA, 22–25 June 2009.









| Reaction Type | Forward, | Backward, |
|---|---|---|
| dissociation | ||
| exchange | ||
| associative ionisation | ||
| electron impact ionisation | ||
| charge exchange |
| Reaction Rate | Arrhenius Law Constants | ||
|---|---|---|---|
| A | β | ||
| Park 1993 | −1.6 | 113,200 | |
| QK | −0.62 | 113,500 | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casseau, V.; Palharini, R.C.; Scanlon, T.J.; Brown, R.E. A Two-Temperature Open-Source CFD Model for Hypersonic Reacting Flows, Part One: Zero-Dimensional Analysis. Aerospace 2016, 3, 34. https://doi.org/10.3390/aerospace3040034
Casseau V, Palharini RC, Scanlon TJ, Brown RE. A Two-Temperature Open-Source CFD Model for Hypersonic Reacting Flows, Part One: Zero-Dimensional Analysis. Aerospace. 2016; 3(4):34. https://doi.org/10.3390/aerospace3040034
Chicago/Turabian StyleCasseau, Vincent, Rodrigo C. Palharini, Thomas J. Scanlon, and Richard E. Brown. 2016. "A Two-Temperature Open-Source CFD Model for Hypersonic Reacting Flows, Part One: Zero-Dimensional Analysis" Aerospace 3, no. 4: 34. https://doi.org/10.3390/aerospace3040034
APA StyleCasseau, V., Palharini, R. C., Scanlon, T. J., & Brown, R. E. (2016). A Two-Temperature Open-Source CFD Model for Hypersonic Reacting Flows, Part One: Zero-Dimensional Analysis. Aerospace, 3(4), 34. https://doi.org/10.3390/aerospace3040034