
Network-Based Prediction of Side Effects of Repurposed Antihypertensive Sartans against COVID-19 via Proteome and Drug-Target Interactomes

 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
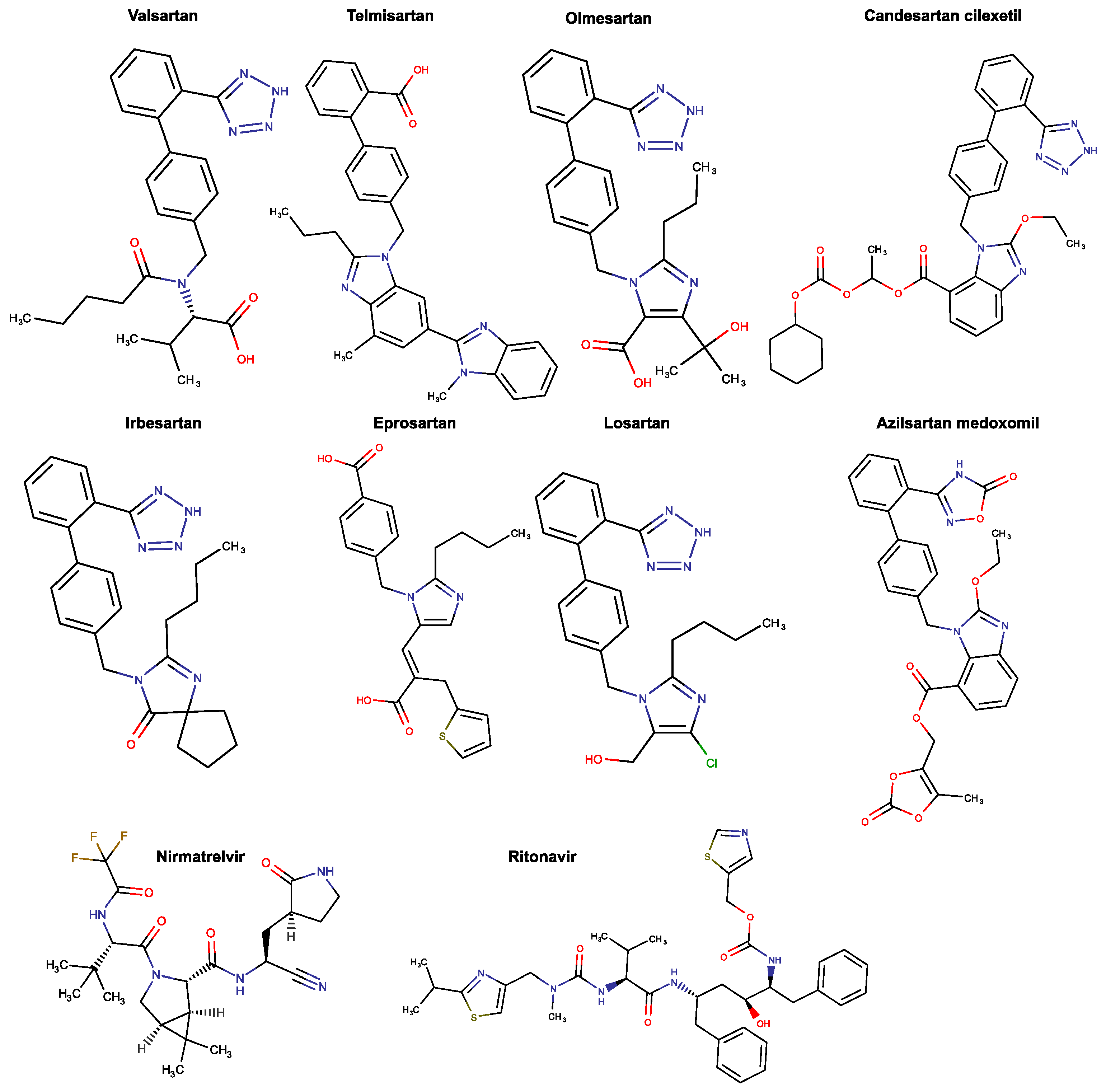
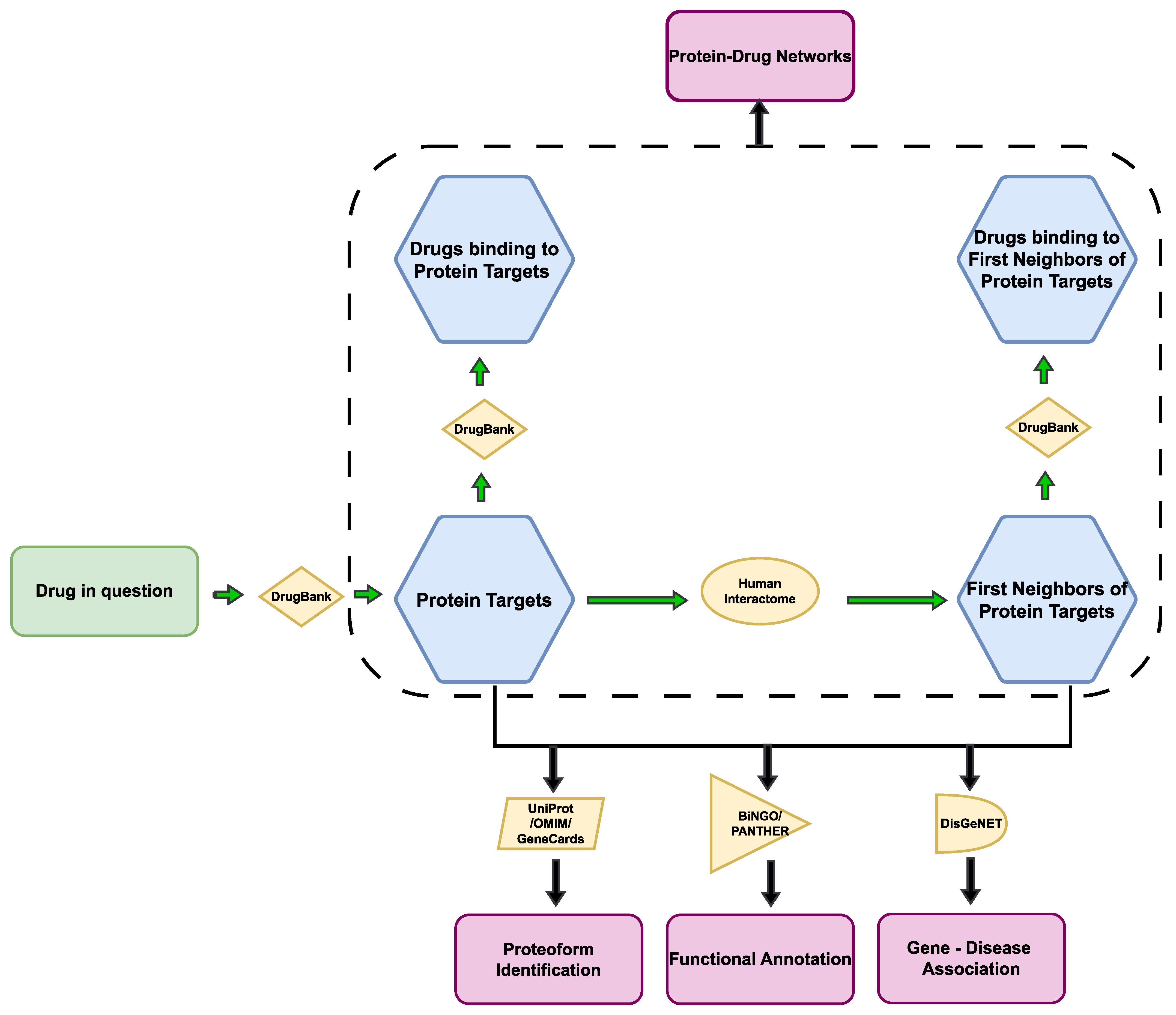
2. Materials and Methods
3. Results
3.1. Retrievement of Drug–Protein Target Associations
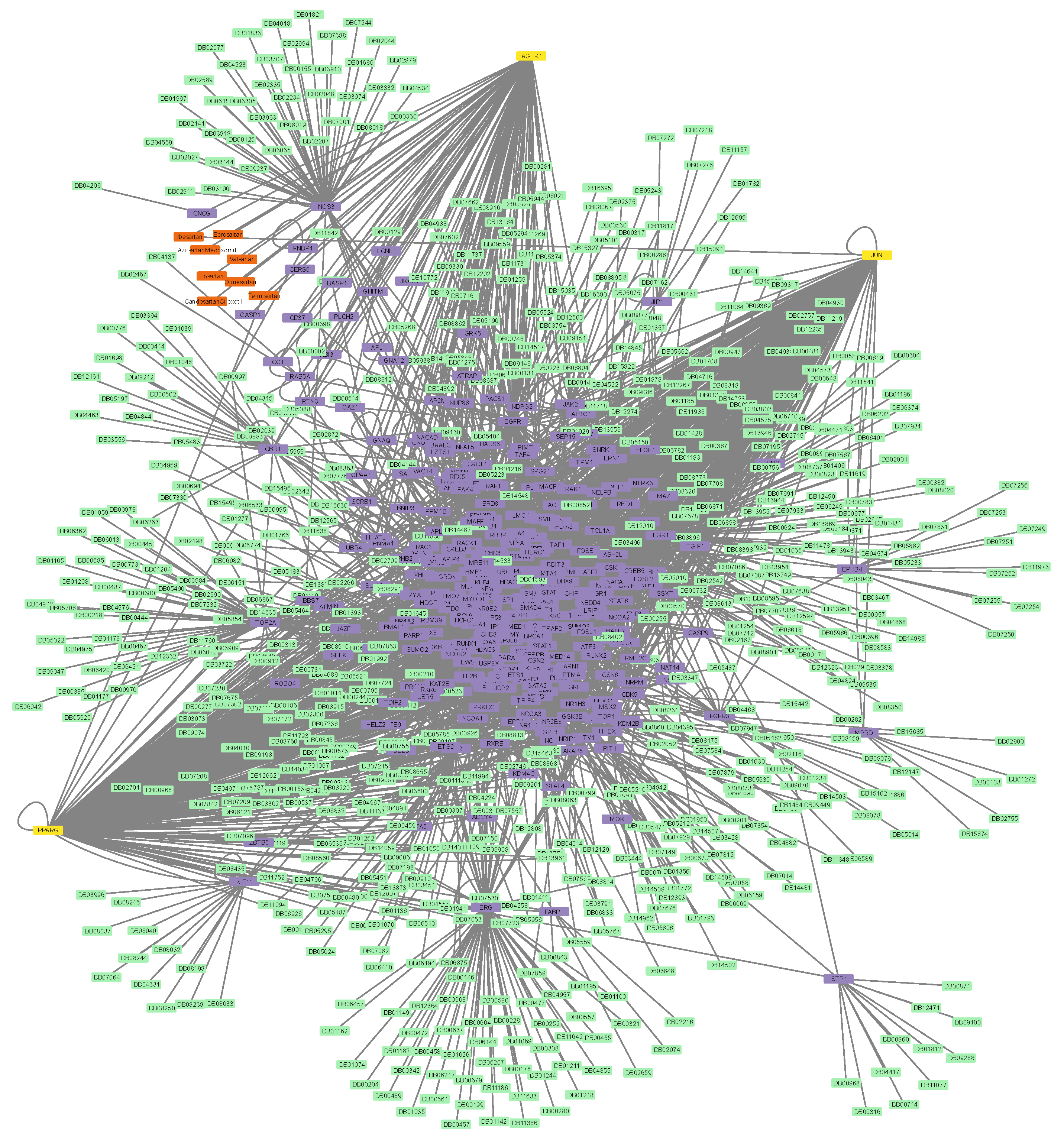
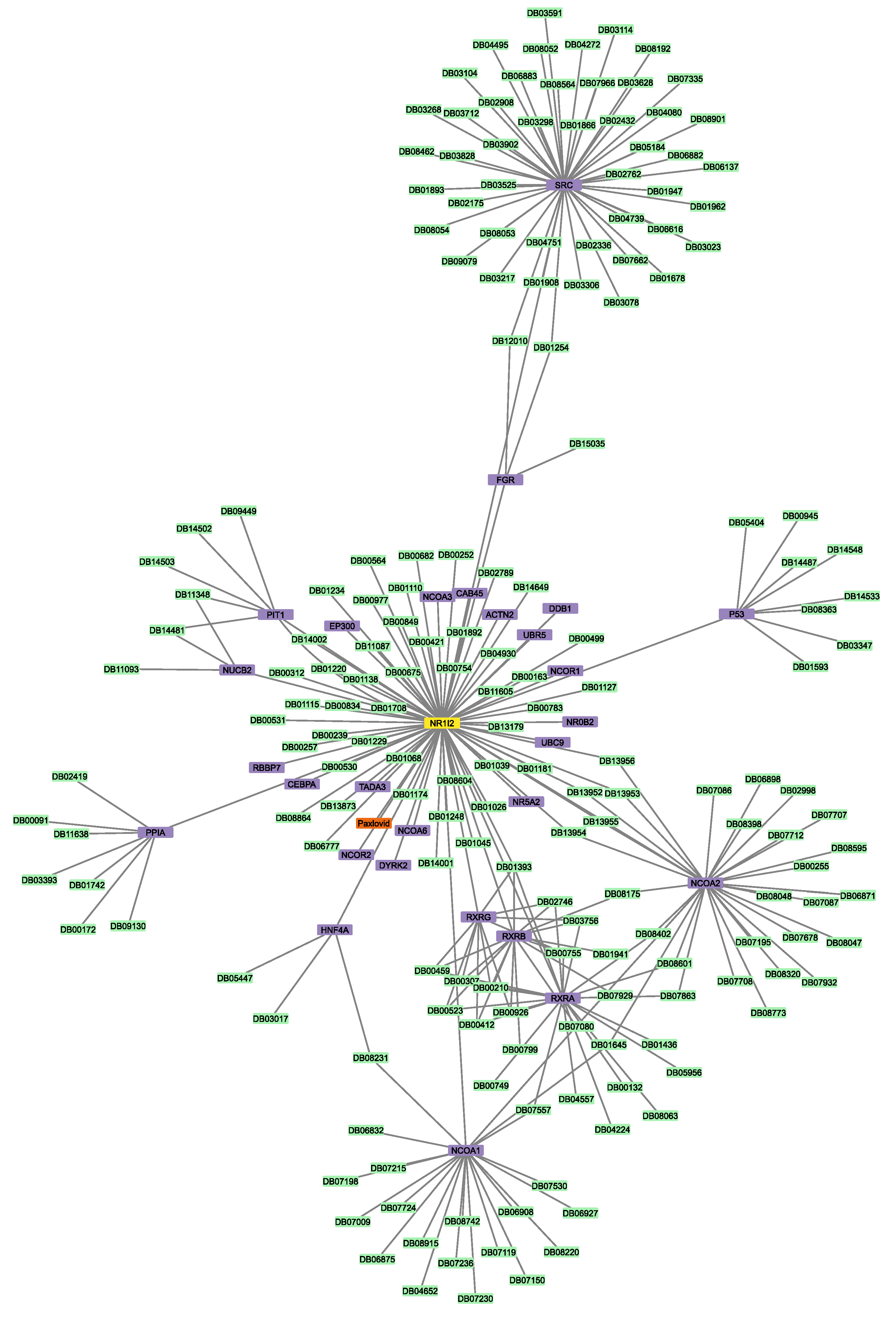
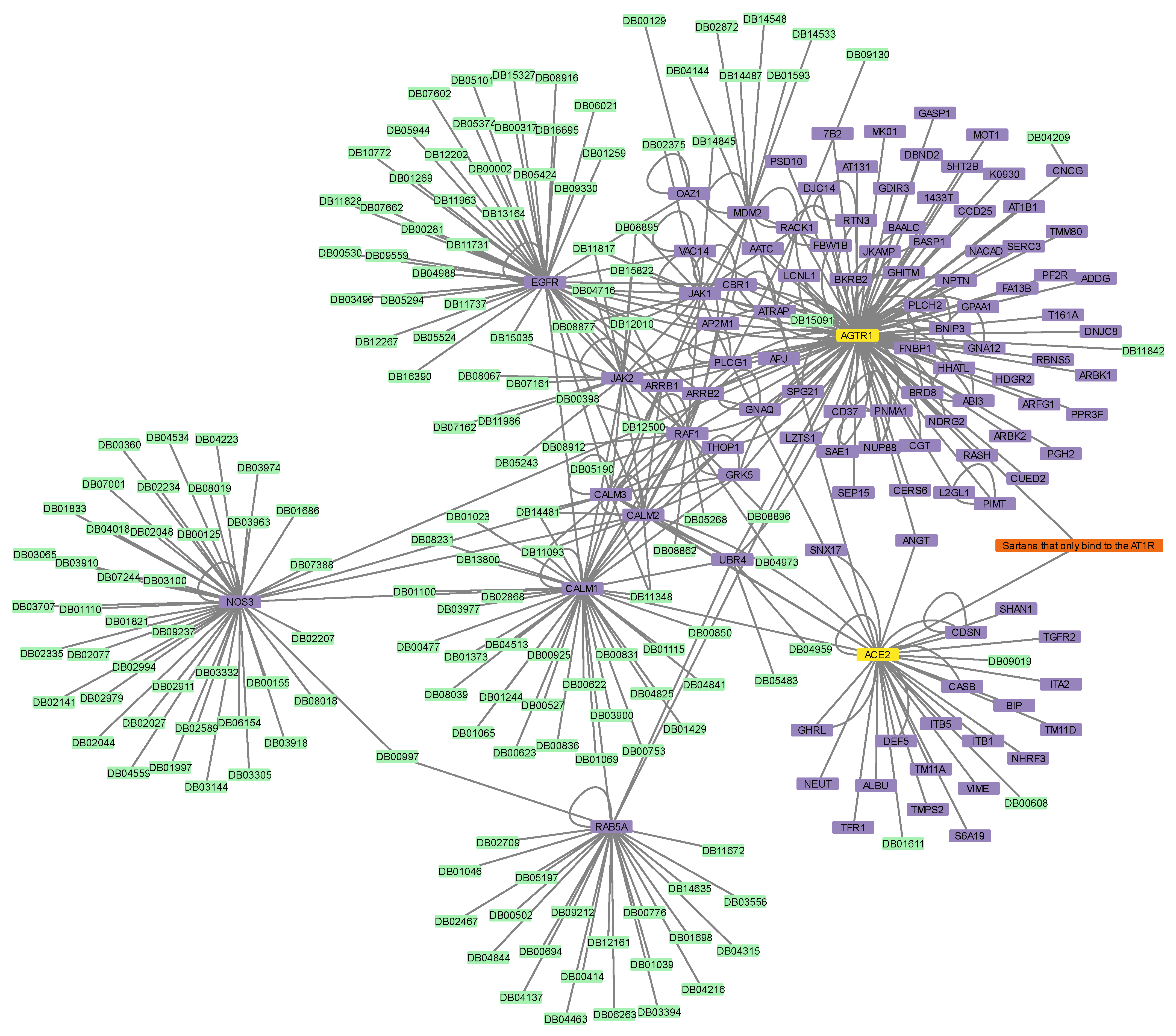
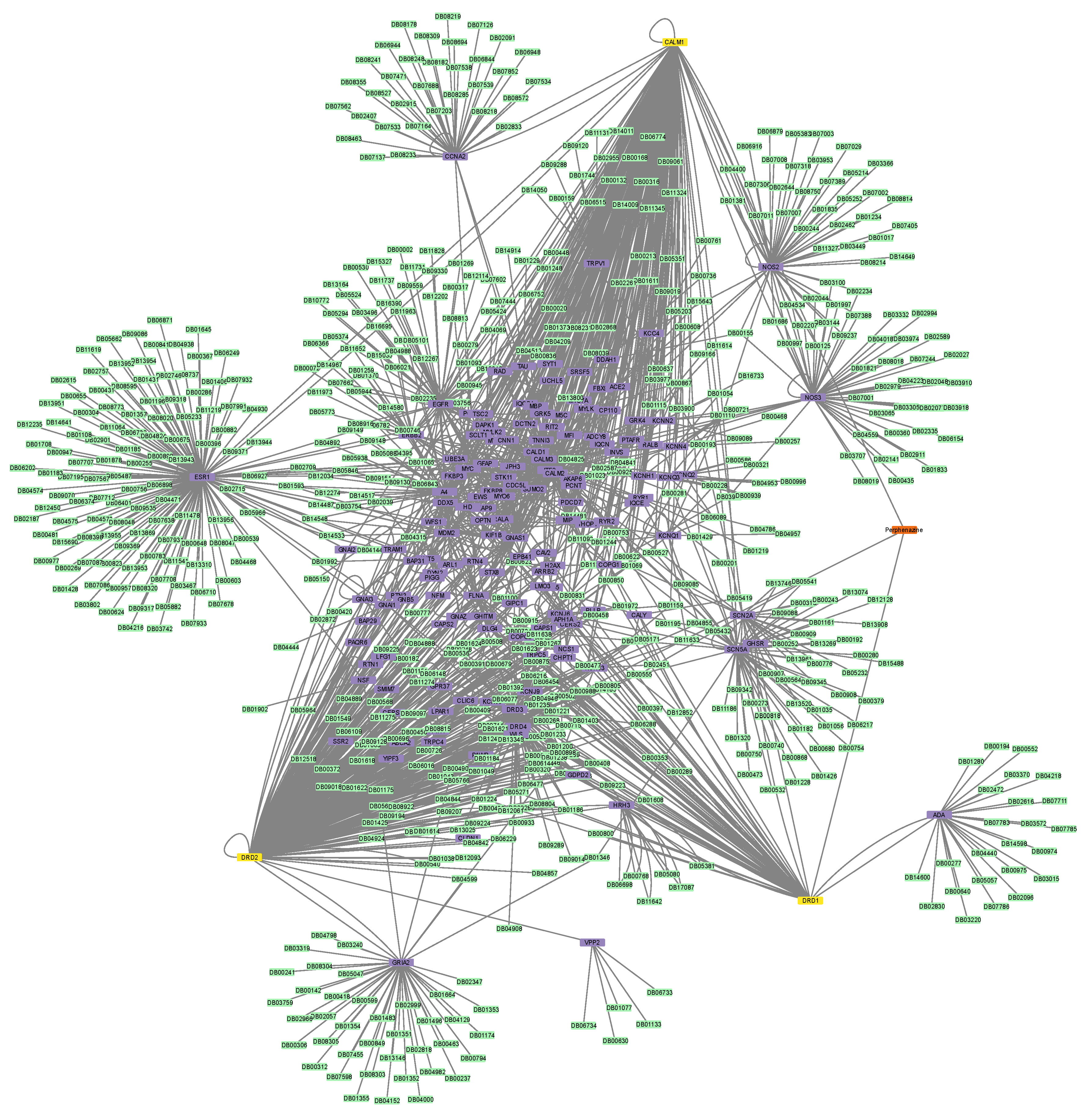
3.2. Construction of Protein–Protein and Protein–Drug Interaction Networks
3.3. Shared First Neighbor Distance Metrics (Jaccard Index)
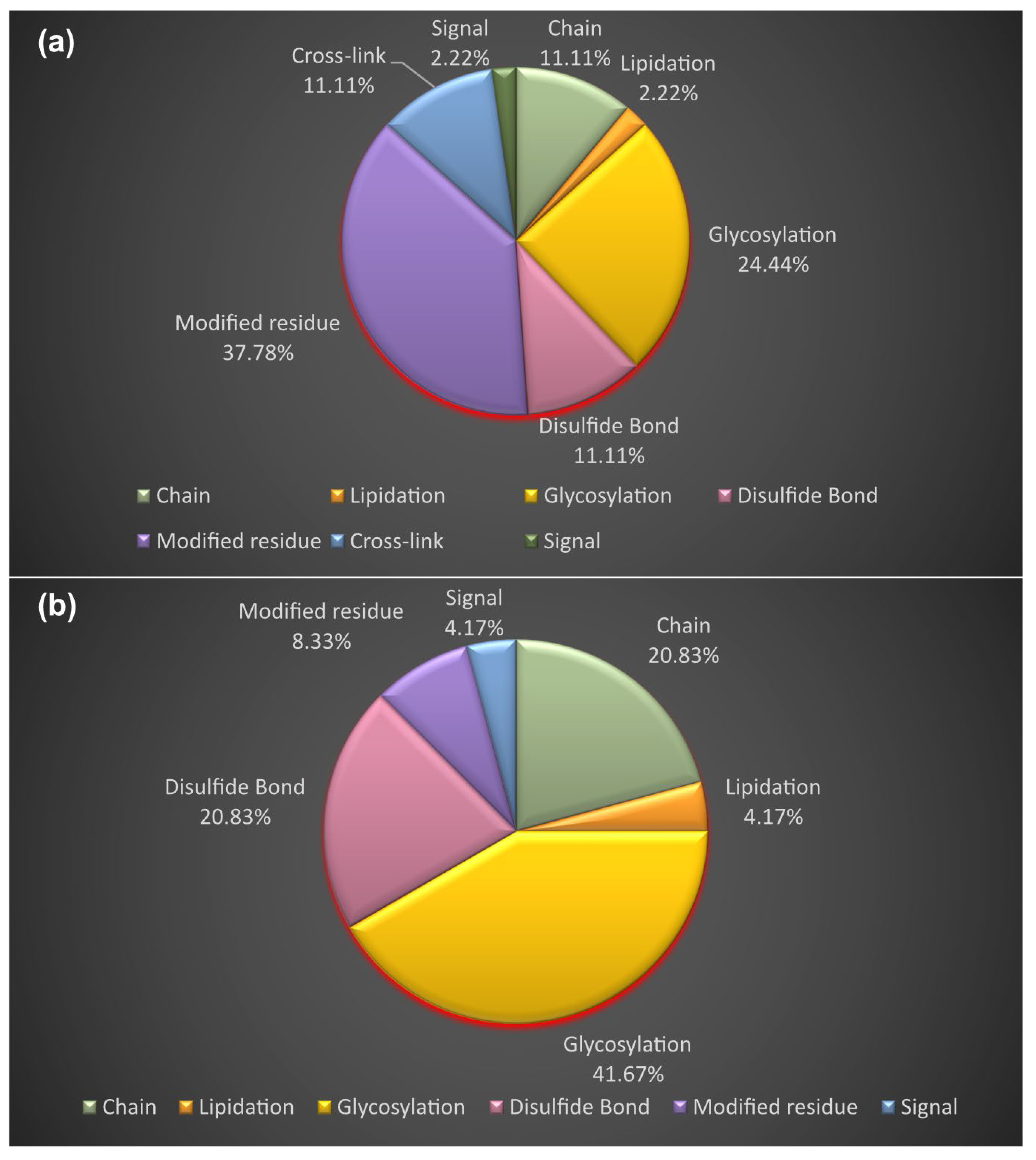
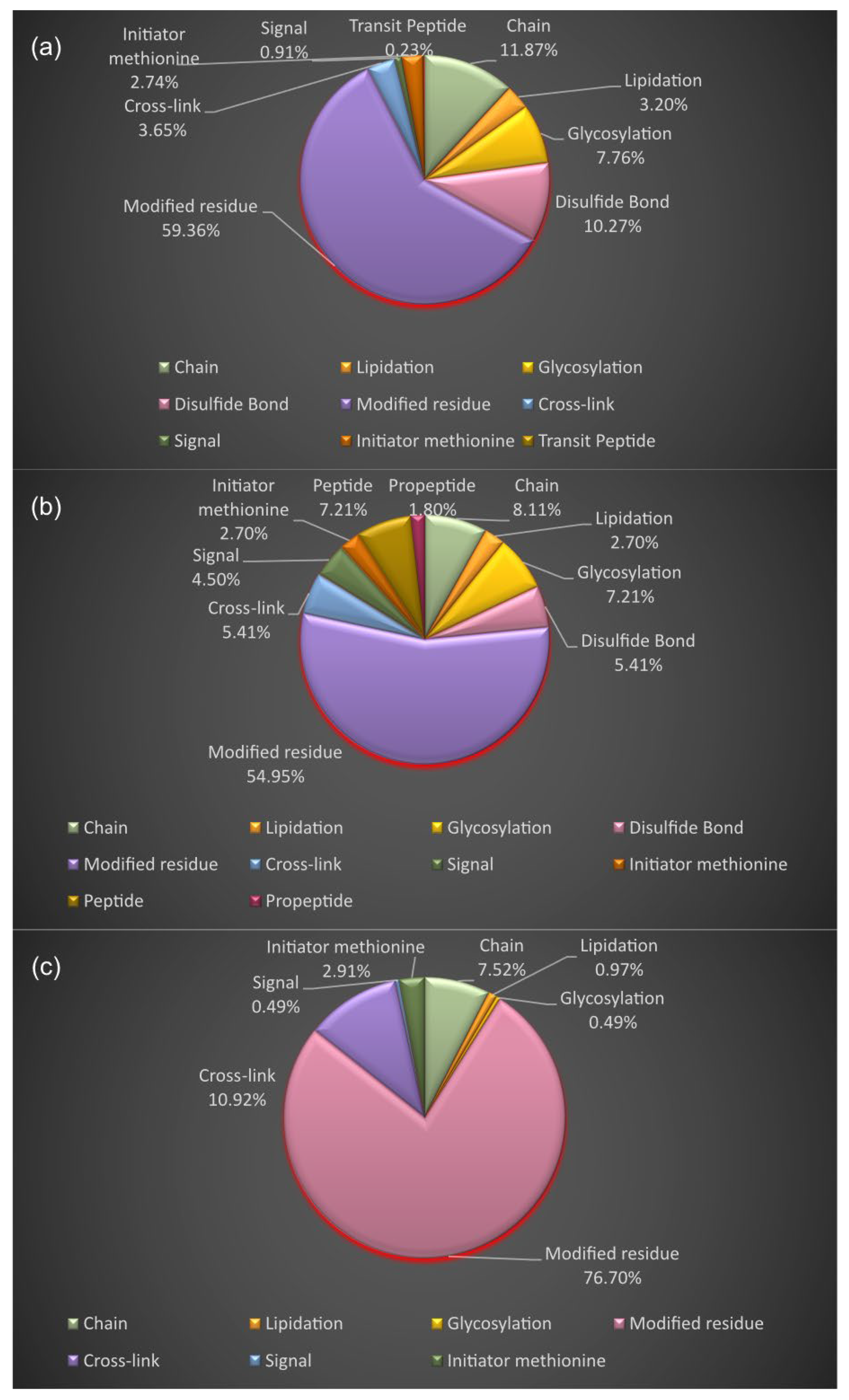
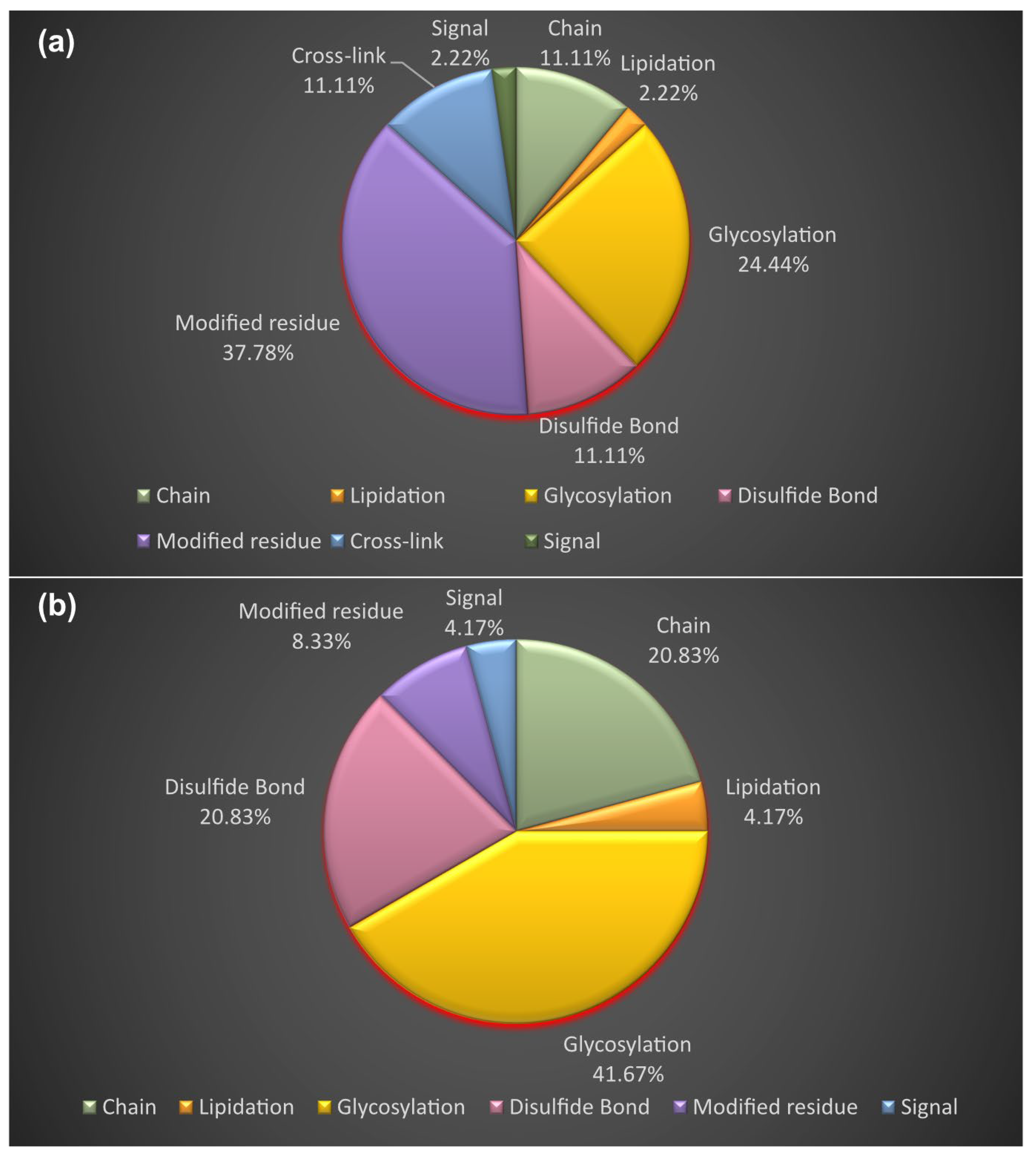
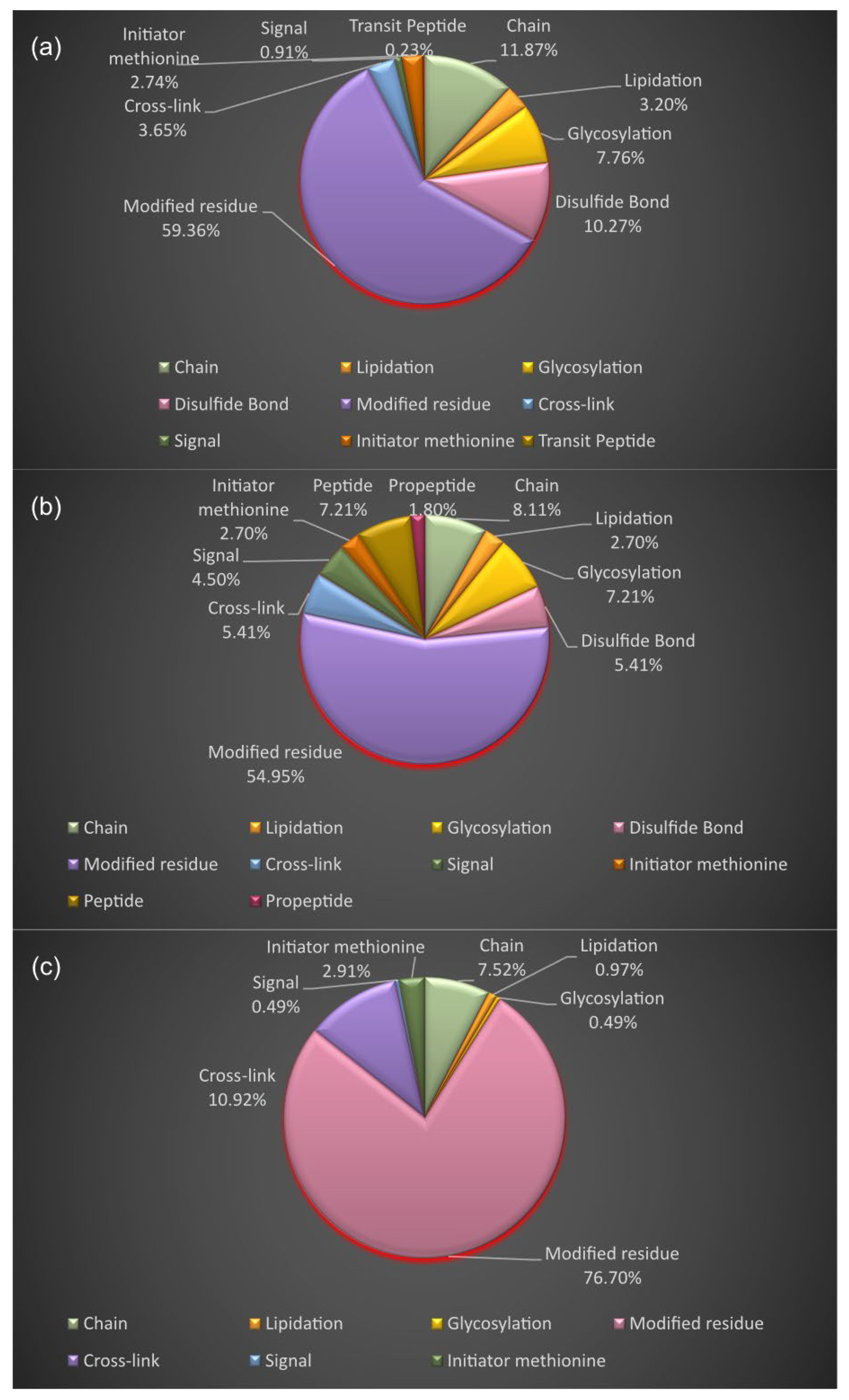
3.4. Proteoform Identification of Protein Drug Targets and Proteins Involved in Sartans’ and Paxlovid’s Based Interactomes
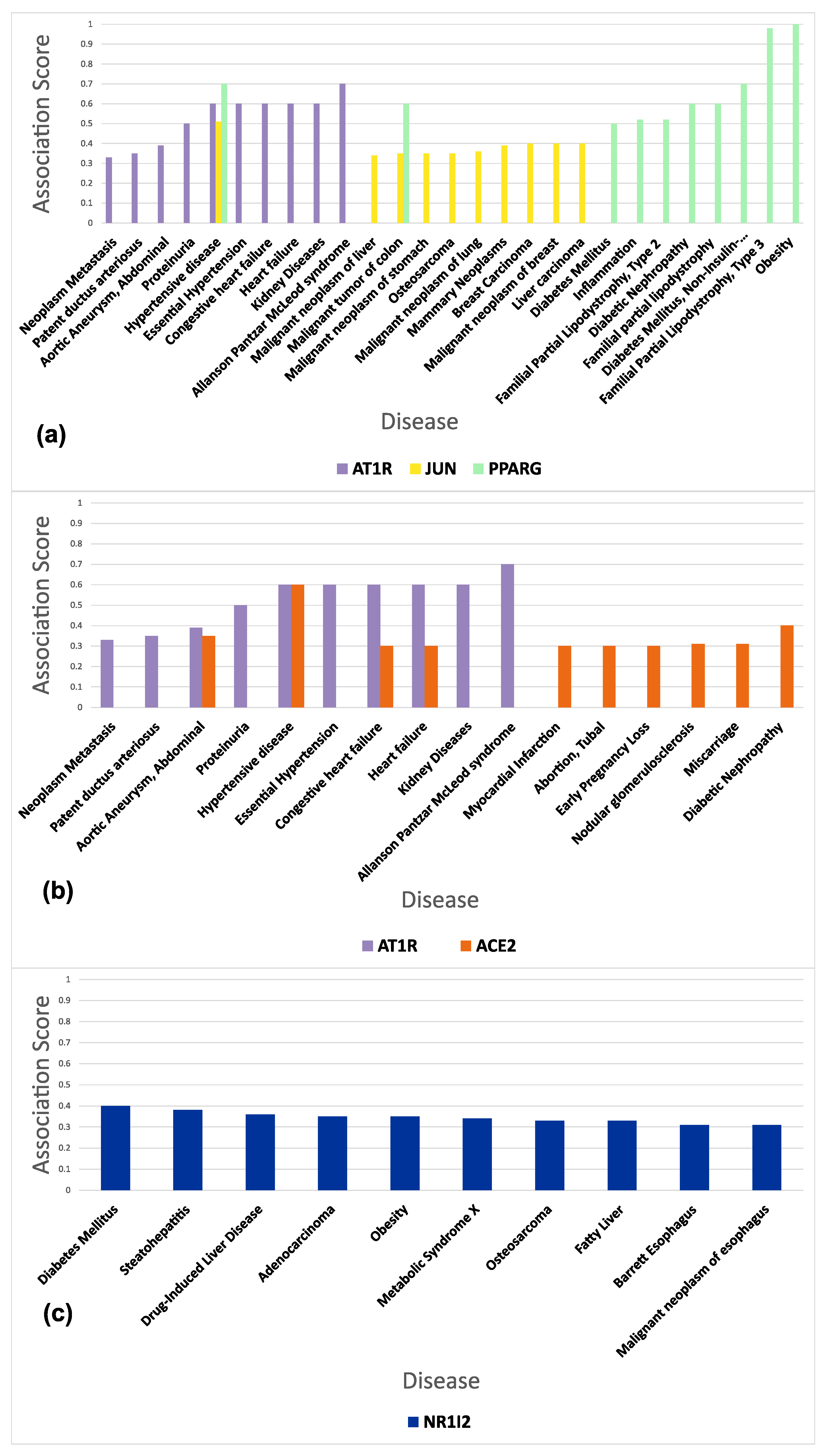
3.5. Functional and Disease Annotation of Proteins Involved in Sartans’ and Paxlovids’ Based Interactomes
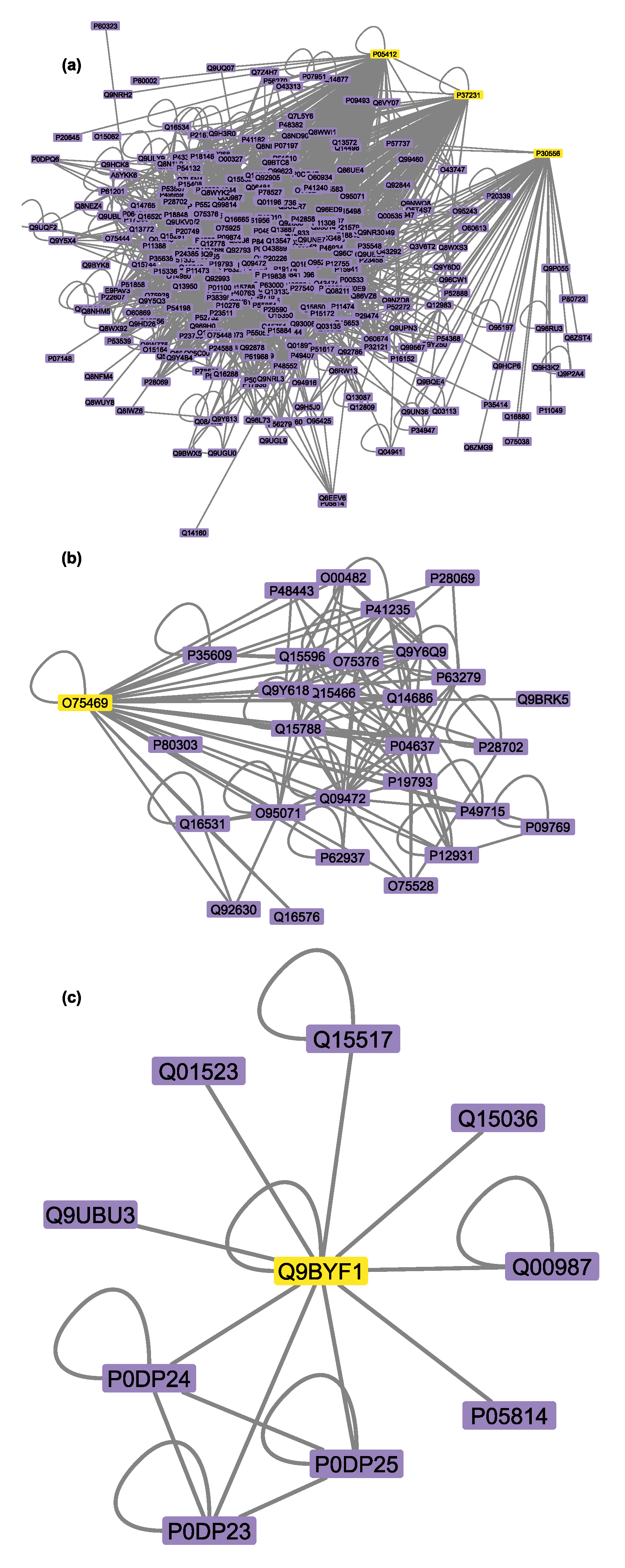
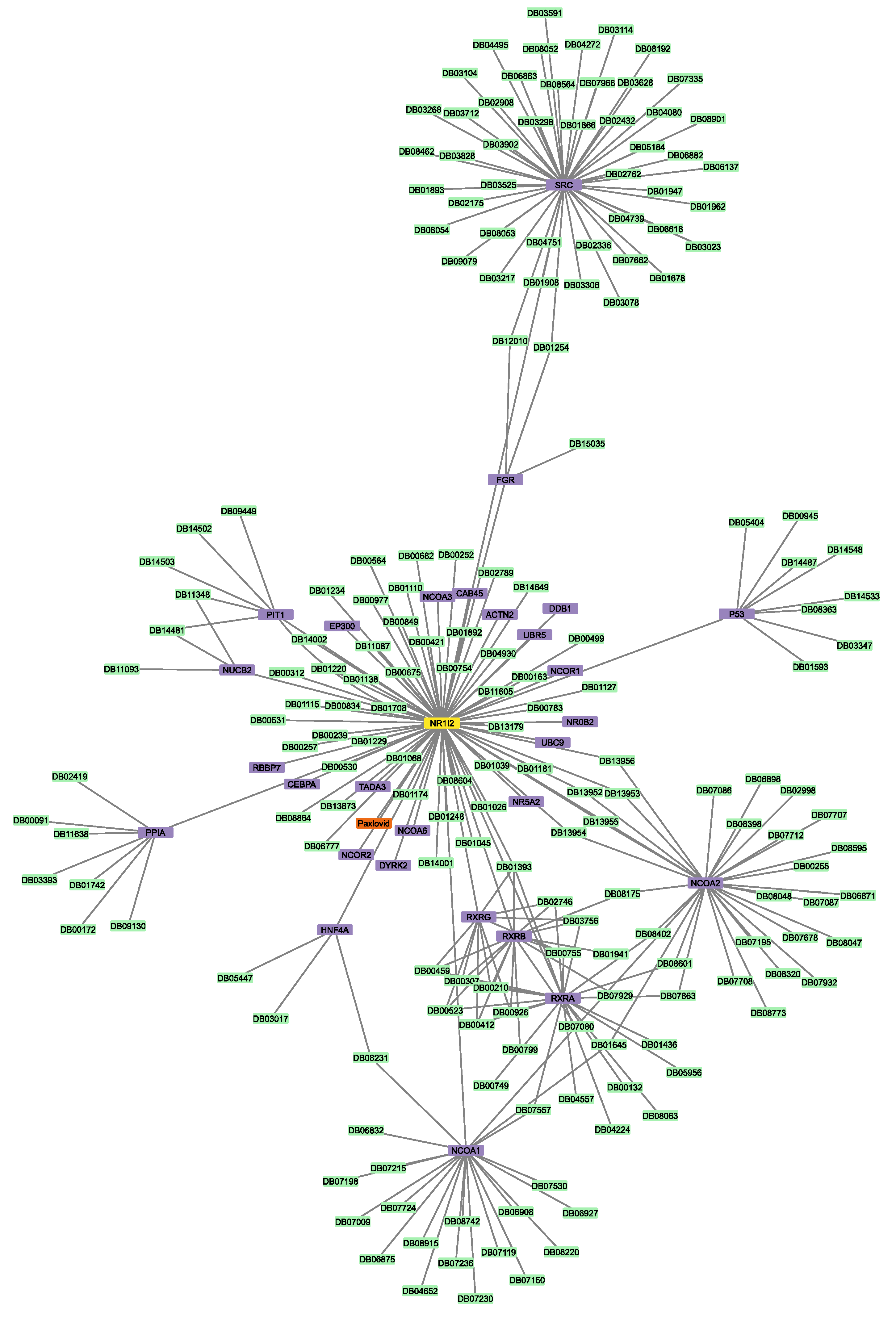
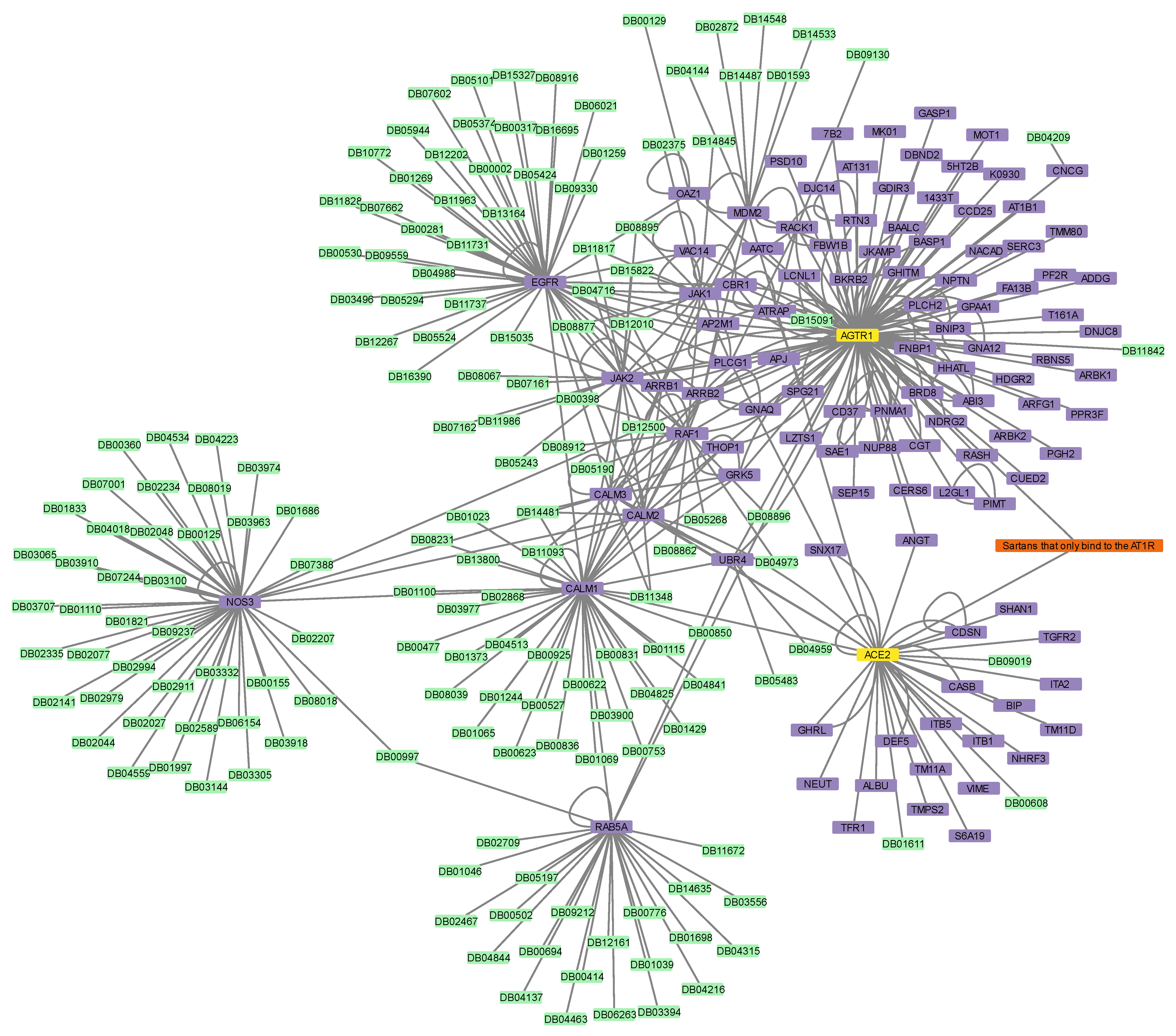
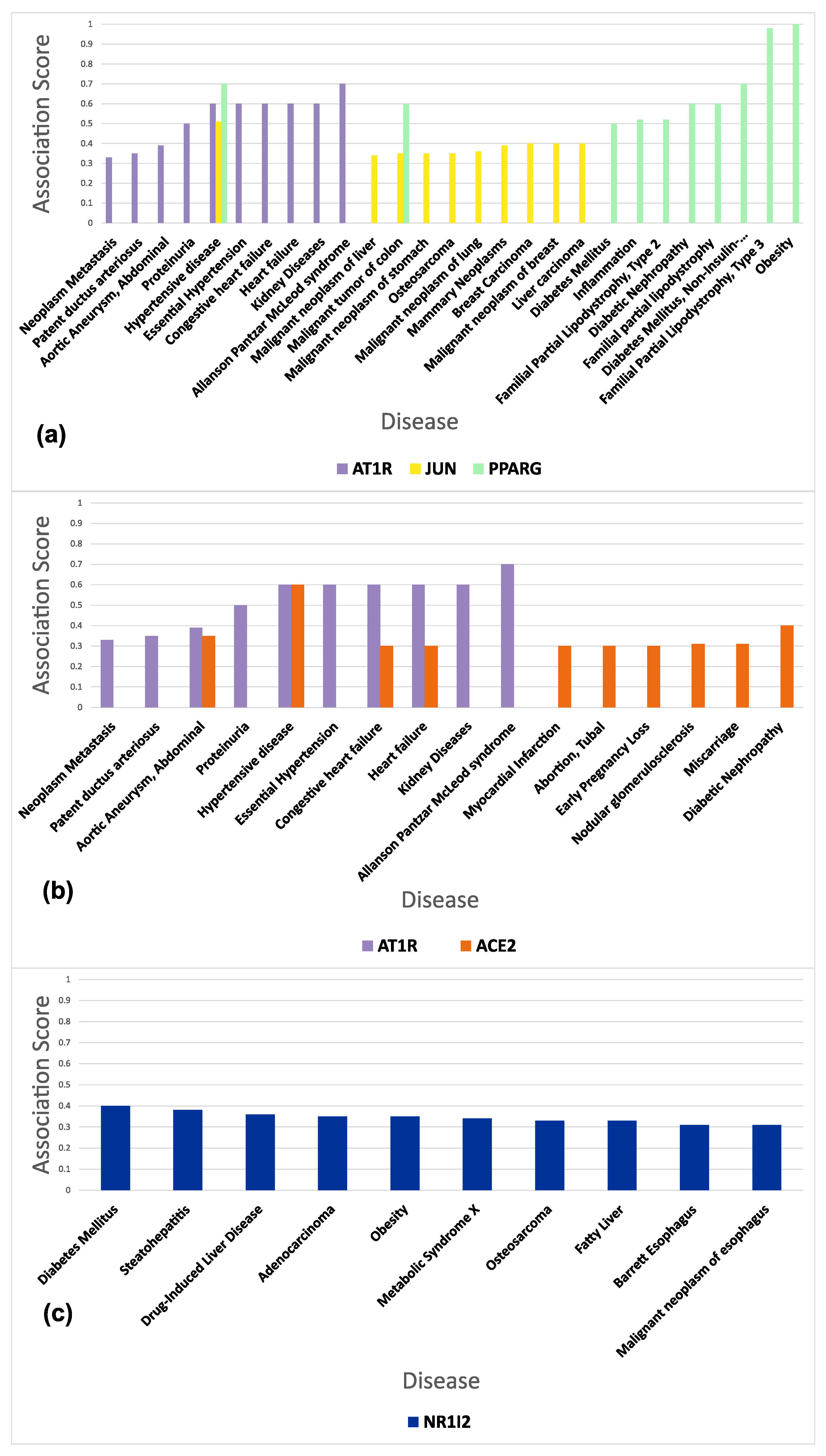
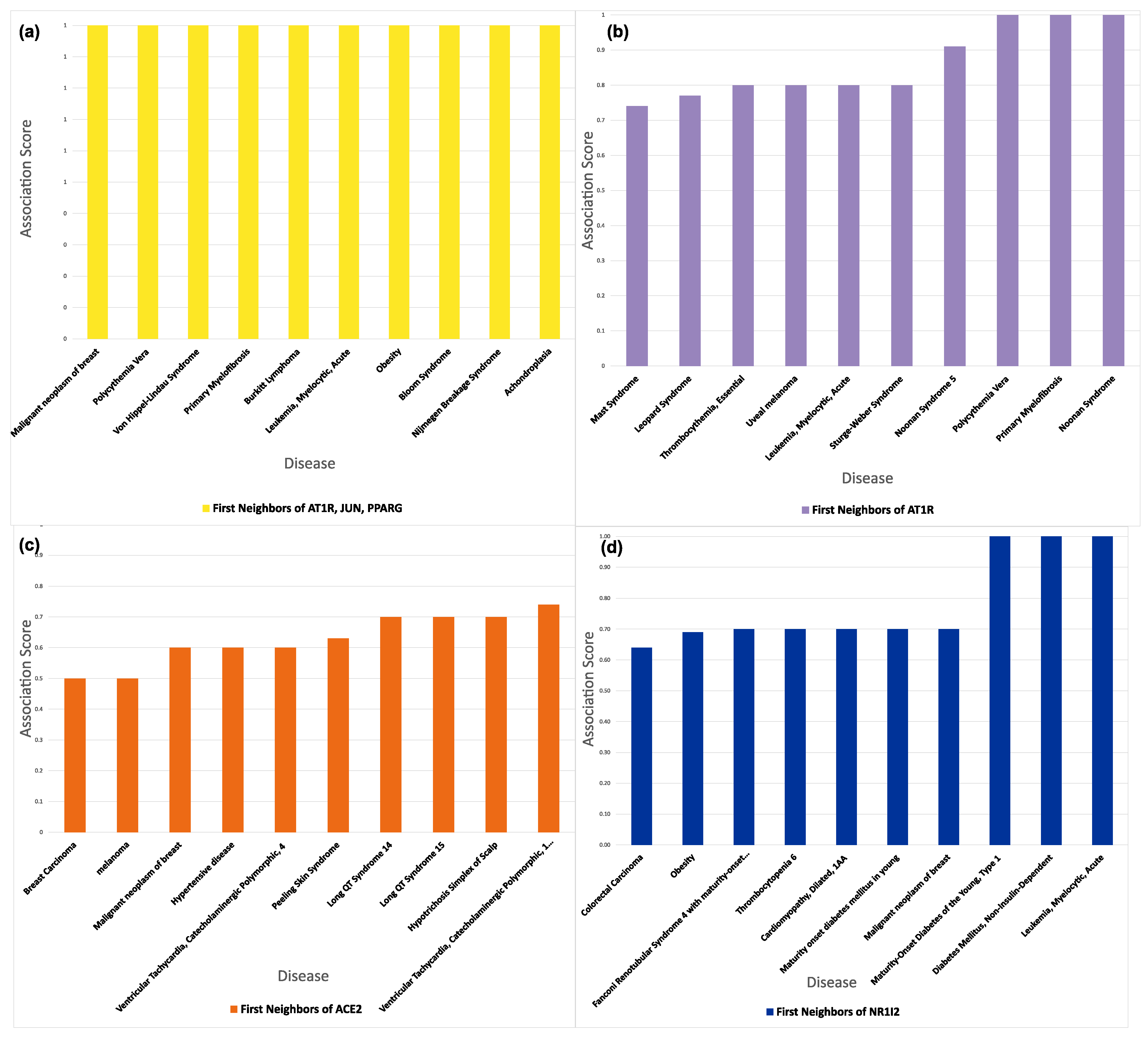
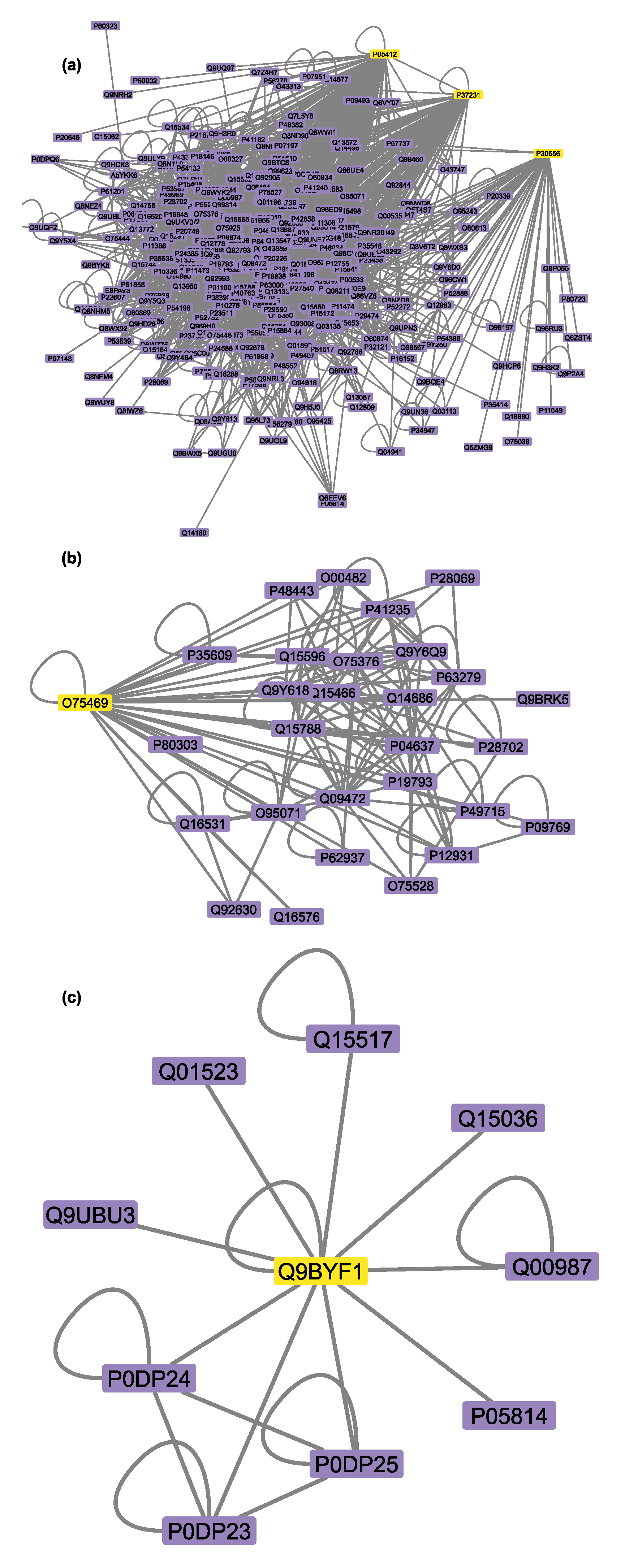
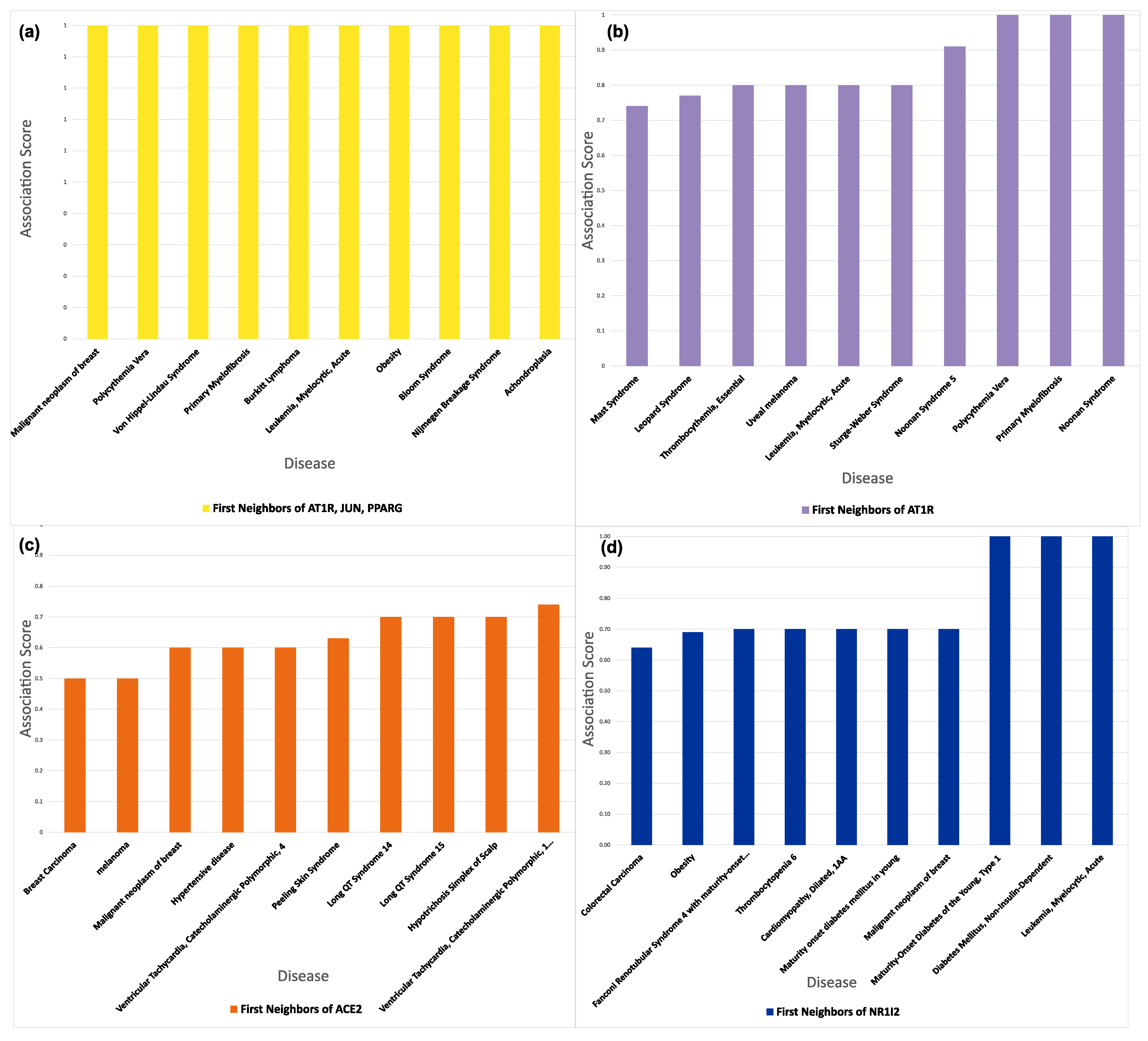
3.6. Connectivity of AT1R, ACE2, and NR1I’s Interactors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Fountain, J.H.; Lappin, S.L. Physiology, Renin Angiotensin System. In StatPearls; StatPearls Publishing Copyright © 2022; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Ma, T.K.; Kam, K.K.; Yan, B.P.; Lam, Y.Y. Renin-angiotensin-aldosterone system blockade for cardiovascular diseases: Current status. Br. J. Pharm. 2010, 160, 1273–1292. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Zhang, J.; Zhuo, J.L. The vasoprotective axes of the renin-angiotensin system: Physiological relevance and therapeutic implications in cardiovascular, hypertensive and kidney diseases. Pharmacol. Res. 2017, 125, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.B. Renin Angiotensin System Inhibition as treatment for Covid-19? eClinicalMedicine 2021, 37, 101023. [Google Scholar] [CrossRef] [PubMed]
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Momoniat, T.; Ilyas, D.; Bhandari, S. ACE inhibitors and ARBs: Managing potassium and renal function. Clevel. Clin. J. Med. 2019, 86, 601–607. [Google Scholar] [CrossRef]
- Gallo, G.; Volpe, M.; Rubattu, S. Angiotensin Receptor Blockers in the Management of Hypertension: A Real-World Perspective and Current Recommendations. Vasc. Health Risk Manag. 2022, 18, 507–515. [Google Scholar] [CrossRef]
- Fedson, D.S. Treating the host response to emerging virus diseases: Lessons learned from sepsis, pneumonia, influenza and Ebola. Ann. Transl. Med. 2016, 4, 421. [Google Scholar] [CrossRef] [Green Version]
- Moore, G.J.; Ridgway, H.; Kelaidonis, K.; Chasapis, C.T.; Ligielli, I.; Mavromoustakos, T.; Bojarska, J.; Matsoukas, J.M. Actions of Novel Angiotensin Receptor Blocking Drugs, Bisartans, Relevant for COVID-19 Therapy: Biased Agonism at Angiotensin Receptors and the Beneficial Effects of Neprilysin in the Renin Angiotensin System. Molecules 2022, 27, 4854. [Google Scholar] [CrossRef]
- Ridgway, H.; Moore, G.J.; Mavromoustakos, T.; Tsiodras, S.; Ligielli, I.; Kelaidonis, K.; Chasapis, C.T.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; et al. Discovery of a new generation of angiotensin receptor blocking drugs: Receptor mechanisms and in silico binding to enzymes relevant to SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 2091–2111. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Marzi, M.; Vakil, M.K.; Bahmanyar, M.; Zarenezhad, E. Paxlovid: Mechanism of Action, Synthesis, and In Silico Study. BioMed Res. Int. 2022, 2022, 7341493. [Google Scholar] [CrossRef]
- Cokley, J.A.; Gidal, B.E.; Keller, J.A.; Vossler, D.G. Paxlovid(TM) Information From FDA and Guidance for AES Members. Epilepsy Curr. 2022, 22, 201–204. [Google Scholar] [CrossRef]
- Azanza, J.R.; Mensa, J.; González Del Castillo, J.; Linares Rufo, M.; Molero, J.M.; Madero Valle, N.; Barberán, J. Interactions listed in the Paxlovid fact sheet, classified according to risks, pharmacological groups, and consequences. Rev. Esp. Quim. 2022, 35, 357–361. [Google Scholar] [CrossRef]
- Marzolini, C.; Kuritzkes, D.R.; Marra, F.; Boyle, A.; Gibbons, S.; Flexner, C.; Pozniak, A.; Boffito, M.; Waters, L.; Burger, D.; et al. Recommendations for the Management of Drug-Drug Interactions Between the COVID-19 Antiviral Nirmatrelvir/Ritonavir (Paxlovid) and Comedications. Clin. Pharmacol. Pharm. 2022, 112, 1191–1200. [Google Scholar] [CrossRef]
- Angeloni, E. Azilsartan medoxomil in the management of hypertension: An evidence-based review of its place in therapy. Core Evid. 2016, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Nagamine, T. Lithium Intoxication in the Elderly: A Possible Interaction between Azilsartan, Fluvoxamine, and Lithium. Innov. Clin. Neurosci. 2020, 17, 45–46. [Google Scholar]
- Yang, R.; Luo, Z.; Liu, Y.; Sun, M.; Zheng, L.; Chen, Y.; Li, Y.; Wang, H.; Chen, L.; Wu, M.; et al. Drug Interactions with Angiotensin Receptor Blockers: Role of Human Cytochromes P450. Curr. Drug Metab. 2016, 17, 681–691. [Google Scholar] [CrossRef]
- Ridgway, H.; Chasapis, C.T.; Kelaidonis, K.; Ligielli, I.; Moore, G.J.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Mavromoustakos, T.; Matsoukas, J.M. Understanding the Driving Forces That Trigger Mutations in SARS-CoV-2: Mutational Energetics and the Role of Arginine Blockers in COVID-19 Therapy. Viruses 2022, 14, 1029. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, H.; Ntallis, C.; Chasapis, C.T.; Kelaidonis, K.; Matsoukas, M.-T.; Plotas, P.; Apostolopoulos, V.; Moore, G.; Tsiodras, S.; Paraskevis, D.; et al. Molecular Epidemiology of SARS-CoV-2: The Dominant Role of Arginine in Mutations and Infectivity. Viruses 2023, 15, 309. [Google Scholar] [CrossRef] [PubMed]
- Marvin Sketch Software. Available online: https://chemaxon.com/marvin (accessed on 16 January 2023).
- Lin, S.F.; Xiao, K.T.; Huang, Y.T.; Chiu, C.C.; Soo, V.W. Analysis of adverse drug reactions using drug and drug target interactions and graph-based methods. Artif. Intell. Med. 2010, 48, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, L.; Iskar, M.; Zeller, G.; van Noort, V.; Bork, P. Network neighbors of drug targets contribute to drug side-effect similarity. PLoS ONE 2011, 6, e22187. [Google Scholar] [CrossRef] [Green Version]
- Hartung, B.; Sampson, S.; Leucht, S. Perphenazine for schizophrenia. Cochrane Database Syst. Rev. 2015, 2015, Cd003443. [Google Scholar] [CrossRef]
- Schnabel, A.; Eberhart, L.H.; Muellenbach, R.; Morin, A.M.; Roewer, N.; Kranke, P. Efficacy of perphenazine to prevent postoperative nausea and vomiting: A quantitative systematic review. Eur. J. Anaesthesiol. 2010, 27, 1044–1051. [Google Scholar] [CrossRef]
- DrugBank. Available online: https://go.drugbank.com/ (accessed on 16 January 2023).
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2011, 39, D1035–D1041. [Google Scholar] [CrossRef] [Green Version]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef]
- Backman, T.W.; Cao, Y.; Girke, T. ChemMine tools: An online service for analyzing and clustering small molecules. Nucleic Acids Res. 2011, 39, W486–W491. [Google Scholar] [CrossRef] [PubMed]
- ChemMine Tools. Available online: https://chemminetools.ucr.edu/ (accessed on 12 May 2023).
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 12 May 2023).
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- PICKLE (Protein InteraCtion KnowLedgebasE). Available online: http://www.pickle.gr/ (accessed on 16 January 2023).
- UniProt (Universal Protein Resource). Available online: https://www.uniprot.org/ (accessed on 16 January 2023).
- Dimitrakopoulos, G.N.; Klapa, M.I.; Moschonas, N.K. PICKLE 3.0: Enriching the human meta-database with the mouse protein interactome extended via mouse–human orthology. Bioinformatics 2020, 37, 145–146. [Google Scholar] [CrossRef]
- Dimitrakopoulos, G.N.; Klapa, M.I.; Moschonas, N.K. How Far Are We from the Completion of the Human Protein Interactome Reconstruction? Biomolecules 2022, 12, 140. [Google Scholar] [CrossRef]
- Gioutlakis, A.; Klapa, M.I.; Moschonas, N.K. PICKLE 2.0: A human protein-protein interaction meta-database employing data integration via genetic information ontology. PLoS ONE 2017, 12, e0186039. [Google Scholar] [CrossRef] [Green Version]
- Klapa, M.I.; Tsafou, K.; Theodoridis, E.; Tsakalidis, A.; Moschonas, N.K. Reconstruction of the experimentally supported human protein interactome: What can we learn? BMC Syst. Biol. 2013, 7, 96. [Google Scholar] [CrossRef] [Green Version]
- Chasapis, C.T.; Kelaidonis, K.; Ridgway, H.; Apostolopoulos, V.; Matsoukas, J.M. The Human Myelin Proteome and Sub-Metalloproteome Interaction Map: Relevance to Myelin-Related Neurological Diseases. Brain Sci. 2022, 12, 434. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Jaccard, P. THE DISTRIBUTION OF THE FLORA IN THE ALPINE ZONE.1. New Phytol. 1912, 11, 37–50. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [Green Version]
- ProteinAnalysis THrough Evolutionary Relationships (PANTHER). Available online: http://www.pantherdb.org/ (accessed on 10 May 2023).
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.-P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef] [Green Version]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [Green Version]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef] [Green Version]
- Safran, M.; Rosen, N.; Twik, M.; BarShir, R.; Stein, T.I.; Dahary, D.; Fishilevich, S.; Lancet, D. The GeneCards Suite. In Practical Guide to Life Science Databases; Abugessaisa, I., Kasukawa, T., Eds.; Springer Nature Singapore: Singapore, 2021; pp. 27–56. [Google Scholar]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.31–1.30.33. [Google Scholar] [CrossRef]
- Schrankl, J.; Fuchs, M.; Broeker, K.; Daniel, C.; Kurtz, A.; Wagner, C. Localization of angiotensin II type 1 receptor gene expression in rodent and human kidneys. Am. J. Physiol.-Ren. Physiol. 2021, 320, F644–F653. [Google Scholar] [CrossRef]
- Meng, Q.; Xia, Y. c-Jun, at the crossroad of the signaling network. Protein Cell 2011, 2, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Dell’Italia, L.J.; Varagic, J. 5—Molecular Signaling Mechanisms of the Renin-Angiotensin System in Heart Failure. In Heart Failure: A Companion to Braunwald’s Heart Disease (Fourth Edition); Felker, G.M., Mann, D.L., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 76–90.e74. [Google Scholar]
- Turner, A.J.; Hooper, N.M. Angiotensin-Converting Enzyme-2 (ACE2). In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–4. [Google Scholar]
- Turner, A.J. Chapter 25—ACE2 Cell Biology, Regulation, and Physiological Functions. In The Protective Arm of the Renin Angiotensin System (RAS); Unger, T., Steckelings, U.M., dos Santos, R.A.S., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 185–189. [Google Scholar]
- Zhang, B.; Xie, W.; Krasowski, M.D. PXR: A xenobiotic receptor of diverse function implicated in pharmacogenetics. Pharmacogenomics 2008, 9, 1695–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Arif, G.; Khazaal, S.; Farhat, A.; Harb, J.; Annweiler, C.; Wu, Y.; Cao, Z.; Kovacic, H.; Abi Khattar, Z.; Fajloun, Z.; et al. Angiotensin II Type I Receptor (AT1R): The Gate towards COVID-19-Associated Diseases. Molecules 2022, 27, 2048. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Huang, J.; Fajas, L.; Li, A.; Welch, J.; Najib, J.; Witztum, J.L.; Auwerx, J.; Palinski, W.; Glass, C.K. Expression of the peroxisome proliferator-activated receptor γ (PPARγ) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1998, 95, 7614–7619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daujat-Chavanieu, M.; Gerbal-Chaloin, S. Regulation of CAR and PXR Expression in Health and Disease. Cells 2020, 9, 2395. [Google Scholar] [CrossRef]
- Qiao, E.; Ji, M.; Wu, J.; Ma, R.; Zhang, X.; He, Y.; Zha, Q.; Song, X.; Zhu, L.W.; Tang, J. Expression of the PXR gene in various types of cancer and drug resistance. Oncol. Lett. 2013, 5, 1093–1100. [Google Scholar] [CrossRef] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-Cell RNA Expression Profiling of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 202, 756–759. [Google Scholar] [CrossRef]
- Smith, L.M.; Kelleher, N.L. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [Green Version]
- Berger, S.I.; Iyengar, R. Role of systems pharmacology in understanding drug adverse events. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.; Poleksic, A.; Yao, Y.; Tong, H.; He, D.; Zhuang, L.; Meng, P.; Xie, L. Large-Scale Off-Target Identification Using Fast and Accurate Dual Regularized One-Class Collaborative Filtering and Its Application to Drug Repurposing. PLoS Comput. Biol. 2016, 12, e1005135. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Gao, M.; Skolnick, J. Comprehensive prediction of drug-protein interactions and side effects for the human proteome. Sci. Rep. 2015, 5, 11090. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Sakagami, H.; Miwa, N. ACE2: The key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel? Viruses 2020, 12, 491. [Google Scholar] [CrossRef]
- Smith, K.P.; Gifford, K.M.; Waitzman, J.S.; Rice, S.E. Survey of phosphorylation near drug binding sites in the Protein Data Bank (PDB) and their effects. Proteins 2015, 83, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Christensen, D.G.; Xie, X.; Basisty, N.; Byrnes, J.; McSweeney, S.; Schilling, B.; Wolfe, A.J. Post-translational Protein Acetylation: An Elegant Mechanism for Bacteria to Dynamically Regulate Metabolic Functions. Front. Microbiol. 2019, 10, 1604. [Google Scholar] [CrossRef] [Green Version]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Et Biophys. Acta (BBA)—Proteins Proteom. 2016, 1864, 1372–1401. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, U.; Ponnuraj, K. Isopeptide bond in collagen- and fibrinogen-binding MSCRAMMs. Biophys. Rev. 2016, 8, 75–83. [Google Scholar] [CrossRef]
- Fu, J.; Gao, J.; Liang, Z.; Yang, D. PDI-Regulated Disulfide Bond Formation in Protein Folding and Biomolecular Assembly. Molecules 2021, 26, 171. [Google Scholar] [CrossRef]
- Siegmund, V.; Piater, B.; Zakeri, B.; Eichhorn, T.; Fischer, F.; Deutsch, C.; Becker, S.; Toleikis, L.; Hock, B.; Betz, U.A.K.; et al. Spontaneous Isopeptide Bond Formation as a Powerful Tool for Engineering Site-Specific Antibody-Drug Conjugates. Sci. Rep. 2016, 6, 39291. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, A.; Wintrode, P.L. Effects of glycosylation on the stability and flexibility of a metastable protein: The human serpin α(1)-antitrypsin. Int. J. Mass Spectrom. 2011, 302, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Shental-Bechor, D.; Levy, Y. Effect of glycosylation on protein folding: A close look at thermodynamic stabilization. Proc. Natl. Acad. Sci. USA 2008, 105, 8256–8261. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Qian, J. Chapter Four—Applications of Functional Protein Microarrays in Basic and Clinical Research. In Advances in Genetics; Friedmann, T., Dunlap, J.C., Goodwin, S.F., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 79, pp. 123–155. [Google Scholar]
- Xiong, Y.; Karuppanan, K.; Bernardi, A.; Li, Q.; Kommineni, V.; Dandekar, A.M.; Lebrilla, C.B.; Faller, R.; McDonald, K.A.; Nandi, S. Effects of N-Glycosylation on the Structure, Function, and Stability of a Plant-Made Fc-Fusion Anthrax Decoy Protein. Front. Plant Sci. 2019, 10, 768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bektas, M.; Rubenstein, D.S. The role of intracellular protein O-glycosylation in cell adhesion and disease. J. Biomed. Res. 2011, 25, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Magalhães, J.P. Every gene can (and possibly will) be associated with cancer. Trends Genet. 2022, 38, 216–217. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.J.; Matsoukas, J.M. Angiotensin as a model for hormone--receptor interactions. Biosci. Rep. 1985, 5, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, P.B.M.W.M. Angiotensin II Receptor Antagonists: An Emerging New Class of Cardiovascular Therapeutics. Hypertens. Res. 1999, 22, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Duarte, M.; Pelorosso, F.; Nicolosi, L.N.; Salgado, M.V.; Vetulli, H.; Aquieri, A.; Azzato, F.; Castro, M.; Coyle, J.; Davolos, I.; et al. Telmisartan for treatment of Covid-19 patients: An open multicenter randomized clinical trial. eClinicalMedicine 2021, 37, 100962. [Google Scholar] [CrossRef]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.J.; Xie, J.; Liu, Y.M.; Zhao, Y.C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality Among Patients With Hypertension Hospitalized With COVID-19. Circ. Res. 2020, 126, 1671–1681. [Google Scholar] [CrossRef] [Green Version]
- Almutlaq, M.; Mansour, F.A.; Alghamdi, J.; Alhendi, Y.; Alamro, A.A.; Alghamdi, A.A.; Alamri, H.S.; Alroqi, F.; Barhoumi, T. Angiotensin II Exaggerates SARS-CoV-2 Specific T-Cell Response in Convalescent Individuals following COVID-19. Int. J. Mol. Sci. 2022, 23, 8669. [Google Scholar] [CrossRef]
- Rysz, S.; Al-Saadi, J.; Sjöström, A.; Farm, M.; Campoccia Jalde, F.; Plattén, M.; Eriksson, H.; Klein, M.; Vargas-Paris, R.; Nyrén, S.; et al. COVID-19 pathophysiology may be driven by an imbalance in the renin-angiotensin-aldosterone system. Nat. Commun. 2021, 12, 2417. [Google Scholar] [CrossRef]
- Platten, M.; Youssef, S.; Hur, E.M.; Ho, P.P.; Han, M.H.; Lanz, T.V.; Phillips, L.K.; Goldstein, M.J.; Bhat, R.; Raine, C.S.; et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc. Natl. Acad. Sci. USA 2009, 106, 14948–14953. [Google Scholar] [CrossRef] [Green Version]
- Stegbauer, J.; Lee, D.H.; Seubert, S.; Ellrichmann, G.; Manzel, A.; Kvakan, H.; Muller, D.N.; Gaupp, S.; Rump, L.C.; Gold, R.; et al. Role of the renin-angiotensin system in autoimmune inflammation of the central nervous system. Proc. Natl. Acad. Sci. USA 2009, 106, 14942–14947. [Google Scholar] [CrossRef] [Green Version]
- Dargahi, N.; Matsoukas, J.; Apostolopoulos, V. Streptococcus thermophilus ST285 Alters Pro-Inflammatory to Anti-Inflammatory Cytokine Secretion against Multiple Sclerosis Peptide in Mice. Brain Sci. 2020, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Katsara, M.; Yuriev, E.; Ramsland, P.A.; Deraos, G.; Tselios, T.; Matsoukas, J.; Apostolopoulos, V. Mannosylation of mutated MBP83–99 peptides diverts immune responses from Th1 to Th2. Mol. Immunol. 2008, 45, 3661–3670. [Google Scholar] [CrossRef]
- Hondrelis, J.; Lonergan, G.; Voliotis, S.; Matsoukas, J. One pot synthesis and conformation of N-t-butyloxycarbonyl, O-Phenacyl derivatives of proline and other secondary amino acids. Tetrahedron 1990, 46, 565–576. [Google Scholar] [CrossRef]
- Matsoukas, J.; Cordopatis, P.; Belte, U.; Goghari, M.H.; Ganter, R.C.; Franklin, K.J.; Moore, G.J. Importance of the N-terminal domain of the type II angiotensin antagonist sarmesin for receptor blockade. J. Med. Chem. 1988, 31, 1418–1421. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Agelis, G.; Hondrelis, J.; Yamdagni, R.; Wu, Q.; Ganter, R.; Smith, J.R.; Moore, D.; Moore, G.J. Synthesis and biological activities of angiotensin II, Sarilesin, and Sarmesin analogues containing Aze or Pip at position 7. J. Med. Chem. 1993, 36, 904–911. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Hondrelis, J.; Keramida, M.; Mavromoustakos, T.; Makriyannis, A.; Yamdagni, R.; Wu, Q.; Moore, G.J. Role of the NH2-terminal domain of angiotensin II (ANG II) and [Sar1]angiotensin II on conformation and activity. NMR evidence for aromatic ring clustering and peptide backbone folding compared with [des-1,2,3]angiotensin II. J. Biol. Chem. 1994, 269, 5303–5312. [Google Scholar] [CrossRef]
- Polevaya, L.; Mavromoustakos, T.; Zoumboulakis, P.; Golic Grdadolnik, S.; Roumelioti, P.; Giatas, N.; Mutule, I.; Keivish, T.; Vlahakos, D.V.; Iliodromitis, E.K.; et al. Synthesis and study of a cyclic angiotensin II antagonist analogue reveals the role of pi*--pi* interactions in the C-terminal aromatic residue for agonist activity and its structure resemblance with AT(1) non-peptide antagonists. Bioorg. Med. Chem. 2001, 9, 1639–1647. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Panagiotopoulos, D.; Keramida, M.; Mavromoustakos, T.; Yamdagni, R.; Wu, Q.; Moore, G.J.; Saifeddine, M.; Hollenberg, M.D. Synthesis and Contractile Activities of Cyclic Thrombin Receptor-Derived Peptide Analogues with a Phe-Leu-Leu-Arg Motif: Importance of the Phe/Arg Relative Conformation and the Primary Amino Group for Activity. J. Med. Chem. 1996, 39, 3585–3591. [Google Scholar] [CrossRef]
- Cheng, Y.W.; Chao, T.L.; Li, C.L.; Chiu, M.F.; Kao, H.C.; Wang, S.H.; Pang, Y.H.; Lin, C.H.; Tsai, Y.M.; Lee, W.H.; et al. Furin Inhibitors Block SARS-CoV-2 Spike Protein Cleavage to Suppress Virus Production and Cytopathic Effects. Cell Rep. 2020, 33, 108254. [Google Scholar] [CrossRef]
- Osman, E.E.A.; Rehemtulla, A.; Neamati, N. Why All the Fury over Furin? J. Med. Chem. 2022, 65, 2747–2784. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zheng, M.; Yang, Y.; Gu, X.; Yang, K.; Li, M.; Liu, Y.; Zhang, Q.; Zhang, P.; Wang, Y.; et al. Furin: A Potential Therapeutic Target for COVID-19. iScience 2020, 23, 101642. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef] [PubMed]
- Brevini, T.; Maes, M.; Webb, G.J.; John, B.V.; Fuchs, C.D.; Buescher, G.; Wang, L.; Griffiths, C.; Brown, M.L.; Scott, W.E.; et al. FXR inhibition may protect from SARS-CoV-2 infection by reducing ACE2. Nature 2022, 615, 134–142. [Google Scholar] [CrossRef]
- Zhang, F.; Jenkins, J.; de Carvalho, R.V.H.; Nakandakari-Higa, S.; Chen, T.; Abernathy, M.E.; Nyakatura, E.; Andrew, D.; Lebedeva, I.; Lorenz, I.C.; et al. Human anti-ACE2 monoclonal antibodies as pan-sarbecovirus prophylactic agents. bioRxiv 2022. [Google Scholar] [CrossRef]
- COVID-19 Drug Interactions. Available online: https://www.covid19-druginteractions.org/ (accessed on 2 February 2023).
- Backman, J.T.; Filppula, A.M.; Niemi, M.; Neuvonen, P.J. Role of Cytochrome P450 2C8 in Drug Metabolism and Interactions. Pharmacol. Rev. 2016, 68, 168–241. [Google Scholar] [CrossRef] [Green Version]
- Drug Interaction Checker (DrugBank). Available online: https://go.drugbank.com/drug-interaction-checker (accessed on 6 February 2023).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | UniProt Entry Name | Organism | Protein Name | Number of Drugs | Experimentally Supported Events of Proteoforms c |
|---|---|---|---|---|---|
| Sartans a | AGTR1_HUMAN | HUMAN | Type-1 angiotensin II receptor | 9 | 19 |
| JUN_HUMAN | HUMAN | Transcription factor AP-1 | 4 | 20 | |
| PPARG_HUMAN | HUMAN | Peroxisome proliferator-activated receptor γ | 30 | 32 | |
| ACE2_HUMAN b | HUMAN | Angiotensin-converting enzyme 2 | 4 | 21 | |
| Nirmatrelvir | NR1I2_HUMAN | HUMAN | Nuclear receptor subfamily 1 group I member 2 | 51 | 4 |
| Ritonavir | R1AB_SARS2 | SARS-CoV-2 | 3CLpro | 2 | - |
| Protein Target | RNA Tissue Specificity | Protein Tissue Expression | Subcellular Location |
|---|---|---|---|
| AGTR1 | Tissue enhanced (liver, placenta) | Cytoplasmic expression in adipocytes and endothelial cells. | Vesicles (Membrane) |
| JUN | Low tissue specificity (overexpressed in cancer tissue) | Nuclear expression in several tissues, mostly in a fraction of the cells. | Nucleoplasm (Intracellular) |
| PPARG | Tissue enhanced (adipose tissue) | Cytoplasmic and nuclear expression in several tissues. | Nucleoplasm, Vesicles (Intracellular) |
| ACE2 | Tissue enhanced (gallbladder, intestine, kidney) | Membranous expression in proximal renal tubules, intestinal tract, seminal vesicle, epididymis, exocrine pancreas, and gallbladder. Expressed in Sertoli and Leydig cells, and trophoblasts. Membranous expression in ciliated cells in nasal mucosa, bronchus, and fallopian tube. Expressed in endothelial cells and pericytes in many tissues. | Membrane, Secreted to blood (different isoforms) |
| NR1I2 | Group enriched (intestine, liver) | Not available | Nucleoplasm (Intracellular) |
| DrugA | DrugB | Shared (M11) | Unique to DrugA (M10) | Unique to DrugB (M01) | Jaccard Index (J) | Jaccard Distance (dJ) |
|---|---|---|---|---|---|---|
| Sartans | Paxlovid | 17 | 303 | 11 | 0.05136 | 0.94864 |
| Sartans (COVID-19) | Paxlovid | 0 | 60 | 28 | 0 | 1 |
| Sartans | Perphenazine | 19 | 301 | 132 | 0.042035 | 0.957965 |
| Sartans (COVID-19) | Perphenazine | 12 | 308 | 139 | 0.026144 | 0.973856 |
| Paxlovid | Perphenazine | 0 | 28 | 151 | 0 | 1 |
| Sartans | Sartans (COVID-19) | 51 | 269 | 9 | 0.155015 | 0.844985 |
| Drug | Go Term | Fold Enrichment | p-Value | FDR |
|---|---|---|---|---|
| Sartans | transcription factor binding (GO:0008134) | 11.23 | 3.09 × 10−80 | 1.56 × 10−76 |
| binding (GO:0005488) | 1.23 | 3.87 × 10−26 | 7.54 × 10−24 | |
| protein domain specific binding (GO:0019904) | 4.47 | 1.08 × 10−17 | 1.65 × 10−15 | |
| acetyltransferase activity (GO:0016407) | 8.22 | 2.43 × 10−8 | 1.81 × 10−6 | |
| phosphothreonine residue binding (GO:0050816) | 60.73 | 8.13 × 10−5 | 3.77 × 10−3 | |
| transferase activity (GO:0016740) | 1.70 | 2.94 × 10−5 | 1.49 × 10−3 | |
| catalytic activity, acting on DNA (GO:0140097) | 3.31 | 1.46 × 10−4 | 6.27 × 10−3 | |
| peptide butyryltransferase activity (GO:0140065) | 60.73 | 1.54 × 10−3 | 5.01 × 10−2 | |
| peptide crotonyltransferase activity (GO:0140064) | 60.73 | 1.54 × 10−3 | 4.98 × 10−2 | |
| histone H2B acetyltransferase activity (GO:0044013) | 60.73 | 1.54 × 10−3 | 4.86 × 10−2 | |
| Paxlovid | transcription factor binding (GO:0008134) | 19.75 | 4.99 × 10−19 | 1.26 × 10-15 |
| transcription regulator activity (GO:0140110) | 7.26 | 8.88 × 10−13 | 8.98 × 10-10 | |
| nuclear steroid receptor activity (GO:0003707) | >100 | 8.43 × 10−8 | 3.55 × 10−5 | |
| DNA binding (GO:0003677) | 4.22 | 3.08 × 10−7 | 8.65 × 10−5 | |
| nuclear thyroid hormone receptor binding (GO:0046966) | 97.93 | 1.26 × 10−7 | 4.53 × 10−5 | |
| nuclear estrogen receptor binding (GO:0030331) | 51.95 | 3.19 × 10−5 | 5.37 × 10−3 | |
| transcription coregulator binding (GO:0001221) | 24.48 | 2.32 × 10−5 | 4.34 × 10−3 | |
| acetyltransferase activity (GO:0016407) | 22.19 | 3.58 × 10−4 | 3.94 × 10−2 | |
| DNA-binding transcription factor activity (GO:0003700) | 4.52 | 2.64 × 10−4 | 3.11 × 10−2 | |
| STAT family protein binding (GO:0097677) | >100 | 1.72 × 10−4 | 2.12 × 10−2 | |
| Sartans (COVID-19) | exogenous protein binding (GO:0140272) | 57.92 | 1.13 × 10−9 | 5.73 × 10−6 |
| G protein-coupled receptor binding (GO:0001664) | 8.74 | 7.10 × 10−8 | 1.20 × 10−4 | |
| adenylate cyclase regulator activity (GO:0010854) | >100 | 3.28 × 10−7 | 3.32 × 10−4 | |
| phosphatase activator activity (GO:0019211) | >100 | 4.52 × 10−6 | 2.08 × 10−3 | |
| protein-containing complex binding (GO:0044877) | 5.85 | 3.31 × 10−6 | 1.68 × 10−3 | |
| beta-adrenergic receptor kinase activity (GO:0047696) | >100 | 2.49 × 10−6 | 2.10 × 10−3 | |
| titin binding (GO:0031432) | >100 | 1.11 × 10−6 | 8.01 × 10−4 | |
| dopamine receptor binding (GO:0050780) | 41.76 | 7.79 × 10−5 | 3.03 × 10−2 | |
| molecular function regulator activity (GO:0098772) | 2.42 | 6.97 × 10−5 | 2.94 × 10−2 | |
| binding (GO:0005488) | 1.19 | 1.09 × 10−4 | 3.92 × 10−2 |
| UniProtID | Protein Name | Degree | Experimentally Supported Events of Proteoforms a | |
|---|---|---|---|---|
| AT1R (Mean Degree: 74) | P00533 | Epidermal growth factor receptor | 752 | 73 |
| P04049 | RAF proto-oncogene serine/threonine-protein kinase | 226 | 21 | |
| P19174 | 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma-1 | 206 | 15 | |
| Q8ND90 | Paraneoplastic antigen Ma1 | 174 | 1 | |
| P63244 | Receptor of activated protein C kinase 1 | 171 | 17 | |
| Q9NZD8 | Maspardin | 164 | 2 | |
| P49407 | Beta-arrestin-1 | 140 | 3 | |
| Q6RW13 | Type-1 angiotensin II receptor-associated protein | 139 | 6 | |
| P32121 | Beta-arrestin-2 | 138 | 6 | |
| Q96CW1 | AP-2 complex subunit mu | 135 | 3 | |
| ACE2 (Mean Degree: 170) | Q00987 | E3 ubiquitin-protein ligase Mdm2 | 513 | 14 |
| P0DP24 | Calmodulin-2 | 401 | 15 | |
| P0DP23 | Calmodulin-1 | 329 | 15 | |
| P0DP25 | Calmodulin-3 | 328 | 15 | |
| Q15517 | Corneodesmosin | 42 | 3 | |
| Q9BYF1 | Angiotensin-converting enzyme 2 | 28 | 15 | |
| Q15036 | Sorting nexin-17 | 28 | 8 | |
| P05814 | Beta-casein | 22 | 7 | |
| Q9UBU3 | Appetite-regulating hormone | 12 | 10 | |
| Q01523 | Defensin alpha 5 | 4 | 9 | |
| NR1I2 (Mean Degree: 153) | P04637 | Cellular tumor antigen p53 | 857 | 33 |
| Q09472 | Histone acetyltransferase p300 | 551 | 34 | |
| P63279 | SUMO-conjugating enzyme UBC9 | 548 | 11 | |
| P12931 | Proto-oncogene tyrosine-protein kinase Src | 492 | 9 | |
| Q16531 | DNA damage-binding protein 1 | 241 | 6 | |
| O75376 | Nuclear receptor corepressor 1 | 168 | 33 | |
| P19793 | Retinoic acid receptor RXR-alpha | 136 | 12 | |
| P35609 | Alpha-actinin-2 | 128 | 2 | |
| O95071 | E3 ubiquitin-protein ligase UBR5 | 126 | 36 | |
| Q9Y6Q9 | Nuclear receptor coactivator 3 | 120 | 21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiouri, D.P.; Ntallis, C.; Kelaidonis, K.; Peana, M.; Tsiodras, S.; Mavromoustakos, T.; Giuliani, A.; Ridgway, H.; Moore, G.J.; Matsoukas, J.M.; et al. Network-Based Prediction of Side Effects of Repurposed Antihypertensive Sartans against COVID-19 via Proteome and Drug-Target Interactomes. Proteomes 2023, 11, 21. https://doi.org/10.3390/proteomes11020021
Kiouri DP, Ntallis C, Kelaidonis K, Peana M, Tsiodras S, Mavromoustakos T, Giuliani A, Ridgway H, Moore GJ, Matsoukas JM, et al. Network-Based Prediction of Side Effects of Repurposed Antihypertensive Sartans against COVID-19 via Proteome and Drug-Target Interactomes. Proteomes. 2023; 11(2):21. https://doi.org/10.3390/proteomes11020021
Chicago/Turabian StyleKiouri, Despoina P., Charalampos Ntallis, Konstantinos Kelaidonis, Massimiliano Peana, Sotirios Tsiodras, Thomas Mavromoustakos, Alessandro Giuliani, Harry Ridgway, Graham J. Moore, John M. Matsoukas, and et al. 2023. "Network-Based Prediction of Side Effects of Repurposed Antihypertensive Sartans against COVID-19 via Proteome and Drug-Target Interactomes" Proteomes 11, no. 2: 21. https://doi.org/10.3390/proteomes11020021
APA StyleKiouri, D. P., Ntallis, C., Kelaidonis, K., Peana, M., Tsiodras, S., Mavromoustakos, T., Giuliani, A., Ridgway, H., Moore, G. J., Matsoukas, J. M., & Chasapis, C. T. (2023). Network-Based Prediction of Side Effects of Repurposed Antihypertensive Sartans against COVID-19 via Proteome and Drug-Target Interactomes. Proteomes, 11(2), 21. https://doi.org/10.3390/proteomes11020021