





Proteomes 2026, 14(3), 35; https://doi.org/10.3390/proteomes14030035 - 15 Jul 2026
Abstract
Background: Multi-drug resistant Gram-negative bacteria (GNB) are major contributors to the antimicrobial resistance (AMR) burden. AMR mechanisms are primarily mediated by proteoforms; therefore, proteomic analyses of GNB offers a significant advantage in understanding the mechanisms of AMR. A large portion of these mechanisms
[...] Read more.
Background: Multi-drug resistant Gram-negative bacteria (GNB) are major contributors to the antimicrobial resistance (AMR) burden. AMR mechanisms are primarily mediated by proteoforms; therefore, proteomic analyses of GNB offers a significant advantage in understanding the mechanisms of AMR. A large portion of these mechanisms are mediated by membrane proteins; however, they are often difficult to extract due to their hydrophobic nature and complex interactions with other components of the cell membrane. To extract the greatest number of proteoforms, an efficient homogenisation protocol is required to effectively disrupt the rigid cell wall and membrane. Methods: Using Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii and Pseudomonas aeruginosa, we systematically compared the extraction efficiency of bead-beating with flash frozen and lyophilized cell pellets. Results: We demonstrate that lyophilization improves bead-beating extraction methods by increasing the detection of membrane proteins. We detected numerous unique membrane proteins in each bacterial isolate, including ABC transporters and proteins involved in lipopolysaccharide synthesis, when lyophilizing prior to bead-beating, compared to only flash-freezing. Conclusions: As membrane proteins play a central role in AMR mechanisms, this improvement in their isolation and identification will aid in understanding the resistance and molecular mechanisms associated with multi-drug resistant GNB.
Full article
(This article belongs to the Section Proteomics Technology and Methodology Development)
►
Show Figures
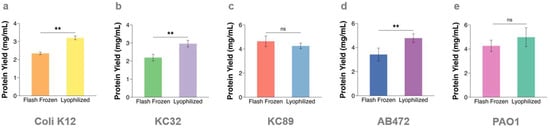
Figure 1





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}