

Key Proteomics Tools for Fundamental and Applied Microalgal Research

Abstract
:1. Introduction
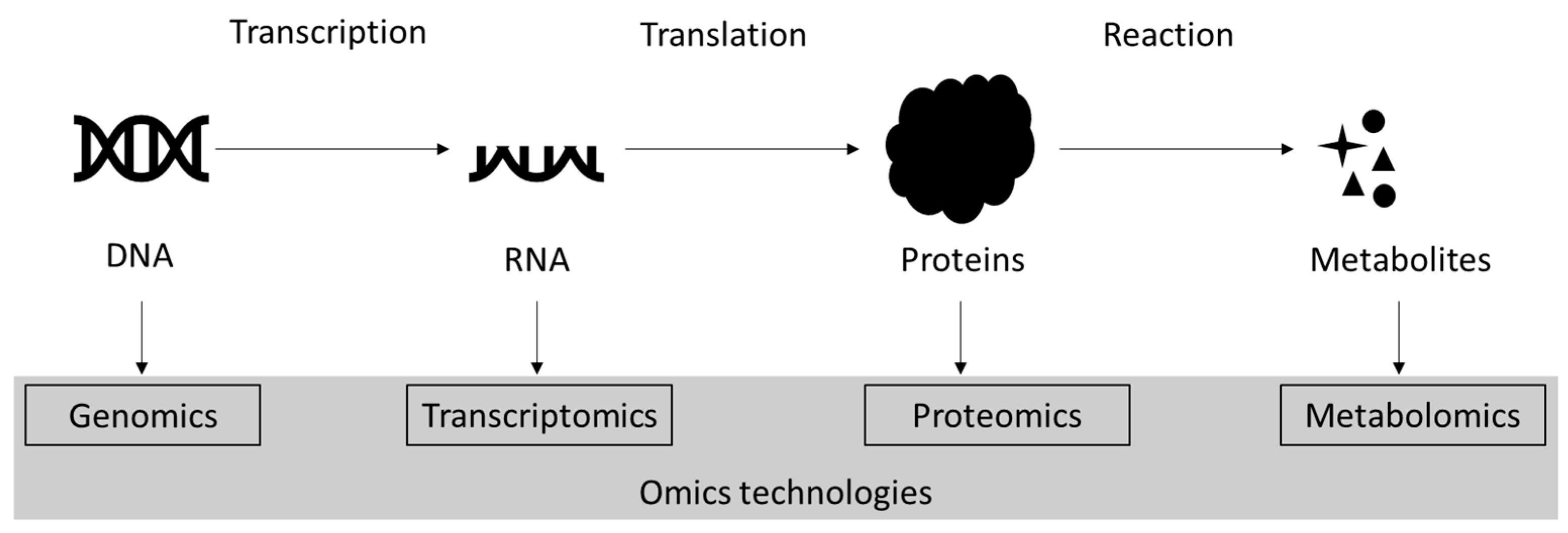
2. Proteomics
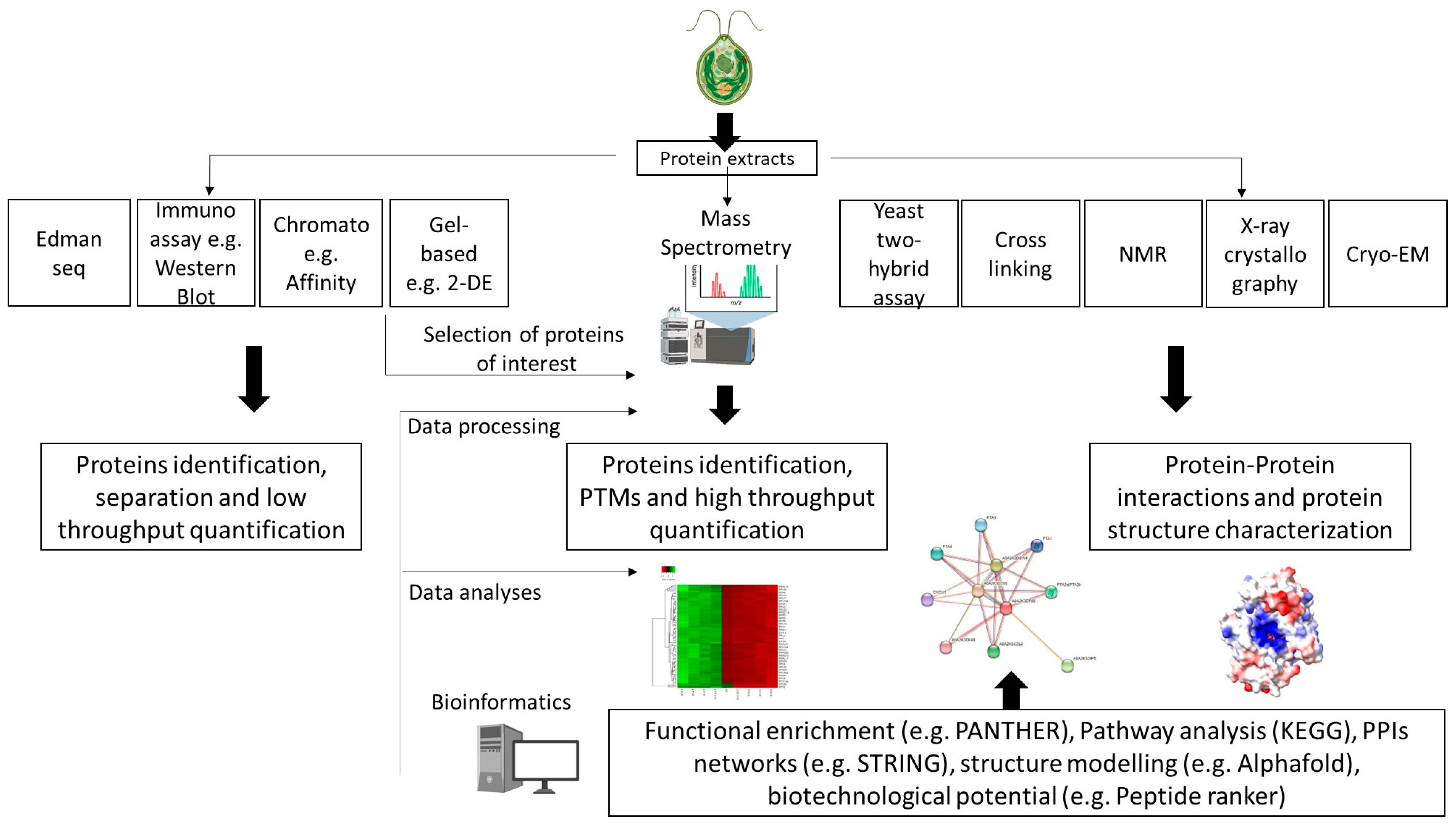
3. Overview of Proteomics Techniques Applied in Microalgae Research
3.1. Quantitative High-Throughput Proteomics: Mass Spectrometry
3.2. Protein–Protein Interaction Techniques
4. Key Considerations for Successful Proteomics
4.1. Protein Sample Preparation
4.1.1. Protein Extraction
4.1.2. Sample Preparation for MS-Based Analysis
4.2. Bioinformatics
4.2.1. MS Data Processing
- Protein samples are prepared for MS, i.e., solubilized proteins are enzymatically digested (usually using trypsin), chemically modified (alkylation) and purified (de-salting) to obtain short MS-accessible peptides;
- Peptides are separated using an LC setup that is coupled to a mass spectrometer (LC-MS);
- Intact peptide masses and the corresponding masses of fragmented peptide ions are measured by mass spectrometry (i.e., tandem LC-MS/MS or MSn setups). As mentioned in Section 3.1, label-based and label-free strategies can be used for protein quantification, and different techniques are available (Table 1). Notably, different data acquisition modes can also be used (e.g., label-free data-dependent acquisition and data-independent acquisition); refer to Schessner et al. [90] for further details;
- Based on a reference proteome, the resulting peptide and fragment ion spectra are used to identify the peptides present in the sample;
- Identified peptide sequences are quantified and assembled to measure protein levels by protein inference.Data from MS analyses are susceptible to systematic, dependent or independent biases (e.g., different handling, equipment calibration) on the measured peptide/protein abundances [102]. Therefore, a key step is to normalize the data to take the bias into account, allowing the data to be comparable and downstream analyses reliable [103,104,105]. Advanced analysis pipeline frameworks are therefore needed for data normalization but also for protein inference and data analysis [88,89,103,104,106]. The latter has been extensively reviewed by Schessner et al. [90] in their guide to interpreting and generate visual representation of bottom-up proteomics data.
4.2.2. MS Data Interpretation
4.2.3. Functional Characterization
5. The Benefit of Proteomics in Microalgal Research
5.1. Fundamental Research
5.2. Applied Research
6. Proteomics in the Post-Omics Era
7. Future Prospects
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Falkowski, P.G. The role of phytoplankton photosynthesis in global biogeochemical cycles. Photosynth. Res. 1994, 39, 235–258. [Google Scholar] [CrossRef] [PubMed]
- Bjorbækmo, M.F.M.; Evenstad, A.; Røsæg, L.L.; Krabberød, A.K.; Logares, R. The planktonic protist interactome: Where do we stand after a century of research? ISME J. 2020, 14, 544–559. [Google Scholar] [CrossRef] [PubMed]
- Falkowski, P.G. The power of plankton. Nature 2012, 483, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Plouviez, M.; Oliveira da Rocha, C.S.; Guieysse, B. Intracellular polyphosphate is a P reserve in Chlamydomonas reinhardtii. Algal Res. 2022, 66, 102779. [Google Scholar] [CrossRef]
- Teuma, L.; Sanz-Luque, E.; Guieysse, B.; Plouviez, M. Are Microalgae New Players in Nitrous Oxide Emissions from Eutrophic Aquatic Environments? Phycology 2023, 3, 356–367. [Google Scholar] [CrossRef]
- Thoré, E.S.J.; Muylaert, K.; Bertram, M.G.; Brodin, T. Microalgae. Curr. Biol. 2023, 33, R91–R95. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Marechal-Drouard, L.; et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Salome, P.A.; Merchant, S.S. A Series of Fortunate Events: Introducing Chlamydomonas as a Reference Organism. Plant Cell 2019, 31, 1682–1707. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, S.; Merchant, S.S. Chlamydomonas reinhardtii: A model for photosynthesis and so much more. Nat. Methods 2023, 20, 1441–1442. [Google Scholar] [CrossRef]
- Findinier, J.; Grossman, A.R. Chlamydomonas: Fast tracking from genomics. J. Phycol. 2023, 59, 644–652. [Google Scholar] [CrossRef]
- Plouviez, M.; Chisti, Y.; Guieysse, B. Practical guide to algal biomass production: What can we learn from past successes and failures? In 3rd Generation Biofuels; Woodhead Publishing: Cambridge, UK, 2022; pp. 979–1000. [Google Scholar] [CrossRef]
- Chen, H.; Li, T.; Wang, Q. Ten years of algal biofuel and bioproducts: Gains and pains. Planta 2019, 249, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Hamzelou, S.; Belobrajdic, D.; Broadbent, J.A.; Juhász, A.; Lee Chang, K.; Jameson, I.; Ralph, P.; Colgrave, M.L. Utilizing proteomics to identify and optimize microalgae strains for high-quality dietary protein: A review. Crit. Rev. Biotechnol. 2023, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Arend, M.; Zimmer, D.; Xu, R.; Sommer, F.; Muhlhaus, T.; Nikoloski, Z. Proteomics and constraint-based modelling reveal enzyme kinetic properties of Chlamydomonas reinhardtii on a genome scale. Nat. Commun. 2023, 14, 4781. [Google Scholar] [CrossRef]
- Anand, V.; Singh, P.K.; Banerjee, C.; Shukla, P. Proteomic approaches in microalgae: Perspectives and applications. 3 Biotech 2017, 7, 197. [Google Scholar] [CrossRef]
- Kumar, G.; Shekh, A.; Jakhu, S.; Sharma, Y.; Kapoor, R.; Sharma, T.R. Bioengineering of Microalgae: Recent Advances, Perspectives, and Regulatory Challenges for Industrial Application. Front. Bioeng. Biotechnol. 2020, 8, 914. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kar, S.; Srivastava, A.; Banerjee, C.; Shukla, P. Algal genomics tools: Technological updates and progress. In Microbial Bioprocesses; Academic Press: Cambridge, MA, USA, 2023; pp. 67–81. [Google Scholar] [CrossRef]
- Chakdar, H.; Hasan, M.; Pabbi, S.; Nevalainen, H.; Shukla, P. High-throughput proteomics and metabolomic studies guide re-engineering of metabolic pathways in eukaryotic microalgae: A review. Bioresour. Technol. 2021, 321, 124495. [Google Scholar] [CrossRef]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Bludau, I.; Aebersold, R. Proteomic and interactomic insights into the molecular basis of cell functional diversity. Nat. Rev. Mol. Cell Biol. 2020, 21, 327–340. [Google Scholar] [CrossRef]
- Cui, M.; Cheng, C.; Zhang, L. High-throughput proteomics: A methodological mini-review. Lab. Investig. 2022, 102, 1170–1181. [Google Scholar] [CrossRef]
- Dai, X.; Shen, L. Advances and Trends in Omics Technology Development. Front. Med. 2022, 9, 911861. [Google Scholar] [CrossRef]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef] [PubMed]
- Flores, J.E.; Claborne, D.M.; Weller, Z.D.; Webb-Robertson, B.M.; Waters, K.M.; Bramer, L.M. Missing data in multi-omics integration: Recent advances through artificial intelligence. Front. Artif. Intell. 2023, 6, 1098308. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, T.D. Omics in Systems Biology: Current Progress and Future Outlook. Proteomics 2021, 21, 2000235. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.F.; Zhou, X.X.; Willems, S.; Ammar, C.; Wahle, M.; Bludau, I.; Voytik, E.; Strauss, M.T.; Mann, M. AlphaPeptDeep: A modular deep learning framework to predict peptide properties for proteomics. Nat. Commun. 2022, 13, 7238. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Kumar, C.; Zeng, W.F.; Strauss, M.T. Artificial intelligence for proteomics and biomarker discovery. Cell Syst. 2021, 12, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Stransky, S.; Sun, Y.; Shi, X.; Sidoli, S. Ten questions to AI regarding the present and future of proteomics. Front. Mol. Biosci. 2023, 10, 1–8. [Google Scholar] [CrossRef]
- Swan, A.L.; Mobasheri, A.; Allaway, D.; Liddell, S.; Bacardit, J. Application of machine learning to proteomics data: Classification and biomarker identification in postgenomics biology. Omics 2013, 17, 595–610. [Google Scholar] [CrossRef]
- Klukowski, P.; Riek, R.; Guntert, P. Rapid protein assignments and structures from raw NMR spectra with the deep learning technique ARTINA. Nat. Commun. 2022, 13, 6151. [Google Scholar] [CrossRef]
- Yan, S.; Bhawal, R.; Yin, Z.; Thannhauser, T.W.; Zhang, S. Recent advances in proteomics and metabolomics in plants. Mol. Hortic. 2022, 2, 17. [Google Scholar] [CrossRef]
- Carrasco-Reinado, R.; Bermudez-Sauco, M.; Escobar-Nino, A.; Cantoral, J.M.; Fernandez-Acero, F.J. Development of the “Applied Proteomics” Concept for Biotechnology Applications in Microalgae: Example of the Proteome Data in Nannochloropsis gaditana. Mar. Drugs 2021, 20, 38. [Google Scholar] [CrossRef]
- Chandramouli, K.; Qian, P.Y. Proteomics: Challenges, techniques and possibilities to overcome biological sample complexity. Hum. Genom. Proteom. 2009, 2009, 239204. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Davey, N.E.; Simonetti, L.; Ivarsson, Y. The next wave of interactomics: Mapping the SLiM-based interactions of the intrinsically disordered proteome. Curr. Opin. Struct. Biol. 2023, 80, 102593. [Google Scholar] [CrossRef] [PubMed]
- Luck, K.; Kim, D.K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef]
- Dyhrman, S.T.; Jenkins, B.D.; Rynearson, T.A.; Saito, M.A.; Mercier, M.L.; Alexander, H.; Whitney, L.P.; Drzewianowski, A.; Bulygin, V.V.; Bertrand, E.M.; et al. The transcriptome and proteome of the diatom Thalassiosira pseudonana reveal a diverse phosphorus stress response. PLoS ONE 2012, 7, e33768. [Google Scholar] [CrossRef]
- Mettler, T.; Muhlhaus, T.; Hemme, D.; Schottler, M.A.; Rupprecht, J.; Idoine, A.; Veyel, D.; Pal, S.K.; Yaneva-Roder, L.; Winck, F.V.; et al. Systems Analysis of the Response of Photosynthesis, Metabolism, and Growth to an Increase in Irradiance in the Photosynthetic Model Organism Chlamydomonas reinhardtii. Plant Cell 2014, 26, 2310–2350. [Google Scholar] [CrossRef] [PubMed]
- de Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef]
- Plouviez, M.; Abyadeh, M.; Hasan, M.; Mirzaei, M.; Paulo, J.A.; Guieysse, B. The proteome of Chlamydomonas reinhardtii during phosphorus depletion and repletion. Algal Res. 2023, 71, 103037. [Google Scholar] [CrossRef]
- Cliff, A.; Guieysse, B.; Brown, N.; Lockhart, P.; Dubreucq, E.; Plouviez, M. Polyphosphate synthesis is an evolutionarily ancient phosphorus storage strategy in microalgae. Algal Res 2023, 73, 103161. [Google Scholar] [CrossRef]
- Plouviez, M.; Fernandez, E.; Grossman, A.R.; Sanz-Luque, E.; Sells, M.; Wheeler, D.; Guieysse, B. Responses of Chlamydomonas reinhardtii during the transition from P-deficient to P-sufficient growth (the P-overplus response): The roles of the vacuolar transport chaperones and polyphosphate synthesis. J. Phycol. 2021, 57, 988–1003. [Google Scholar] [CrossRef]
- Sithtisarn, S.; Yokthongwattana, K.; Mahong, B.; Roytrakul, S.; Paemanee, A.; Phaonakrop, N.; Yokthongwattana, C. Comparative proteomic analysis of Chlamydomonas reinhardtii control and a salinity-tolerant strain revealed a differential protein expression pattern. Planta 2017, 246, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Terashima, M.; Specht, M.; Hippler, M. The chloroplast proteome: A survey from the Chlamydomonas reinhardtii perspective with a focus on distinctive features. Curr. Genet. 2011, 57, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Gessner, G.; Luff, M.; Heiland, I.; Wagner, V.; Kaminski, M.; Geimer, S.; Eitzinger, N.; Reissenweber, T.; Voytsekh, O.; et al. Proteomic analysis of the eyespot of Chlamydomonas reinhardtii provides novel insights into its components and tactic movements. Plant Cell 2006, 18, 1908–1930. [Google Scholar] [CrossRef] [PubMed]
- Perez-Perez, M.E.; Mauries, A.; Maes, A.; Tourasse, N.J.; Hamon, M.; Lemaire, S.D.; Marchand, C.H. The Deep Thioredoxome in Chlamydomonas reinhardtii: New Insights into Redox Regulation. Mol. Plant 2017, 10, 1107–1125. [Google Scholar] [CrossRef]
- Morisse, S.; Zaffagnini, M.; Gao, X.H.; Lemaire, S.D.; Marchand, C.H. Insight into protein S-nitrosylation in Chlamydomonas reinhardtii. Antioxid. Redox Signal. 2014, 21, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Werth, E.G.; McConnell, E.W.; Gilbert, T.S.K.; Couso Lianez, I.; Perez, C.A.; Manley, C.K.; Graves, L.M.; Umen, J.G.; Hicks, L.M. Probing the global kinome and phosphoproteome in Chlamydomonas reinhardtii via sequential enrichment and quantitative proteomics. Plant J. 2017, 89, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Wienkoop, S.; Weiss, J.; May, P.; Kempa, S.; Irgang, S.; Recuenco-Munoz, L.; Pietzke, M.; Schwemmer, T.; Rupprecht, J.; Egelhofer, V.; et al. Targeted proteomics for Chlamydomonas reinhardtii combined with rapid subcellular protein fractionation, metabolomics and metabolic flux analyses. Mol. Biosyst. 2010, 6, 1018–1031. [Google Scholar] [CrossRef] [PubMed]
- Mastrobuoni, G.; Irgang, S.; Pietzke, M.; Aßmus, H.E.; Wenzel, M.; Schulze, W.X.; Kempa, S. Proteome dynamics and early salt stress response of the photosynthetic organism Chlamydomonas reinhardtii. BMC Genom. 2012, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Mathieu-Rivet, E.; Scholz, M.; Arias, C.; Dardelle, F.; Schulze, S.; Le Mauff, F.; Teo, G.; Hochmal, A.K.; Blanco-Rivero, A.; Loutelier-Bourhis, C.; et al. Exploring the N-glycosylation pathway in Chlamydomonas reinhardtii unravels novel complex structures. Mol. Cell. Proteom. 2013, 12, 3160–3183. [Google Scholar] [CrossRef]
- McConnell, E.W.; Werth, E.G.; Hicks, L.M. The phosphorylated redox proteome of Chlamydomonas reinhardtii: Revealing novel means for regulation of protein structure and function. Redox Biol. 2018, 17, 35–46. [Google Scholar] [CrossRef]
- Wang, H.; Gau, B.; Slade, W.O.; Juergens, M.; Li, P.; Hicks, L.M. The global phosphoproteome of Chlamydomonas reinhardtii reveals complex organellar phosphorylation in the flagella and thylakoid membrane. Mol. Cell. Proteom. 2014, 13, 2337–2353. [Google Scholar] [CrossRef] [PubMed]
- Gurrieri, L.; Del Giudice, A.; Demitri, N.; Falini, G.; Pavel, N.V.; Zaffagnini, M.; Polentarutti, M.; Crozet, P.; Marchand, C.H.; Henri, J.; et al. Arabidopsis and Chlamydomonas phosphoribulokinase crystal structures complete the redox structural proteome of the Calvin&Benson cycle. Proc. Natl. Acad. Sci. USA 2019, 116, 8048–8053. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Huang, Z.; Chang, S.; Wang, W.; Wang, J.; Kuang, T.; Han, G.; Shen, J.-R.; Zhang, X. Structure of a C2S2M2N2-type PSII;LHCII supercomplex from the green alga Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 2019, 116, 21246–21255. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, E.; Borner, G.H.H. Spatial proteomics: A powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Patena, W.; Van Baalen, K.A.; Xie, Y.; Singer, E.R.; Gavrilenko, S.; Warren-Williams, M.; Han, L.; Harrigan, H.R.; Hartz, L.D.; et al. A chloroplast protein atlas reveals punctate structures and spatial organization of biosynthetic pathways. Cell 2023, 186, 3499–3518.e3414. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Marchand, C.H.; Maes, A.; Mauries, A.; Sun, Y.; Dhaliwal, J.S.; Uniacke, J.; Arragain, S.; Jiang, H.; Gold, N.D.; et al. Pyrenoid functions revealed by proteomics in Chlamydomonas reinhardtii. PLoS ONE 2018, 13, e0185039. [Google Scholar] [CrossRef] [PubMed]
- Gerin, S.; Mathy, G.; Blomme, A.; Franck, F.; Sluse, F.E. Plasticity of the mitoproteome to nitrogen sources (nitrate and ammonium) in Chlamydomonas reinhardtii: The logic of Aox1 gene localization. Biochim. Biophys. Acta 2010, 1797, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Manjasetty, B.A.; Büssow, K.; Panjikar, S.; Turnbull, A.P. Current methods in structural proteomics and its applications in biological sciences. 3 Biotech 2011, 2, 89–113. [Google Scholar] [CrossRef]
- Meyer, J.G. Qualitative and Quantitative Shotgun Proteomics Data Analysis from Data-Dependent Acquisition Mass Spectrometry. Methods Mol. Biol. 2021, 2259, 297–308. [Google Scholar] [CrossRef]
- Stauber, E.J.; Fink, A.; Markert, C.; Kruse, O.; Johanningmeier, U.; Hippler, M. Proteomics of Chlamydomonas reinhardtii light-harvesting proteins. Eukaryot Cell 2003, 2, 978–994. [Google Scholar] [CrossRef]
- Quisel, J.D.; Wykoff, D.D.; Grossman, A.R. Biochemical Characterization of the Extracellular Phosphatases Produced by Phosphorus-Deprived Chlamydomonas reinhardtii. Plant Physiol. 1996, 111, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Kohinata, T.; Nishino, H.; Fukuzawa, H. Significance of zinc in a regulatory protein, CCM1, which regulates the carbon-concentrating mechanism in Chlamydomonas reinhardtii. Plant Cell Physiol. 2008, 49, 273–283. [Google Scholar] [CrossRef]
- Yang, S.-Y.; Tsuzuki, M.; Muiyachi, S. Carbonic Anhydrase of Chlamydomonas: Purification and Studies on its Induction Using Antiserum against Chlamydomonas Carbonic Anhydrase. Plant Cell Physiol. 1985, 26, 25–34. [Google Scholar] [CrossRef]
- Kuchitsu, K.; Tsuzuki, M.; Miyachi, S. Characterization of the Pyrenoid Isolated from Unicellular Green Alga Chlamydomonas reinhardtii: Particulate Form of RuBisCO Protein. Protoplasma 1988, 144, 17–24. [Google Scholar] [CrossRef]
- Jinag, W.; Cossey, S.; Rosenberg, J.N.; Oyler, G.A.; Olson, B.J.S.C.; Weeks, D.P. A rapid live-cell ELISA for characterizing antibodies against cell surface antigens of Chlamydomonas reinhardtii and its use in isolating algae from natural environments with related cell wall components. BMC Plant Biol. 2014, 14, 244. [Google Scholar] [CrossRef] [PubMed]
- Scmitter, J.M.; Jacquot, J.P.; de Lamotte-Guery, F.; Beauvallet, C.; Dutka, S.; Gadal, P.; Decottignies, P. Purification, properties and complete amino acid sequence of the ferredoxin from a green alga, Chlamydomonas reinhardtii. Eur. J. Biochem. 1988, 172, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Cid, C.; Garcia-Descalzo, L.; Casado-Lafuente, V.; Amils, R.; Aguilera, A. Proteomic analysis of the response of an acidophilic strain of Chlamydomonas sp. (Chlorophyta) to natural metal-rich water. Proteomics 2010, 10, 2026–2036. [Google Scholar] [CrossRef]
- Wang, H.; Alvarez, S.; Hicks, L.M. Comprehensive comparison of iTRAQ and label-free LC-based quantitative proteomics approaches using two Chlamydomonas reinhardtii strains of interest for biofuels engineering. J. Proteome Res. 2012, 11, 487–501. [Google Scholar] [CrossRef]
- Helliwell, K.E.; Pandhal, J.; Cooper, M.B.; Longworth, J.; Kudahl, U.J.; Russo, D.A.; Tomsett, E.V.; Bunbury, F.; Salmon, D.L.; Smirnoff, N.; et al. Quantitative proteomics of a B12-dependent alga grown in coculture with bacteria reveals metabolic tradeoffs required for mutualism. N. Phytol. 2018, 217, 599–612. [Google Scholar] [CrossRef]
- Hammel, A.; Zimmer, D.; Sommer, F.; Muhlhaus, T.; Schroda, M. Absolute Quantification of Major Photosynthetic Protein Complexes in Chlamydomonas reinhardtii Using Quantification Concatamers (QconCATs). Front. Plant Sci. 2018, 9, 1265. [Google Scholar] [CrossRef]
- Rommelfanger, S.R.; Zhou, M.; Shaghasi, H.; Tzeng, S.C.; Evans, B.S.; Pasa-Tolic, L.; Umen, J.G.; Pesavento, J.J. An Improved Top-Down Mass Spectrometry Characterization of Chlamydomonas reinhardtii Histones and Their Post-translational Modifications. J. Am. Soc. Mass Spectrom. 2021, 32, 1671–1688. [Google Scholar] [CrossRef] [PubMed]
- Preimesberger, M.R.; Majumdar, A.; Lecomte, J.T. Dynamics of Lysine as a Heme Axial Ligand: NMR Analysis of the Chlamydomonas reinhardtii Hemoglobin THB1. Biochemistry 2017, 56, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.C.; Backlund, A.; Bjorhall, K.; Spreitzer, R.J.; Andersson, I. First crystal structure of Rubisco from a green alga, Chlamydomonas reinhardtii. J. Biol. Chem. 2001, 276, 48159–48164. [Google Scholar] [CrossRef] [PubMed]
- Melby, J.A.; Roberts, D.S.; Larson, E.J.; Brown, K.A.; Bayne, E.F.; Jin, S.; Ge, Y. Novel Strategies to Address the Challenges in Top-Down Proteomics. J. Am. Soc. Mass Spectrom. 2021, 32, 1278–1294. [Google Scholar] [CrossRef] [PubMed]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of this Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Samuel, M.; Ang, C.-S.; Keerthikumar, S.; Mathivanan, S. Label-Based and Label-Free Strategies for Protein Quantitation. In Methods in Molecular Biology; Keerthikumar, S., Mathivanan, S., Eds.; Humana Press: New York, NY, USA, 2016; Volume 1549, pp. 31–43. [Google Scholar]
- Rao, V.S.; Srinivas, K.; Sujini, G.N.; Kumar, G.N. Protein-protein interaction detection: Methods and analysis. Int. J. Proteom. 2014, 2014, 147648. [Google Scholar] [CrossRef] [PubMed]
- Strotmann, V.I.; Stahl, Y. Visualization of in vivo protein-protein interactions in plants. J. Exp. Bot. 2022, 73, 3866–3880. [Google Scholar] [CrossRef] [PubMed]
- Behal, R.H.; Miller, M.S.; Qin, H.; Lucker, B.F.; Jones, A.; Cole, D.G. Subunit interactions and organization of the Chlamydomonas reinhardtii intraflagellar transport complex A proteins. J. Biol. Chem. 2012, 287, 11689–11703. [Google Scholar] [CrossRef]
- Atkinson, N.; Velanis, C.N.; Wunder, T.; Clarke, D.J.; Mueller-Cajar, O.; McCormick, A.J. The pyrenoidal linker protein EPYC1 phase separates with hybrid Arabidopsis-Chlamydomonas Rubisco through interactions with the algal Rubisco small subunit. J. Exp. Bot. 2019, 70, 5271–5285. [Google Scholar] [CrossRef]
- Mackinder, L.C.M.; Chen, C.; Leib, R.D.; Patena, W.; Blum, S.R.; Rodman, M.; Ramundo, S.; Adams, C.M.; Jonikas, M.C. A Spatial Interactome Reveals the Protein Organization of the Algal CO(2)-Concentrating Mechanism. Cell 2017, 171, 133–147.e114. [Google Scholar] [CrossRef]
- Blatti, J.L.; Beld, J.; Behnke, C.A.; Mendez, M.; Mayfield, S.P.; Burkart, M.D. Manipulating Fatty Acid Biosynthesis in Microalgae for Biofuel through Protein-Protein Interactions. PLoS ONE 2012, 7, e42949. [Google Scholar] [CrossRef] [PubMed]
- Slocombe, S.P.; Ross, M.; Thomas, N.; McNeill, S.; Stanley, M.S. A rapid and general method for measurement of protein in micro-algal biomass. Bioresour. Technol. 2013, 129, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Q.; Jensen, O.N.; Moller, I.M.; Hebelstrup, K.H.; Rogowska-Wrzesinska, A. Evaluation of sample preparation methods for mass spectrometry-based proteomic analysis of barley leaves. Plant Methods 2018, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, M.; Sakata, M.; Ohnishi, K.; Tokumaru, Y.; Kato, Y.; Tokutsu, R.; Sakamoto, W.; Minagawa, J.; Matsuda, F.; Shimizu, H. Targeted proteome analysis of microalgae under high-light conditions by optimized protein extraction of photosynthetic organisms. J. Biosci. Bioeng. 2019, 127, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Bichmann, L.; Gupta, S.; Rosenberger, G.; Kuchenbecker, L.; Sachsenberg, T.; Ewels, P.; Alka, O.; Pfeuffer, J.; Kohlbacher, O.; Rost, H. DIAproteomics: A Multifunctional Data Analysis Pipeline for Data-Independent Acquisition Proteomics and Peptidomics. J. Proteome Res. 2021, 20, 3758–3766. [Google Scholar] [CrossRef] [PubMed]
- Pedrioli, P.G.A. Trans-Proteomic Pipeline: A Pipeline for Proteomic Analysis. In Proteome Bioinformatics, Methods in Molecular Biology; Hubbard, S.J., Jones, A.R., Eds.; Humana Press: New York, NY, USA, 2010; Volume 604. [Google Scholar] [CrossRef]
- Schessner, J.P.; Voytik, E.; Bludau, I. A practical guide to interpreting and generating bottom-up proteomics data visualizations. Proteomics 2022, 22, 2100103. [Google Scholar] [CrossRef]
- Anjos, L.; Estevao, J.; Infante, C.; Mantecon, L.; Power, D.M. Extracting protein from microalgae (Tetraselmis chuii) for proteome analysis. MethodsX 2022, 9, 101637. [Google Scholar] [CrossRef] [PubMed]
- Graves, P.R.; Haystead, T.A. Molecular biologist’s guide to proteomics. Microbiol. Mol. Biol. Rev. 2002, 66, 39–63. [Google Scholar] [CrossRef] [PubMed]
- Mear, H.; Gillon, P.; Gifuni, I.; Lavenant, L.; Poidevin, A.; Couallier, E. Extraction of soluble proteins by bead milling from Tetraselmis chui in two different physiological states. Algal Res 2023, 74, 103180. [Google Scholar] [CrossRef]
- Dixon, C.; Wilken, L.R. Green microalgae biomolecule separations and recovery. Bioresour. Bioprocess. 2018, 5, 14. [Google Scholar] [CrossRef]
- 5 Steps to Fundamental Protein Preparation. Available online: https://www.thermofisher.com/nz/en/home/life-science/protein-biology/protein-purification-isolation/5-steps-fundamental-protein-preparation.html (accessed on 11 December 2023).
- 5 Steps to Protein Isolation and Purification. Available online: https://www.thermofisher.com/nz/en/home/life-science/protein-biology/protein-purification-isolation/5-steps-protein-purification.html (accessed on 11 December 2023).
- Knoshaug, E.P.; Gerristen, A.T.; Henard, C.A.; Guarnieri, M.T. Methods for Algal Protein Isolation and Proteome Analysis. In Metabolic Pathway Engineering; Himmel, M.E., Bomble, Y.J., Eds.; Humana Press: New York, NY, USA, 2020. [Google Scholar] [CrossRef]
- Bandeira, N.; Clauser, K.R.; Pevzner, P.A. Shotgun protein sequencing: Assembly of peptide tandem mass spectra from mixtures of modified proteins. Mol. Cell. Proteom. 2007, 6, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.D.M.; Lima, D.B.; Fischer, J.S.G.; Clasen, M.A.; Kurt, L.U.; Camillo-Andrade, A.C.; Monteiro, L.C.; de Aquino, P.F.; Neves-Ferreira, A.G.C.; Valente, R.H.; et al. Simple, efficient and thorough shotgun proteomic analysis with PatternLab V. Nat. Protoc. 2022, 17, 1553–1578. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.G.; Niemi, N.M.; Pagliarini, D.J.; Coon, J.J. Quantitative shotgun proteome analysis by direct infusion. Nat. Methods 2020, 17, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Oye, O.K.; Jorgensen, K.M.; Hjelle, S.M.; Sulen, A.; Ulvang, D.M.; Gjertsen, B.T. Gel2DE—A software tool for correlation analysis of 2D gel electrophoresis data. BMC Bioinf. 2013, 14, 215. [Google Scholar] [CrossRef] [PubMed]
- Urban, P.L. Quantitative mass spectrometry: An overview. Philos. Trans. A Math. Phys. Eng. Sci. 2016, 374, 20150382. [Google Scholar] [CrossRef] [PubMed]
- Dressler, F.F.; Bragelmann, J.; Reischl, M.; Perner, S. Normics: Proteomic Normalization by Variance and Data-Inherent Correlation Structure. Mol. Cell. Proteom. 2022, 21, 100269. [Google Scholar] [CrossRef] [PubMed]
- Graw, S.; Tang, J.; Zafar, M.K.; Byrd, A.K.; Bolden, C.; Peterson, E.C.; Byrum, S.D. proteiNorm—A User-Friendly Tool for Normalization and Analysis of TMT and Label-Free Protein Quantification. ACS Omega 2020, 5, 25625–25633. [Google Scholar] [CrossRef] [PubMed]
- Valikangas, T.; Suomi, T.; Elo, L.L. A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief. Bioinform. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Wang, J.; Yu, W.; He, Z. Protein inference: A review. Brief. Bioinform. 2012, 13, 586–614. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Koh, C.W.T.; Ooi, J.S.G.; Ong, E.Z.; Chan, K.R. STAGEs: A web-based tool that integrates data visualization and pathway enrichment analysis for gene expression studies. Sci. Rep. 2023, 13, e7535. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, 587–592. [Google Scholar] [CrossRef] [PubMed]
- May, P.; Wienkoop, S.; Kempa, S.; Usadel, B.; Christian, N.; Rupprecht, J.; Weiss, J.; Recuenco-Munoz, L.; Ebenhoh, O.; Weckwerth, W.; et al. Metabolomics- and proteomics-assisted genome annotation and analysis of the draft metabolic network of Chlamydomonas reinhardtii. Genetics 2008, 179, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cong, Y.; Wang, Y.; Guo, Z.; Yue, J.; Xing, Z.; Gao, X.; Chai, X. Identification of Early Salinity Stress-Responsive Proteins in Dunaliella salina by isobaric tags for relative and absolute quantitation (iTRAQ)-Based Quantitative Proteomic Analysis. Int. J. Mol. Sci. 2019, 20, 599. [Google Scholar] [CrossRef] [PubMed]
- Kurotani, A.; Yamada, Y.; Sakurai, T. Alga-PrAS (Algal Protein Annotation Suite): A Database of Comprehensive Annotation in Algal Proteomes. Plant Cell Physiol. 2017, 58, e6. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.W.B.; Wong, L. Integrating Networks and Proteomics: Moving Forward. Trends Biotechnol. 2016, 34, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Hasan, M.A.; Chen, J.Y. Pathway and network analysis in proteomics. J. Theor. Biol. 2014, 362, 44–52. [Google Scholar] [CrossRef]
- Montone, C.M.; Capriotti, A.L.; Cavaliere, C.; La Barbera, G.; Piovesana, S.; Zenezini Chiozzi, R.; Lagana, A. Peptidomic strategy for purification and identification of potential ACE-inhibitory and antioxidant peptides in Tetradesmus obliquus microalgae. Anal. Bioanal. Chem. 2018, 410, 3573–3586. [Google Scholar] [CrossRef]
- Carrasco-Reinado, R.; Escobar-Nino, A.; Fajardo, C.; Morano, I.M.; Amil-Ruiz, F.; Martinez-Rodriguez, G.; Fuentes-Almagro, C.; Capilla, V.; Tomas-Cobos, L.; Soriano-Romani, L.; et al. Development of New Antiproliferative Compound against Human Tumor Cells from the Marine Microalgae Nannochloropsis gaditana by Applied Proteomics. Int. J. Mol. Sci. 2020, 22, 96. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Cao, P.; Su, X.; Liu, Z.; Li, M. Structural analysis and comparison of light-harvesting complexes I and II. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148038. [Google Scholar] [CrossRef] [PubMed]
- Bujaldon, S.; Kodama, N.; Rappaport, F.; Subramanyam, R.; de Vitry, C.; Takahashi, Y.; Wollman, F.A. Functional Accumulation of Antenna Proteins in Chlorophyll b-Less Mutants of Chlamydomonas reinhardtii. Mol. Plant 2017, 10, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.M.; Marriboina, S.; Zamal, M.Y.; Pandey, J.; Subramanyam, R. High light-induced changes in whole-cell proteomic profile and its correlation with the organization of thylakoid super-complex in cyclic electron transport mutants of Chlamydomonas reinhardtii. Front. Plant. Sci. 2023, 14, 1198474. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.I.; Castruita, M.; Malasarn, D.; Urzica, E.; Erde, J.; Page, M.D.; Yamasaki, H.; Casero, D.; Pellegrini, M.; Merchant, S.S.; et al. The proteome of copper, iron, zinc, and manganese micronutrient deficiency in Chlamydomonas reinhardtii. Mol. Cell. Proteom. 2013, 12, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Schmollinger, S.; Muhlhaus, T.; Boyle, N.R.; Blaby, I.K.; Casero, D.; Mettler, T.; Moseley, J.L.; Kropat, J.; Sommer, F.; Strenkert, D.; et al. Nitrogen-Sparing Mechanisms in Chlamydomonas Affect the Transcriptome, the Proteome, and Photosynthetic Metabolism. Plant Cell 2014, 26, 1410–1435. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Ning, Z.; Wang, Y.; Zheng, Y.; Zhang, C.; Figeys, D. Quantitative proteomic analysis of Dunaliella salina upon acute arsenate exposure. Chemosphere 2016, 145, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Yang, L.; Wang, Y.; Jiang, F.; Lai, J.; Pan, K. Proteomics Provide Insight into the Interaction between Selenite and the Microalgae Dunaliella salina. Processes 2023, 11, 563. [Google Scholar] [CrossRef]
- Leon-Vaz, A.; Romero, L.C.; Gotor, C.; Leon, R.; Vigara, J. Effect of cadmium in the microalga Chlorella sorokiniana: A proteomic study. Ecotoxicol. Environ. Saf. 2021, 207, 111301. [Google Scholar] [CrossRef]
- Guarnieri, M.T.; Nag, A.; Smolinski, S.L.; Darzins, A.; Seibert, M.; Pienkos, P.T. Examination of triacylglycerol biosynthetic pathways via de novo transcriptomic and proteomic analyses in an unsequenced microalga. PLoS ONE 2011, 6, e25851. [Google Scholar] [CrossRef]
- Patel, A.K.; Huang, E.L.; Low-Decarie, E.; Lefsrud, M.G. Comparative Shotgun Proteomic Analysis of Wastewater-Cultured Microalgae: Nitrogen Sensing and Carbon Fixation for Growth and Nutrient Removal in Chlamydomonas reinhardtii. J. Proteome Res. 2015, 14, 3051–3067. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Muthuraj, M.; Gandhi, M.N.; Das, D.; Srivastava, S. Real-time iTRAQ-based proteome profiling revealed the central metabolism involved in nitrogen starvation induced lipid accumulation in microalgae. Sci. Rep. 2017, 7, 45732. [Google Scholar] [CrossRef] [PubMed]
- Ben Amor, F.; Elleuch, F.; Ben Hlima, H.; Garnier, M.; Saint-Jean, B.; Barkallah, M.; Pichon, C.; Abdelkafi, S.; Fendri, I. Proteomic Analysis of the Chlorophyta Dunaliella New Strain AL-1 Revealed Global Changes of Metabolism during High Carotenoid Production. Mar. Drugs 2017, 15, 293. [Google Scholar] [CrossRef]
- Wei, S.; Bian, Y.; Zhao, Q.; Chen, S.; Mao, J.; Song, C.; Cheng, K.; Xiao, Z.; Zhang, C.; Ma, W.; et al. Salinity-Induced Palmella Formation Mechanism in Halotolerant Algae Dunaliella salina Revealed by Quantitative Proteomics and Phosphoproteomics. Front. Plant Sci. 2017, 8, 810. [Google Scholar] [CrossRef]
- Wang, B.; Pan, X.; Wang, F.; Liu, L.; Jia, J. Photoprotective carbon redistribution in mixotrophic Haematococcus pluvialis under high light stress. Bioresour. Technol. 2022, 362, 127761. [Google Scholar] [CrossRef]
- Chen, C.; Harst, A.; You, W.; Xu, J.; Ning, K.; Poetsch, A. Proteomic study uncovers molecular principles of single-cell-level phenotypic heterogeneity in lipid storage of Nannochloropsis oceanica. Biotechnol. Biofuels 2019, 12, 21. [Google Scholar] [CrossRef]
- Roccuzzo, S.; Couto, N.; Karunakaran, E.; Kapoore, R.V.; Butler, T.O.; Mukherjee, J.; Hansson, E.M.; Beckerman, A.P.; Pandhal, J. Metabolic Insights into Infochemicals Induced Colony Formation and Flocculation in Scenedesmus subspicatus Unraveled by Quantitative Proteomics. Front. Microbiol. 2020, 11, 792. [Google Scholar] [CrossRef] [PubMed]
- Salama, E.S.; Govindwar, S.P.; Khandare, R.V.; Roh, H.S.; Jeon, B.H.; Li, X. Can Omics Approaches Improve Microalgal Biofuels under Abiotic Stress? Trends Plant. Sci. 2019, 24, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Service, R.B. 2021 Breakthrough of the year: Protein structures for all. Science 2021, 374, eabm4805. Available online: https://www.science.org/content/article/breakthrough-2021 (accessed on 1 April 2024). [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Callaway, E. ‘The entire protein universe’: AI predicts shape of nearly every known protein. Nature 2022, 608, 15–16. Available online: https://www.nature.com/articles/d41586-022-02083-2 (accessed on 1 April 2024). [CrossRef] [PubMed]
- Plouviez, M.; Bolot, P.; Shilton, A.; Guieysse, B. Phosphorus uptake and accumulation in Chlamydomonas reinhardtii: Influence of biomass concentration, phosphate concentration, phosphorus depletion time, and light supply. Algal Res. 2023, 71, 103085. [Google Scholar] [CrossRef]
- Karbalaei, M.; Rezaee, S.A.; Farsiani, H. Pichia pastoris: A highly successful expression system for optimal synthesis of heterologous proteins. J. Cell Physiol. 2020, 235, 5867–5881. [Google Scholar] [CrossRef] [PubMed]
- Arísticas Ribalta, R.C.; Valdés, L.M.; Lafargue Gámez, M.; Rodríguez Davydenko, S.; Dubreucq, E.; Perrier, V.; Moreau, B.; Vidal, R.F. Constitutive high expression level of a synthetic deleted encoding gene of Talaromyces minioluteus endodextranase variant (r–TmDEX49A–ΔSP–ΔN30) in Komagataella phaffii (Pichia pastoris). Appl. Sci. 2022, 12, 7562. [Google Scholar] [CrossRef]
- Hothorn, M.; Neumann, H.; Lenherr, E.D.; Wehner, M.; Rybin, V.; Hassa, P.O.; Uttenweiler, A.; Reinhardt, M.; Schmidt, A.; Seiler, J.; et al. Catalytic core of a membrane-associated eukaryotic polyphosphate polymerase. Science 2009, 24, 513–516. [Google Scholar] [CrossRef]
- Stoffels, L.; Finlan, A.; Mannall, G.; Purton, S.; Parker, B. Downstream Processing of Chlamydomonas reinhardtii TN72 for Recombinant Protein Recovery. Front. Bioeng. Biotechnol. 2019, 7, 383. [Google Scholar] [CrossRef]
- Rosales-Mendoza, S.; Paz-Maldonado, L.M.; Soria-Guerra, R.E. Chlamydomonas reinhardtii as a viable platform for the production of recombinant proteins: Current status and perspectives. Plant Cell Rep. 2012, 31, 479–494. [Google Scholar] [CrossRef]
- Masi, A.; Leonelli, F.; Scognamiglio, V.; Gasperuzzo, G.; Antonacci, A.; Terzidis, M.A. Chlamydomonas reinhardtii: A Factory of Nutraceutical and Food Supplements for Human Health. Molecules 2023, 28, 1185. [Google Scholar] [CrossRef]
- Grigoriev, I.V.; Hayes, R.D.; Calhoun, S.; Kamel, B.; Wang, A.; Ahrendt, S.; Dusheyko, S.; Nikitin, R.; Mondo, S.J.; Salamov, A.; et al. PhycoCosm, a comparative algal genomics resource. Nucleic Acids Res. 2021, 49, D1004–D1011. [Google Scholar] [CrossRef]
- Nguyen, T.H.T.; Park, S.; Jeong, J.; Shin, Y.S.; Sim, S.J.; Jin, E. Enhancing lipid productivity by modulating lipid catabolism using the CRISPR-Cas9 system in Chlamydomonas. J. Appl. Phycol. 2020, 32, 2829–2840. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Method | Description | Example(s) | |
|---|---|---|---|
| Conventional | Chromatography | Chromatography-based techniques are used for protein separation and purification. In brief, proteins can be separated/purified based on charges (IEC: ion-exchange chromatography), size (SEC: size-exclusion chromatography) or (bio)chemical affinity with a matrix (AC: affinity chromatography). | [63,64,65] |
| Immuno assay/blotting | The enzyme-linked immunosorbent assay (ELISA) detects the presence of a protein by measuring the enzymatic activity of an enzyme-labeled antigen or antibody binding to an immobilized target protein (or antibody). ELISA is widely used for diagnostics. Western blotting allows the separation of proteins based on molecular weight through gel electrophoresis and identification of protein from binding of a labeled antibody to its target antigen (i.e., protein) on a membrane. | [66,67] | |
| Edman sequencing | The Edman method (or Edman degradation) determines the amino acid sequence in peptides/proteins by sequentially identifying and cleaving amino acids from the N-terminal side of a peptide/protein. | [68] | |
| Advanced | Gel-based | The conventional polyacrylamide gel-based method used for protein separation and identification based on proteins’ mass (SDS-PAGE: sodium dodecyl sulfate–polyacrylamide gel electrophoresis) has evolved into more advanced 2D methods based on both mass and charge separation (2D-PAGE: two-dimensional polyacrylamide gel electrophoresis) or using labels with a fluorescent dye (2D-DIGE: two-dimensional differential gel electrophoresis). 2D-PAGE and 2D-DIGE can resolve and investigate the abundance of several thousand proteins in a single sample. | [59,62,69] |
| Mass spectrometry | MS is one of the most used analytical techniques to identify and, coupled with (ultra)-high performance chromatography, quantitatively measure protein levels. MS of peptides/proteins ionized via matrix-assisted laser desorption ionization (MALDI), surface-enhanced laser desorption/ionization (SELDI) or, more classically, electrospray ionization (ESI), allow the determination, through deconvolution of the mass spectra obtained, of their molecular mass. In the context of proteomics and peptide analysis, multi-stage tandem mass spectrometry (MSn) is also a powerful technique for obtaining detailed information on the structure and sequence of peptides and proteins, particularly with regard to the localization of post-translational modifications (PTMs). Label-free or labeled approaches can be used for quantification (ICAT: isotope-coded affinity tag; iTRAQ: isobaric tagging for relative and absolute quantification; SILAC: stable isotope labeling by/with amino acids in cell culture; QconCAT: quantification concatemer; TMT: Tandem Mass Tag). MS is often combined with separations and fractionation techniques to identify target proteins or subproteomes. Sample fractionation and enrichment are also important when identifying PTMs (phosphorylation, oxidation, nitrosylation, glycosylation, methylation, etc.). | ICAT: [46] iTRAQ and label-free: [70] iTRAQ: [71] QconCAT: [72] SILAC: [50] TMT: [40] PTMs: [46,47,51,53,73] | |
| Nuclear Magnetic Resonance | Protein structural determination in solution or solid phase by measuring chemical shifts. NMR structural determination involved several steps, each requiring significant expertise/techniques. | [74] | |
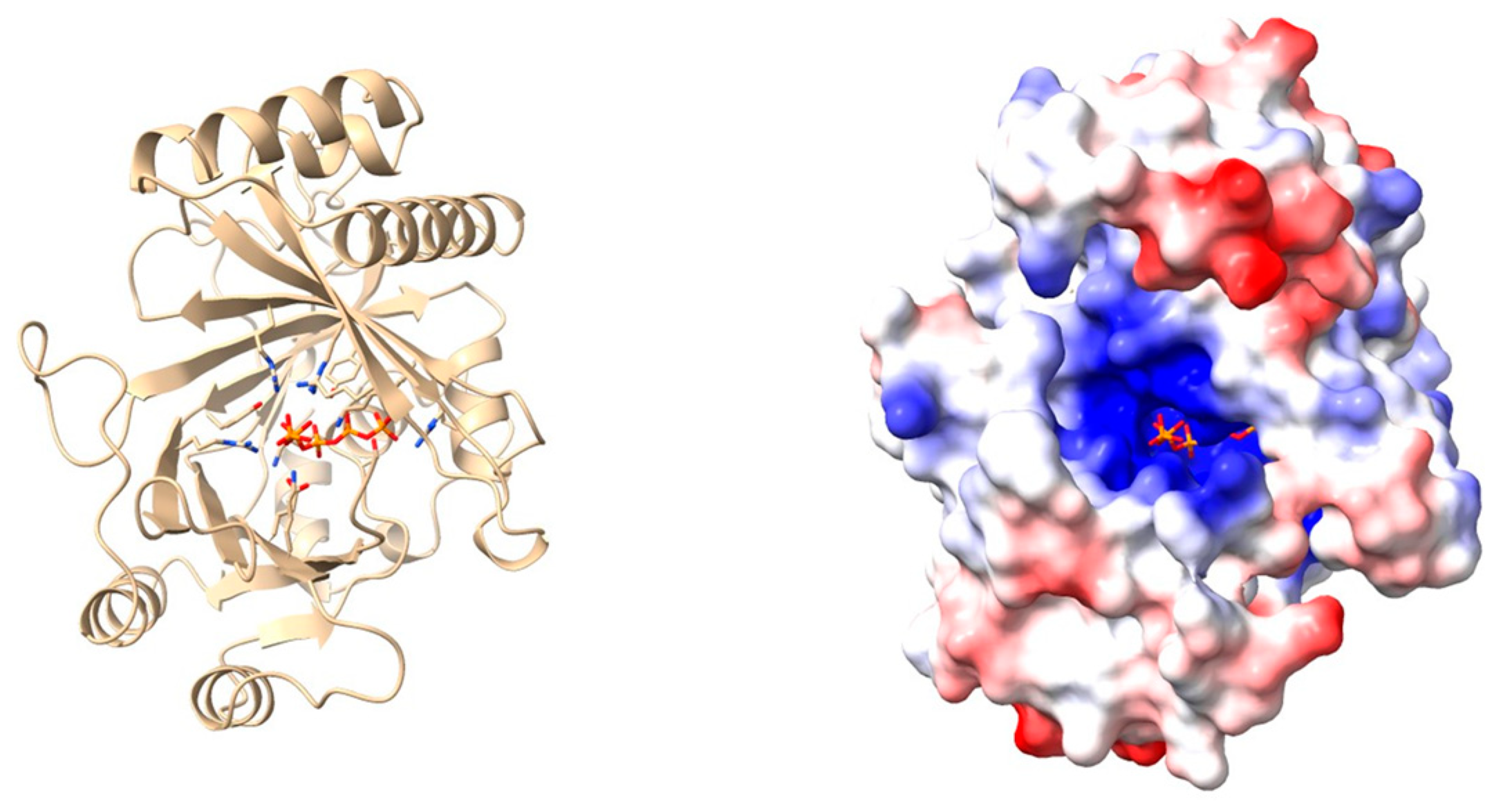
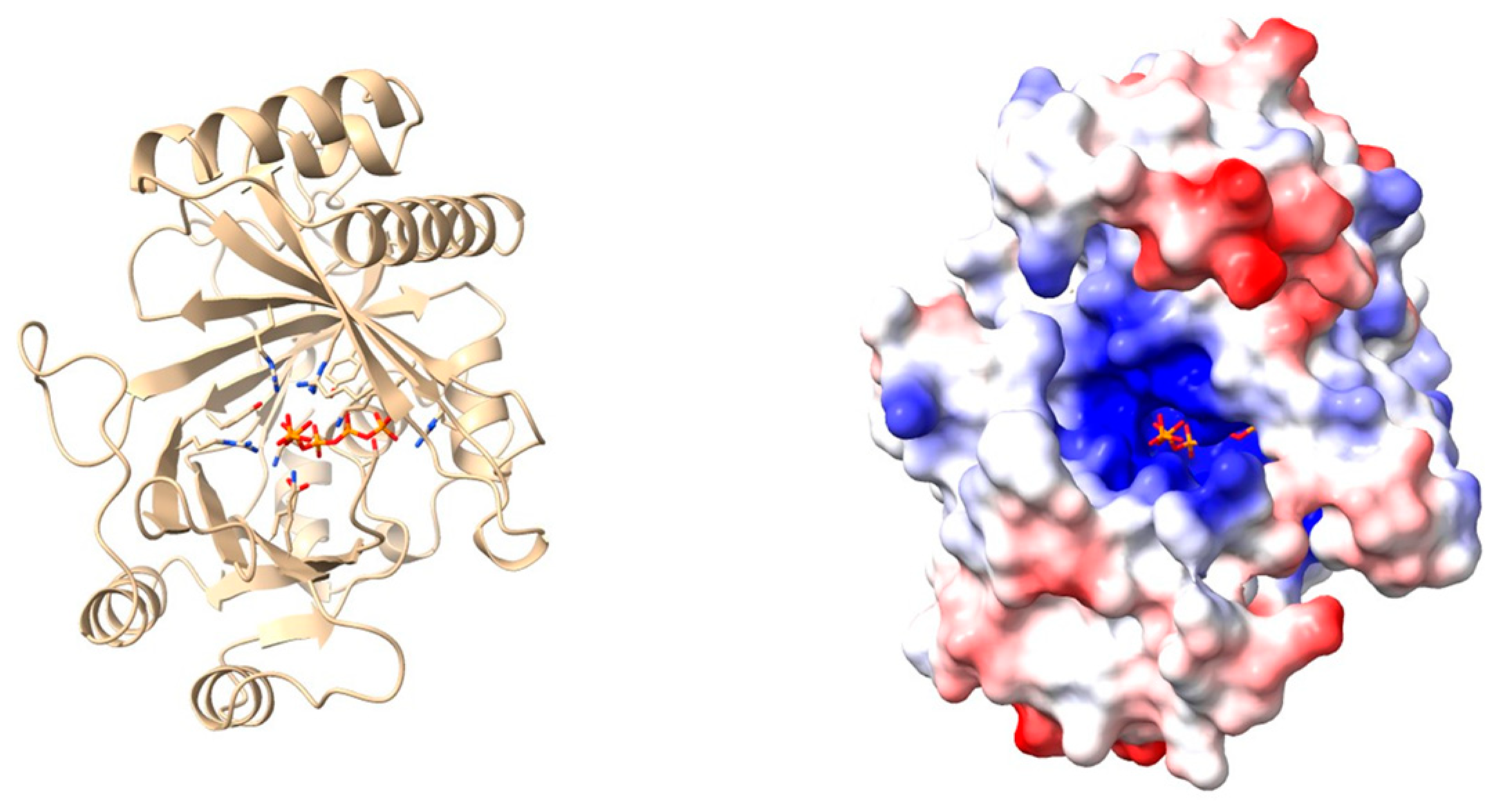
| X-ray crystallography | Three-dimensional protein structures are determined by exposing highly purified crystallized protein samples to X-rays and measuring diffraction patterns. | [75] | |
| Cryogenic electron microscopy | Microscopy techniques used to determine 3D structure of proteins or protein complexes via flash freezing and electron bombardment of samples in solution. | [55] | |
| In silico modelling | Generation and study of protein 3D models using homology modelling or deep-learning-based predictions. | [41] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plouviez, M.; Dubreucq, E. Key Proteomics Tools for Fundamental and Applied Microalgal Research. Proteomes 2024, 12, 13. https://doi.org/10.3390/proteomes12020013
Plouviez M, Dubreucq E. Key Proteomics Tools for Fundamental and Applied Microalgal Research. Proteomes. 2024; 12(2):13. https://doi.org/10.3390/proteomes12020013
Chicago/Turabian StylePlouviez, Maxence, and Eric Dubreucq. 2024. "Key Proteomics Tools for Fundamental and Applied Microalgal Research" Proteomes 12, no. 2: 13. https://doi.org/10.3390/proteomes12020013