
Role of Protein Carbonylation in Skeletal Muscle Mass Loss Associated with Chronic Conditions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Production of Oxidants and in Vivo Protein Carbonylation
3. Oxidants and Skeletal Muscle Contraction
4. Antioxidants in Skeletal Muscle Fibers
5. Protein Carbonylation in Disuse Muscle Atrophy
6. Protein Carbonylation in Aging Muscles
7. Protein Carbonylation in Muscles Exposed to Chronic Cigarette Smoke
8. Protein Carbonylation in Skeletal Muscle Dysfunction and Mass Loss in COPD
8.1. Muscle Dysfunction and Mass Loss in COPD
8.2. Biological Significance of Muscle Protein Carbonylation in COPD
9. Protein Carbonylation in Cancer Cachexia Models
10. Protein Oxidation in Cancer Cachectic Muscles
11. Protein Carbonylation and Muscle Dysfunction in Sepsis
12. Carbonylated Proteins in Septic Muscles
13. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| COPD | Chronic obstructive pulmonary disease |
| DNA | Deoxyribonucleic acid |
| DNP | 2,4-dinitrophenylhydrazone |
| DNPH | 2,4-dinitrophenylhydrazine |
| O2−· | Superoxide anion |
| ELISA | Enzyme-linked immunosorbent assay |
| FEV1 | Forced expiratory volume in one second |
| GSH/GSSG | Ratio of reduced to oxidized glutathione |
| HNE | Hydroxynonenal |
| H2O2 | Hydrogen peroxide |
| HOO· | Hydroperoxyl radicals |
| iTRAQ | Isobaric tags for relative and absolute quantitation |
| MIP | Maximal inspiratory pressure |
| MDA | Malondialdehyde |
| NADPH | Nicotinamide adenine dinucleotide phosphate hydrogen |
| NAC | N-acetyl cysteine |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| NO2 | Nitrogen dioxide |
| OH∙ | Hydroxyl radicals |
| ROS | Reactive oxygen species |
| RNS | Reactive nitrogen species |
| SOD | Superoxide dismutase |
| UCP | Uncoupling protein |
References
- Barreiro, E.; Bustamante, V.; Cejudo, P.; Galdiz, J.B.; Gea, J.; de Lucas, P.; Martinez-Llorens, J.; Ortega, F.; Puente-Maestu, L.; Roca, J.; et al. Guidelines for the Evaluation and Treatment of Muscle Dysfunction in Patients With Chronic Obstructive Pulmonary Disease. Arch. Bronconeumol. 2015, 51, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Maltais, F.; Decramer, M.; Casaburi, R.; Barreiro, E.; Burelle, Y.; Debigare, R.; Dekhuijzen, P.N.; Franssen, F.; Gayan-Ramirez, G.; Gea, J.; et al. An official American Thoracic Society/European Respiratory Society statement: Update on limb muscle dysfunction in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care. Med. 2014, 189, e15–e62. [Google Scholar] [CrossRef] [PubMed]
- Miravitlles, M.; Soler-Cataluna, J.J.; Calle, M.; Molina, J.; Almagro, P.; Antonio, Q.J.; Antonio, R.J.; Antonio, T.J.; Pinera, P.; Simon, A.; et al. Spanish Guideline for COPD (GesEPOC). Update 2014. Arch. Bronconeumol. 2014, 50, 1–16. [Google Scholar] [CrossRef]
- Vestbo, J.; Hurd, S.S.; Agusti, A.G.; Jones, P.W.; Vogelmeier, C.; Anzueto, A.; Barnes, P.J.; Fabbri, L.M.; Martinez, F.J.; Nishimura, M.; et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care. Med. 2013, 187, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Miravitlles, M. What was the impact of the Spanish COPD guidelines (GesEPOC) and how can they be improved? Arch. Bronconeumol. 2016, 52, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Marquis, K.; Debigare, R.; Lacasse, Y.; LeBlanc, P.; Jobin, J.; Carrier, G.; Maltais, F. Midthigh muscle cross-sectional area is a better predictor of mortality than body mass index in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care. Med. 2002, 166, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.M.; Spruit, M.A.; Hopkinson, N.S.; Natanek, S.A.; Man, W.D.; Jackson, A.; Gosker, H.R.; Schols, A.M.; Moxham, J.; Polkey, M.I.; et al. The prevalence of quadriceps weakness in COPD and the relationship with disease severity. Eur. Respir. J. 2010, 36, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Shrikrishna, D.; Patel, M.; Tanner, R.J.; Seymour, J.M.; Connolly, B.A.; Puthucheary, Z.A.; Walsh, S.L.; Bloch, S.A.; Sidhu, P.S.; Hart, N.; et al. Quadriceps wasting and physical inactivity in patients with COPD. Eur. Respir. J. 2012, 40, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Swallow, E.B.; Reyes, D.; Hopkinson, N.S.; Man, W.D.; Porcher, R.; Cetti, E.J.; Moore, A.J.; Moxham, J.; Polkey, M.I. Quadriceps strength predicts mortality in patients with moderate to severe chronic obstructive pulmonary disease. Thorax. 2007, 62, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Ponikowski, P.; Varney, S.; Chua, T.P.; Clark, A.L.; Webb-Peploe, K.M.; Harrington, D.; Kox, W.J.; Poole-Wilson, P.A.; Coats, A.J. Wasting as independent risk factor for mortality in chronic heart failure. Lancet 1997, 349, 1050–1053. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Muscaritoli, M.; Bossola, M.; Aversa, Z.; Bellantone, R.; Rossi, F.F. Prevention and treatment of cancer cachexia: new insights into an old problem. Eur. J. Cancer 2006, 42, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Von Haehling, S.; Anker, S.D. Cachexia as a major underestimated and unmet medical need: facts and numbers. J. Cachexia Sarcopenia Muscle 2010, 1, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; de la Puente, B.; Minguella, J.; Corominas, J.M.; Serrano, S.; Hussain, S.N.; Gea, J. Oxidative stress and respiratory muscle dysfunction in severe chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care. Med. 2005, 171, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
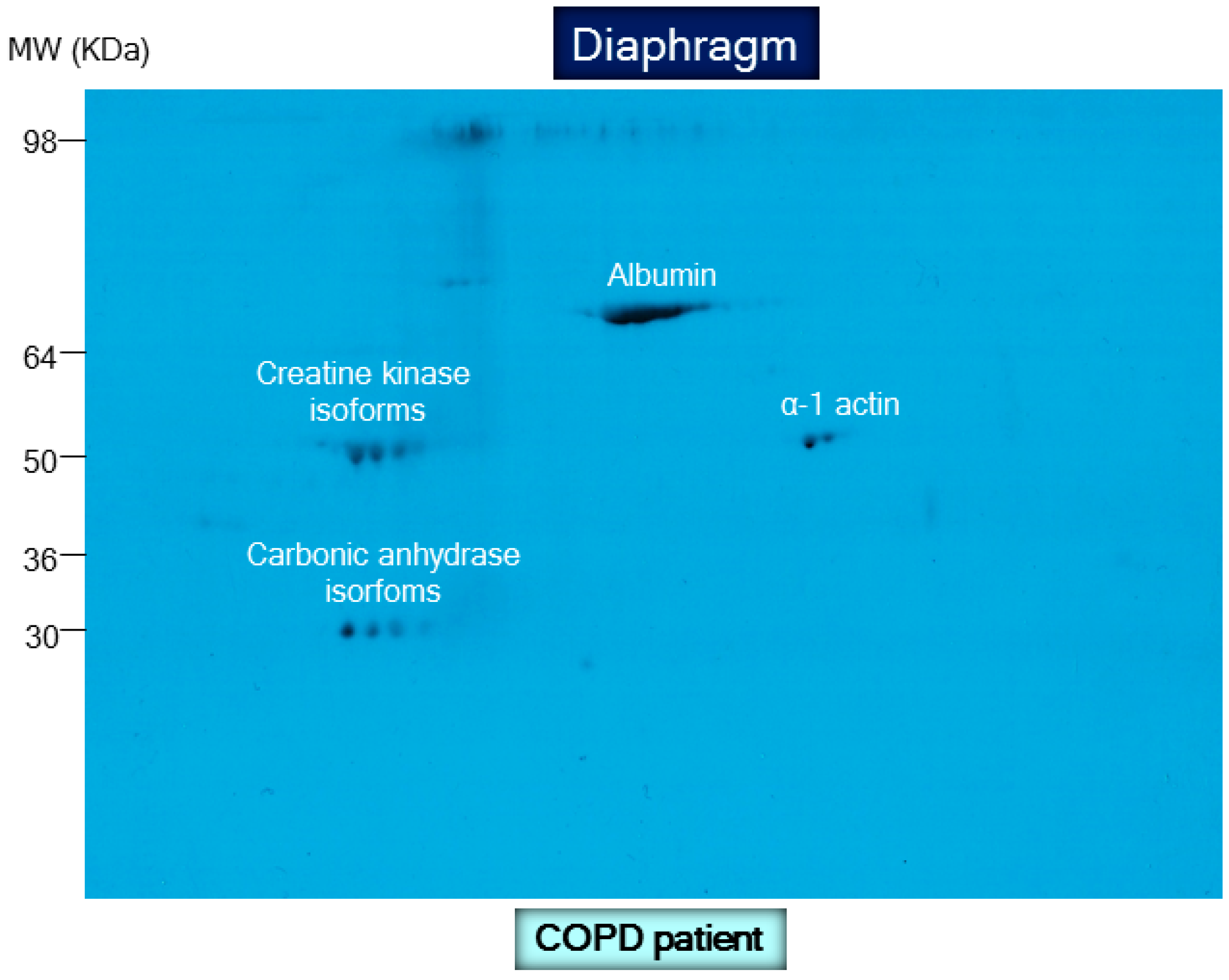
- Barreiro, E.; Gea, J.; Matar, G.; Hussain, S.N. Expression and carbonylation of creatine kinase in the quadriceps femoris muscles of patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2005, 33, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Rabinovich, R.; Marin-Corral, J.; Barbera, J.A.; Gea, J.; Roca, J. Chronic endurance exercise induces quadriceps nitrosative stress in patients with severe COPD. Thorax 2009, 64, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Peinado, V.I.; Galdiz, J.B.; Ferrer, E.; Marin-Corral, J.; Sanchez, F.; Gea, J.; Barbera, J.A. Cigarette smoke-induced oxidative stress: A role in chronic obstructive pulmonary disease skeletal muscle dysfunction. Am. J. Respir. Crit. Care Med. 2010, 182, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, V.; Casanova, J.; de Lopez, S.E.; Mas, S.; Sellares, J.; Gea, J.; Galdiz, J.B.; Barreiro, E. Redox balance following magnetic stimulation training in the quadriceps of patients with severe COPD. Free Radic. Res. 2008, 42, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Fermoselle, C.; Rabinovich, R.; Ausin, P.; Puig-Vilanova, E.; Coronell, C.; Sanchez, F.; Roca, J.; Gea, J.; Barreiro, E. Does oxidative stress modulate limb muscle atrophy in severe COPD patients? Eur. Respir. J. 2012, 40, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Koechlin, C.; Couillard, A.; Simar, D.; Cristol, J.P.; Bellet, H.; Hayot, M.; Prefaut, C. Does oxidative stress alter quadriceps endurance in chronic obstructive pulmonary disease? Am. J. Respir. Crit. Care Med. 2004, 169, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Koechlin, C.; Couillard, A.; Cristol, J.P.; Chanez, P.; Hayot, M.; Le, G.D.; Prefaut, C. Does systemic inflammation trigger local exercise-induced oxidative stress in COPD? Eur. Respir. J. 2004, 23, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Koechlin, C.; Maltais, F.; Saey, D.; Michaud, A.; LeBlanc, P.; Hayot, M.; Prefaut, C. Hypoxaemia enhances peripheral muscle oxidative stress in chronic obstructive pulmonary disease. Thorax 2005, 60, 834–841. [Google Scholar] [CrossRef] [PubMed]
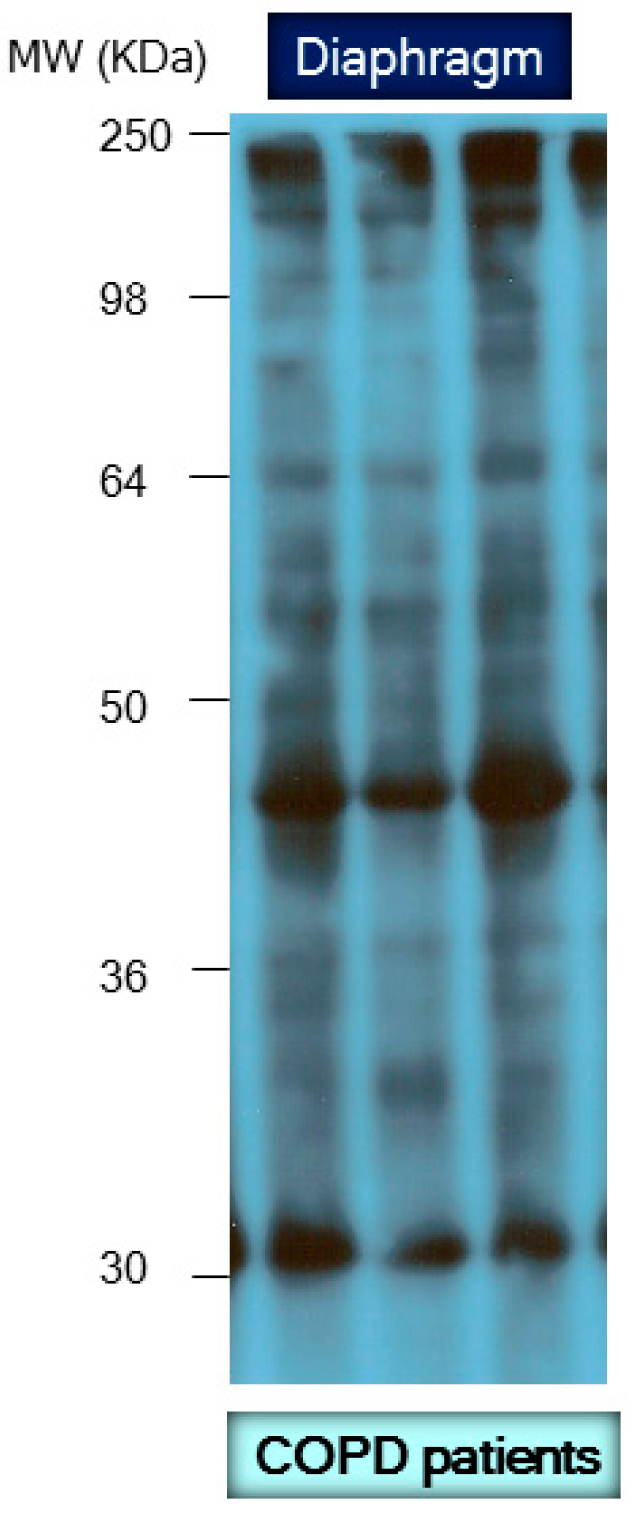
- Marin-Corral, J.; Minguella, J.; Ramirez-Sarmiento, A.L.; Hussain, S.N.; Gea, J.; Barreiro, E. Oxidised proteins and superoxide anion production in the diaphragm of severe COPD patients. Eur. Respir. J. 2009, 33, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Mercken, E.M.; Hageman, G.J.; Schols, A.M.; Akkermans, M.A.; Bast, A.; Wouters, E.F. Rehabilitation decreases exercise-induced oxidative stress in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2005, 172, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Mercken, E.M.; Hageman, G.J.; Langen, R.C.; Wouters, E.F.; Schols, A.M. Decreased exercise-induced expression of nuclear factor-kappaB-regulated genes in muscle of patients with COPD. Chest 2011, 139, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Guardia, S.; Wodja, E.; Gorostiza, A.; de Lopez, S.E.; Gea, J.; Galdiz, J.B.; Sliwinski, P.; Barreiro, E. (Improvement in quality of life and exercise capacity without muscular biology changes after general training in patients with severe chronic obstructive pulmonary disease). Med. Clin. 2013, 140, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Puente-Maestu, L.; Tejedor, A.; Lázaro, A.; de Miguel, J.; Alvarez-Sala, L.; Gonzalez-Aragoneses, F.; Simon, C.; Agusti, A. Site of mitochondrial reactive oxygen species production in skeletal muscle of chronic obstructive pulmonary disease and its relationship with exercise oxidative stress. Am. J. Respir. Cell Mol. Biol. 2012, 47, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, R.A.; Ardite, E.; Troosters, T.; Carbo, N.; Alonso, J.; de Suso, G.J.M.; Vilaro, J.; Barbera, J.A.; Polo, M.F.; Argiles, J.M.; et al. Reduced muscle redox capacity after endurance training in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 164, 1114–1118. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, R.A.; Ardite, E.; Mayer, A.M.; Polo, M.F.; Vilaro, J.; Argiles, J.M.; Roca, J. Training depletes muscle glutathione in patients with chronic obstructive pulmonary disease and low body mass index. Respiration 2006, 73, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.A.; Kalko, S.; Puig-Vilanova, E.; Perez-Olabarria, M.; Falciani, F.; Gea, J.; Cascante, M.; Barreiro, E.; Roca, J. Muscle and blood redox status after exercise training in severe COPD patients. Free Radic. Biol. Med. 2012, 52, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; de la Puente, B.; Busquets, S.; Lopez-Soriano, F.J.; Gea, J.; Argiles, J.M. Both oxidative and nitrosative stress are associated with muscle wasting in tumour-bearing rats. FEBS Lett. 2005, 579, 1646–1652. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Garcia-Martinez, C.; Mas, S.; Ametller, E.; Gea, J.; Argiles, J.M.; Busquets, S.; Lopez-Soriano, F.J. UCP3 overexpression neutralizes oxidative stress rather than nitrosative stress in mouse myotubes. FEBS Lett. 2009, 583, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Marin-Corral, J.; Fontes, C.C.; Pascual-Guardia, S.; Sanchez, F.; Olivan, M.; Argiles, J.M.; Busquets, S.; Lopez-Soriano, F.J.; Barreiro, E. Redox balance and carbonylated proteins in limb and heart muscles of cachectic rats. Antioxid. Redox Signal 2010, 12, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Comtois, A.S.; Mohammed, S.; Lands, L.C.; Hussain, S.N. Role of heme oxygenases in sepsis-induced diaphragmatic contractile dysfunction and oxidative stress. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L476–L484. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Sanchez, D.; Galdiz, J.B.; Hussain, S.N.; Gea, J. N-acetylcysteine increases manganese superoxide dismutase activity in septic rat diaphragms. Eur. Respir. J. 2005, 26, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Gea, J.; di Falco, M.; Kriazhev, L.; James, S.; Hussain, S.N. Protein carbonyl formation in the diaphragm. Am. J. Respir. Cell Mol. Biol. 2005, 32, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.; Chojkier, M. Muscle wasting and dedifferentiation induced by oxidative stress in a murine model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxidants. EMBO. J. 1996, 15, 1753–1765. [Google Scholar] [PubMed]
- Gomes-Marcondes, M.C.; Tisdale, M.J. Induction of protein catabolism and the ubiquitin-proteasome pathway by mild oxidative stress. Cancer Lett. 2002, 180, 69–74. [Google Scholar] [CrossRef]
- Pascual-Guardia, S.; Arbol, F.; Sanchez, E.; Casadevall, C.; Merlo, V.; Gea, J.; Barreiro, E. (Inflammation and oxidative stress in respiratory and limb muscles of patients with severe sepsis). Med. Clin. 2013, 141, 194–200. [Google Scholar]
- Taille, C.; Foresti, R.; Lanone, S.; Zedda, C.; Green, C.; Aubier, M.; Motterlini, R.; Boczkowski, J. Protective role of heme oxygenases against endotoxin-induced diaphragmatic dysfunction in rats. Am. J. Respir. Crit. Care Med. 2001, 163, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Coronell, C.; Lavina, B.; Ramirez-Sarmiento, A.; Orozco-Levi, M.; Gea, J. Aging, sex differences, and oxidative stress in human respiratory and limb muscles. Free Radic. Biol. Med. 2006, 41, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Pansarasa, O.; Bertorelli, L.; Vecchiet, J.; Felzani, G.; Marzatico, F. Age-dependent changes of antioxidant activities and markers of free radical damage in human skeletal muscle. Free Radic. Biol. Med. 1999, 27, 617–622. [Google Scholar] [CrossRef]
- Pansarasa, O.; Castagna, L.; Colombi, B.; Vecchiet, J.; Felzani, G.; Marzatico, F. Age and sex differences in human skeletal muscle: Role of reactive oxygen species. Free Radic. Res. 2000, 33, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Quintanilha, A.T.; Brooks, G.A.; Packer, L. Free radicals and tissue damage produced by exercise. Biochem. Biophys. Res. Commun. 1982, 107, 1198–1205. [Google Scholar] [CrossRef]
- Souza, J.M.; Choi, I.; Chen, Q.; Weisse, M.; Daikhin, E.; Yudkoff, M.; Obin, M.; Ara, J.; Horwitz, J.; Ischiropoulos, H. Proteolytic degradation of tyrosine nitrated proteins. Arch. Biochem. Biophys. 2000, 380, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.P.; Dean, R.T. Fragmentation of proteins by free radicals and its effect on their susceptibility to enzymic hydrolysis. Biochem. J. 1986, 234, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Lu, B.; Monroe, R.K.; Ward, S.M.; Halvorsen, S.W. Inducers of oxidative stress block ciliary neurotrophic factor activation of Jak/STAT signaling in neurons. J. Neurochem. 2005, 92, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- 48 Starke, P.E.; Oliver, C.N.; Stadtman, E.R. Modification of hepatic proteins in rats exposed to high oxygen concentration. FASEB. J. 1987, 1, 36–39. [Google Scholar] [PubMed]
- Weiss, S.J. Tissue destruction by neutrophils. N. Engl. J. Med. 1989, 320, 365–376. [Google Scholar] [PubMed]
- Zweier, J.L.; Kuppusamy, P.; Williams, R.; Rayburn, B.K.; Smith, D.; Weisfeldt, M.L.; Flaherty, J.T. Measurement and characterization of postischemic free radical generation in the isolated perfused heart. J. Biol. Chem. 1989, 264, 18890–18895. [Google Scholar] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Duarte, J.; Kavazis, A.N.; Talbert, E.E. Reactive oxygen species are signalling molecules for skeletal muscle adaptation. Exp. Physiol. 2010, 95, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Grandhee, S.K.; Monnier, V.M. Mechanism of formation of the Maillard protein cross-link pentosidine. Glucose, fructose, and ascorbate as pentosidine precursors. J. Biol. Chem. 1991, 266, 11649–11653. [Google Scholar] [PubMed]
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino. Acids. 2003, 25, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Friguet, B.; Stadtman, E.R.; Szweda, L.I. Modification of glucose-6-phosphate dehydrogenase by 4-hydroxy-2-nonenal. Formation of cross-linked protein that inhibits the multicatalytic protease. J. Biol. Chem. 1994, 269, 21639–21643. [Google Scholar]
- Requena, J.R.; Fu, M.X.; Ahmed, M.U.; Jenkins, A.J.; Lyons, T.J.; Thorpe, S.R. Lipoxidation products as biomarkers of oxidative damage to proteins during lipid peroxidation reactions. Nephrol. Dial. Transplant 1996, 11, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Meany, D.L.; Xie, H.; Thompson, L.V.; Arriaga, E.A.; Griffin, T.J. Identification of carbonylated proteins from enriched rat skeletal muscle mitochondria using affinity chromatography-stable isotope labeling and tandem mass spectrometry. Proteomics 2007, 7, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Regnier, F. Affinity chromatographic selection of carbonylated proteins followed by identification of oxidation sites using tandem mass spectrometry. Anal. Chem. 2005, 77, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.S.; Regnier, F.E. Proteomic analysis of carbonylated proteins in two-dimensional gel electrophoresis using avidin-fluorescein affinity staining. Electrophoresis 2004, 25, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Williams, J.A.; Stadtman, E.R.; Shacter, E. Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol. 1994, 233, 346–357. [Google Scholar] [PubMed]
- Barreiro, E.; Gea, J.; Corominas, J.M.; Hussain, S.N. Nitric oxide synthases and protein oxidation in the quadriceps femoris of patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2003, 29, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Galdiz, J.B.; Marinan, M.; Alvarez, F.J.; Hussain, S.N.; Gea, J. Respiratory loading intensity and diaphragm oxidative stress: N-acetyl-cysteine effects. J. Appl. Physiol. 2006, 100, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Schols, A.M.; Polkey, M.I.; Galdiz, J.B.; Gosker, H.R.; Swallow, E.B.; Coronell, C.; Gea, J. Cytokine profile in quadriceps muscles of patients with severe COPD. Thorax 2008, 63, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; del Puerto-Nevado, L.; Puig-Vilanova, E.; Perez-Rial, S.; Sanchez, F.; Martinez-Galan, L.; Rivera, S.; Gea, J.; Gonzalez-Mangado, N.; Peces-Barba, G. Cigarette smoke-induced oxidative stress in skeletal muscles of mice. Respir. Physiol. Neurobiol. 2012, 182, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Couillard, A.; Maltais, F.; Saey, D.; Debigare, R.; Michaud, A.; Koechlin, C.; LeBlanc, P.; Prefaut, C. Exercise-induced quadriceps oxidative stress and peripheral muscle dysfunction in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2003, 167, 1664–1669. [Google Scholar] [CrossRef] [PubMed]
- Fermoselle, C.; Sanchez, F.; Barreiro, E. (Reduction of muscle mass mediated by myostatin in an experimental model of pulmonary emphysema). Arch. Bronconeumol. 2011, 47, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Van Den, B.B.; Slot, I.G.; Hellwig, V.A.; Vosse, B.A.; Kelders, M.C.; Barreiro, E.; Schols, A.M.; Gosker, H.R. Loss of quadriceps muscle oxidative phenotype and decreased endurance in patients with mild-to-moderate COPD. J. Appl. Physiol. 2013, 114, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Puig-Vilanova, E.; Rodriguez, D.A.; Lloreta, J.; Ausin, P.; Pascual-Guardia, S.; Broquetas, J.; Roca, J.; Gea, J.; Barreiro, E. Oxidative stress, redox signaling pathways, and autophagy in cachectic muscles of male patients with advanced COPD and lung cancer. Free Radic. Biol. Med. 2015, 79, 91–108. [Google Scholar] [CrossRef] [PubMed]
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997, 272, 20313–20316. [Google Scholar] [CrossRef] [PubMed]
- Botto, N.; Masetti, S.; Petrozzi, L.; Vassalle, C.; Manfredi, S.; Biagini, A.; Andreassi, M.G. Elevated levels of oxidative DNA damage in patients with coronary artery disease. Coron. Artery Dis. 2002, 13, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J.; Pye, D.; Palomero, J. The production of reactive oxygen and nitrogen species by skeletal muscle. J. Appl. Physiol. 2007, 102, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B.; Shoji, T.; Moody, M.R.; Entman, M.L. Reactive oxygen in skeletal muscle. II. Extracellular release of free radicals. J. Appl. Physiol. 1992, 73, 1805–1809. [Google Scholar] [PubMed]
- Reid, M.B.; Khawli, F.A.; Moody, M.R. Reactive oxygen in skeletal muscle. III. Contractility of unfatigued muscle. J. Appl. Physiol. 1993, 75, 1081–1087. [Google Scholar] [PubMed]
- Reid, M.B. Nitric oxide, reactive oxygen species, and skeletal muscle contraction. Med. Sci. Sports. Exerc. 2001, 33, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B. Invited Review: Redox modulation of skeletal muscle contraction: what we know and what we don't. J. Appl. Physiol. 2001, 90, 724–731. [Google Scholar] [PubMed]
- Javesghani, D.; Magder, S.A.; Barreiro, E.; Quinn, M.T.; Hussain, S.N. Molecular characterization of a superoxide-generating NAD(P)H oxidase in the ventilatory muscles. Am. J. Respir. Crit. Care Med. 2002, 165, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Kavazis, A.N.; DeRuisseau, K.C. Mechanisms of disuse muscle atrophy: Role of oxidative stress. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R337–R344. [Google Scholar] [CrossRef] [PubMed]
- Supinski, G. Free radical induced respiratory muscle dysfunction. Mol. Cell Biochem. 1998, 179, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Vassilakopoulos, T.; Deckman, G.; Kebbewar, M.; Rallis, G.; Harfouche, R.; Hussain, S.N. Regulation of nitric oxide production in limb and ventilatory muscles during chronic exercise training. Am. J. Physiol. Lung. Cell Mol. Physiol. 2003, 284, L452–L457. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.F.; Reid, M.B. Muscle-derived ROS and thiol regulation in muscle fatigue. J. Appl. Physiol. 2008, 104, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Booth, F.W. Effect of limb immobilization on skeletal muscle. J. Appl. Physiol. 1982, 52, 1113–1118. [Google Scholar] [PubMed]
- Kondo, H.; Miura, M.; Itokawa, Y. Oxidative stress in skeletal muscle atrophied by immobilization. Acta. Physiol. Scand. 1991, 142, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Miura, M.; Nakagaki, I.; Sasaki, S.; Itokawa, Y. Trace element movement and oxidative stress in skeletal muscle atrophied by immobilization. Am. J. Physiol. 1992, 262, E583–E590. [Google Scholar] [PubMed]
- Kondo, H.; Nakagaki, I.; Sasaki, S.; Hori, S.; Itokawa, Y. Mechanism of oxidative stress in skeletal muscle atrophied by immobilization. Am. J. Physiol. 1993, 265, E839–E844. [Google Scholar] [PubMed]
- Lawler, J.M.; Song, W.; Demaree, S.R. Hindlimb unloading increases oxidative stress and disrupts antioxidant capacity in skeletal muscle. Free Radic. Biol. Med. 2003, 35, 9–16. [Google Scholar] [CrossRef]
- Shanely, R.A.; Zergeroglu, M.A.; Lennon, S.L.; Sugiura, T.; Yimlamai, T.; Enns, D.; Belcastro, A.; Powers, S.K. Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am. J. Respir. Crit. Care Med. 2002, 166, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Zergeroglu, M.A.; McKenzie, M.J.; Shanely, R.A.; van Gammeren, D.; DeRuisseau, K.C.; Powers, S.K. Mechanical ventilation-induced oxidative stress in the diaphragm. J. Appl. Physiol. 2003, 95, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Levine, S.; Nguyen, T.; Taylor, N.; Friscia, M.E.; Budak, M.T.; Rothenberg, P.; Zhu, J.; Sachdeva, R.; Sonnad, S.; Kaiser, L.R.; et al. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N. Engl. J. Med. 2008, 358, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Kavazis, A.N.; Talbert, E.E.; Smuder, A.J.; Hudson, M.B.; Nelson, W.B.; Powers, S.K. Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic. Biol. Med. 2009, 46, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Whidden, M.A.; McClung, J.M.; Falk, D.J.; Hudson, M.B.; Smuder, A.J.; Nelson, W.B.; Powers, S.K. Xanthine oxidase contributes to mechanical ventilation-induced diaphragmatic oxidative stress and contractile dysfunction. J. Appl. Physiol. 2009, 106, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Gumuslu, S. Stress-dependent induction of protein oxidation, lipid peroxidation and anti-oxidants in peripheral tissues of rats: comparison of three stress models (immobilization, cold and immobilization-cold). Clin. Exp. Pharmacol. Physiol. 2007, 34, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Gumuslu, S. Immobilization stress in rat tissues: alterations in protein oxidation, lipid peroxidation and antioxidant defense system. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2007, 144, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Primeau, A.J.; Adhihetty, P.J.; Hood, D.A. Apoptosis in heart and skeletal muscle. Can. J. Appl. Physiol. 2002, 27, 349–395. [Google Scholar] [CrossRef]
- Li, Y.P.; Chen, Y.; Li, A.S.; Reid, M.B. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am. J. Physiol. Cell Physiol. 2003, 285, C806–C812. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Orlander, J.; Kiessling, K.H.; Larsson, L.; Karlsson, J.; Aniansson, A. Skeletal muscle metabolism and ultrastructure in relation to age in sedentary men. Acta. Physiol. Scand. 1978, 104, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; Fano, G.; Fulle, S.; MacGarvey, U.; Shinobu, L.; Polidori, M.C.; Cherubini, A.; Vecchiet, J.; Senin, U.; Beal, M.F. Age-dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle. Free Radic. Biol. Med. 1999, 26, 303–308. [Google Scholar] [CrossRef]
- Marzani, B.; Felzani, G.; Bellomo, R.G.; Vecchiet, J.; Marzatico, F. Human muscle aging: ROS-mediated alterations in rectus abdominis and vastus lateralis muscles. Exp. Gerontol 2005, 40, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Fulle, S.; di Donna, S.; Puglielli, C.; Pietrangelo, T.; Beccafico, S.; Bellomo, R.; Protasi, F.; Fano, G. Age-dependent imbalance of the antioxidative system in human satellite cells. Exp. Gerontol 2005, 40, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Kamel, H.K.; Maas, D.; Duthie, E.H., Jr. Role of hormones in the pathogenesis and management of sarcopenia. Drugs Aging 2002, 19, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Carmeli, E.; Coleman, R.; Reznick, A.Z. The biochemistry of aging muscle. Exp. Gerontol 2002, 37, 477–489. [Google Scholar] [CrossRef]
- Leeuwenburgh, C.; Hansen, P.; Shaish, A.; Holloszy, J.O.; Heinecke, J.W. Markers of protein oxidation by hydroxyl radical and reactive nitrogen species in tissues of aging rats. Am. J. Physiol. 1998, 274, R453–R461. [Google Scholar] [PubMed]
- Stadtman, E.R. Role of oxidant species in aging. Curr. Med. Chem. 2004, 11, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Lawler, J.; Criswell, D.; Lieu, F.K.; Dodd, S. Alterations in diaphragmatic oxidative and antioxidant enzymes in the senescent Fischer 344 rat. J. Appl. Physiol. 1992, 72, 2317–2321. [Google Scholar] [PubMed]
- Oh-ishi, S.; Toshinai, K.; Kizaki, T.; Haga, S.; Fukuda, K.; Nagata, N.; Ohno, H. Effects of aging and/or training on antioxidant enzyme system in diaphragm of mice. Respir. Physiol. 1996, 105, 195–202. [Google Scholar] [CrossRef]
- Schoneich, C.; Viner, R.I.; Ferrington, D.A.; Bigelow, D.J. Age-related chemical modification of the skeletal muscle sarcoplasmic reticulum Ca-ATPase of the rat. Mech. Ageing Dev. 1999, 107, 221–231. [Google Scholar] [CrossRef]
- Figueiredo, P.A.; Powers, S.K.; Ferreira, R.M.; Appell, H.J.; Duarte, J.A. Aging impairs skeletal muscle mitochondrial bioenergetic function. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Xie, H.; Meany, D.L.; Thompson, L.V.; Arriaga, E.A.; Griffin, T.J. Quantitative proteomic profiling of muscle type-dependent and age-dependent protein carbonylation in rat skeletal muscle mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 1137–1152. [Google Scholar] [CrossRef] [PubMed]
- Guerassimov, A.; Hoshino, Y.; Takubo, Y.; Turcotte, A.; Yamamoto, M.; Ghezzo, H.; Triantafillopoulos, A.; Whittaker, K.; Hoidal, J.R.; Cosio, M.G. The development of emphysema in cigarette smoke-exposed mice is strain dependent. Am. J. Respir. Crit. Care Med. 2004, 170, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Ardite, E.; Peinado, V.I.; Rabinovich, R.A.; Fernandez-Checa, J.C.; Roca, J.; Barbera, J.A. Systemic effects of cigarette smoke exposure in the guinea pig. Respir. Med. 2006, 100, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Frei, B.; Forte, T.M.; Ames, B.N.; Cross, C.E. Gas phase oxidants of cigarette smoke induce lipid peroxidation and changes in lipoprotein properties in human blood plasma. Protective effects of ascorbic acid. Biochem. J. 1991, 277, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.X.; Fusch, C.; Jaeger, P.; Peheim, E.; Horber, F.F. Impact of chronic cigarette smoking on body composition and fuel metabolism. J. Clin. Endocrinol. Metab. 1995, 80, 2181–2185. [Google Scholar]
- Kalra, J.; Chaudhary, A.K.; Prasad, K. Increased production of oxygen free radicals in cigarette smokers. Int. J. Exp. Pathol. 1991, 72, 1–7. [Google Scholar] [PubMed]
- Morrow, J.D.; Frei, B.; Longmire, A.W.; Gaziano, J.M.; Lynch, S.M.; Shyr, Y.; Strauss, W.E.; Oates, J.A.; Roberts, L.J. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. N. Engl. J. Med. 1995, 332, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Park, E.M.; Park, Y.M.; Gwak, Y.S. Oxidative damage in tissues of rats exposed to cigarette smoke. Free Radic. Biol. Med. 1998, 25, 79–86. [Google Scholar] [CrossRef]
- Reznick, A.Z.; Cross, C.E.; Hu, M.L.; Suzuki, Y.J.; Khwaja, S.; Safadi, A.; Motchnik, P.A.; Packer, L.; Halliwell, B. Modification of plasma proteins by cigarette smoke as measured by protein carbonyl formation. Biochem. J. 1992, 286, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Colombo, G.; Clerici, M.; Giustarini, D.; Rossi, R.; Milzani, A.; Dalle-Donne, I. Redox albuminomics: oxidized albumin in human diseases. Antioxid. Redox. Signal 2012, 17, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Colombo, G.; Rossi, R.; Gagliano, N.; Portinaro, N.; Clerici, M.; Annibal, A.; Giustarini, D.; Colombo, R.; Milzani, A.; Dalle-Donne, I. Red blood cells protect albumin from cigarette smoke-induced oxidation. PLoS ONE 2012, 7, e29930. [Google Scholar] [CrossRef] [PubMed]
- Gosselink, R.; Troosters, T.; Decramer, M. Peripheral muscle weakness contributes to exercise limitation in COPD. Am. J. Respir. Crit. Care Med. 1996, 153, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Gea, J. Respiratory and Limb Muscle Dysfunction in COPD. COPD 2015, 12, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Mador, M.J.; Bozkanat, E. Skeletal muscle dysfunction in chronic obstructive pulmonary disease. Respir. Res. 2001, 2, 216–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, S.; Bashir, M.H.; Clanton, T.L.; Powers, S.K.; Singhal, S. COPD elicits remodeling of the diaphragm and vastus lateralis muscles in humans. J. Appl. Physiol. 2013, 114, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Levine, S.; Kaiser, L.; Leferovich, J.; Tikunov, B. Cellular adaptations in the diaphragm in chronic obstructive pulmonary disease. N. Engl. J. Med. 1997, 337, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Levine, S.; Gregory, C.; Nguyen, T.; Shrager, J.; Kaiser, L.; Rubinstein, N.; Dudley, G. Bioenergetic adaptation of individual human diaphragmatic myofibers to severe COPD. J. Appl. Physiol. 2002, 92, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Levine, S.; Nguyen, T.; Kaiser, L.R.; Rubinstein, N.A.; Maislin, G.; Gregory, C.; Rome, L.C.; Dudley, G.A.; Sieck, G.C.; Shrager, J.B. Human diaphragm remodeling associated with chronic obstructive pulmonary disease: Clinical implications. Am. J. Respir. Crit. Care Med. 2003, 168, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Testelmans, D.; Crul, T.; Maes, K.; Agten, A.; Crombach, M.; Decramer, M.; Gayan-Ramirez, G. Atrophy and hypertrophy signalling in the diaphragm of patients with COPD. Eur. Respir. J. 2010, 35, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Ottenheijm, C.A.; Heunks, L.M.; Sieck, G.C.; Zhan, W.Z.; Jansen, S.M.; Degens, H.; de Boo, T.; Dekhuijzen, P.N. Diaphragm dysfunction in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2005, 172, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Ottenheijm, C.A.; Heunks, L.M.; Li, Y.P.; Jin, B.; Minnaard, R.; van Hees, H.W.; Dekhuijzen, P.N. Activation of the ubiquitin-proteasome pathway in the diaphragm in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2006, 174, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Ottenheijm, C.A.; Heunks, L.M.; Dekhuijzen, P.N. Diaphragm muscle fiber dysfunction in chronic obstructive pulmonary disease: Toward a pathophysiological concept. Am. J. Respir. Crit. Care Med. 2007, 175, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Puente-Maestu, L.; Perez-Parra, J.; Godoy, R.; Moreno, N.; Tejedor, A.; Gonzalez-Aragoneses, F.; Bravo, J.L.; Alvarez, F.V.; Camano, S.; Agusti, A. Abnormal mitochondrial function in locomotor and respiratory muscles of COPD patients. Eur. Respir. J. 2009, 33, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.; Ferrer, D.; Sanchez, F.; Minguella, J.; Marin-Corral, J.; Martinez-Llorens, J.; Lloreta, J.; Gea, J. Inflammatory cells and apoptosis in respiratory and limb muscles of patients with COPD. J. Appl. Physiol. 2011, 111, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Van Helvoort, H.A.; Heijdra, Y.F.; Thijs, H.M.; Vina, J.; Wanten, G.J.; Dekhuijzen, P.N. Exercise-induced systemic effects in muscle-wasted patients with COPD. Med. Sci. Sports. Exerc. 2006, 38, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- MacMillan-Crow, L.A.; Crow, J.P.; Kerby, J.D.; Beckman, J.S.; Thompson, J.A. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc. Natl. Acad. Sci. USA 1996, 93, 11853–11858. [Google Scholar] [CrossRef]
- MacMillan-Crow, L.A.; Crow, J.P.; Thompson, J.A. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 1998, 37, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Mihm, M.J.; Coyle, C.M.; Schanbacher, B.L.; Weinstein, D.M.; Bauer, J.A. Peroxynitrite induced nitration and inactivation of myofibrillar creatine kinase in experimental heart failure. Cardiovasc. Res. 2001, 49, 798–807. [Google Scholar] [CrossRef]
- Souza, J.M.; Daikhin, E.; Yudkoff, M.; Raman, C.S.; Ischiropoulos, H. Factors determining the selectivity of protein tyrosine nitration. Arch. Biochem. Biophys. 1999, 371, 169–178. [Google Scholar] [CrossRef] [PubMed]
- White, M.Y.; Cordwell, S.J.; McCarron, H.C.; Prasan, A.M.; Craft, G.; Hambly, B.D.; Jeremy, R.W. Proteomics of ischemia/reperfusion injury in rabbit myocardium reveals alterations to proteins of essential functional systems. Proteomics 2005, 5, 1395–1410. [Google Scholar] [CrossRef] [PubMed]
- Panda, K.; Chattopadhyay, R.; Ghosh, M.K.; Chattopadhyay, D.J.; Chatterjee, I.B. Vitamin C prevents cigarette smoke induced oxidative damage of proteins and increased proteolysis. Free Radic. Biol. Med. 1999, 27, 1064–1079. [Google Scholar] [CrossRef]
- Requena, J.R.; Levine, R.L.; Stadtman, E.R. Recent advances in the analysis of oxidized proteins. Amino. Acids. 2003, 25, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Puente-Maestu, L.; Tena, T.; Trascasa, C.; Perez-Parra, J.; Godoy, R.; Garcia, M.J.; Stringer, W.W. Training improves muscle oxidative capacity and oxygenation recovery kinetics in patients with chronic obstructive pulmonary disease. Eur. J. Appl. Physiol. 2003, 88, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Momken, I.; Lechene, P.; Koulmann, N.; Fortin, D.; Mateo, P.; Doan, B.T.; Hoerter, J.; Bigard, X.; Veksler, V.; Ventura-Clapier, R. Impaired voluntary running capacity of creatine kinase-deficient mice. J. Physiol. 2005, 565, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Spindler, M.; Engelhardt, S.; Niebler, R.; Wagner, H.; Hein, L.; Lohse, M.J.; Neubauer, S. Alterations in the myocardial creatine kinase system precede the development of contractile dysfunction in β1-adrenergic receptor transgenic mice. J. Mol. Cell Cardiol. 2003, 35, 389–397. [Google Scholar] [CrossRef]
- Harvey, K.B.; Bothe, A., Jr.; Blackburn, G.L. Nutritional assessment and patient outcome during oncological therapy. Cancer 1979, 43, 2065–2069. [Google Scholar] [CrossRef]
- Brand, M.D.; Affourtit, C.; Esteves, T.C.; Green, K.; Lambert, A.J.; Miwa, S.; Pakay, J.L.; Parker, N. Mitochondrial superoxide: Production, biological effects, and activation of uncoupling proteins. Free Radic. Biol. Med. 2004, 37, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Esteves, T.C. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005, 2, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Esteves, T.C.; Pakay, J.L.; Jekabsons, M.B.; Lambert, A.J.; Portero-Otin, M.; Pamplona, R.; Vidal-Puig, A.J.; Wang, S.; Roebuck, S.J.; et al. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003, 22, 4103–4110. [Google Scholar] [CrossRef] [PubMed]
- Lexell, J.; Taylor, C.C.; Sjostrom, M. What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J. Neurol. Sci. 1988, 84, 275–294. [Google Scholar] [CrossRef]
- Lexell, J.; Downham, D. What determines the muscle cross-sectional area? J. Neurol. Sci. 1992, 111, 113–114. [Google Scholar] [CrossRef]
- Lexell, J.; Downham, D. What is the effect of ageing on type 2 muscle fibres? J. Neurol. Sci. 1992, 107, 250–251. [Google Scholar] [CrossRef]
- Lexell, J. Human aging, muscle mass, and fiber type composition. J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50, 11–16. [Google Scholar] [PubMed]
- Anderson, E.J.; Neufer, P.D. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H2O2 generation. Am. J. Physiol. Cell Physiol. 2006, 290, C844–C851. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R. Metal ion-catalyzed oxidation of proteins: biochemical mechanism and biological consequences. Free Radic. Biol. Med. 1990, 9, 315–325. [Google Scholar] [CrossRef]
- Hussain, S.N. Respiratory muscle dysfunction in sepsis. Mol. Cell Biochem. 1998, 179, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.N.; Simkus, G.; Roussos, C. Respiratory muscle fatigue: A cause of ventilatory failure in septic shock. J. Appl. Physiol. 1985, 58, 2033–2040. [Google Scholar] [PubMed]
- Hussain, S.N.; Matar, G.; Barreiro, E.; Florian, M.; Divangahi, M.; Vassilakopoulos, T. Modifications of proteins by 4-hydroxy-2-nonenal in the ventilatory muscles of rats. Am. J. Physiol. Lung. Cell Mol. Physiol. 2006, 290, L996–L1003. [Google Scholar] [CrossRef] [PubMed]
- Lanone, S.; Taille, C.; Boczkowski, J.; Aubier, M. Diaphragmatic fatigue during sepsis and septic shock. Intensive Care Med. 2005, 31, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Colzani, M.; Aldini, G.; Carini, M. Mass spectrometric approaches for the identification and quantification of reactive carbonyl species protein adducts. J. Proteomics 2013, 92, 28–50. [Google Scholar] [CrossRef] [PubMed]
- Grimsrud, P.A.; Xie, H.; Griffin, T.J.; Bernlohr, D.A. Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 2008, 283, 21837–21841. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Smuder, A.J.; Kavazis, A.N.; Hudson, M.B. Experimental guidelines for studies designed to investigate the impact of antioxidant supplementation on exercise performance. Int. J. Sport Nutr. Exerc. Metab. 2010, 20, 2–14. [Google Scholar]
- Butterfield, D.A.; Dalle-Donne, I. Redox proteomics. Antioxid. Redox. Signal 2012, 17, 1487–1489. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barreiro, E. Role of Protein Carbonylation in Skeletal Muscle Mass Loss Associated with Chronic Conditions. Proteomes 2016, 4, 18. https://doi.org/10.3390/proteomes4020018
Barreiro E. Role of Protein Carbonylation in Skeletal Muscle Mass Loss Associated with Chronic Conditions. Proteomes. 2016; 4(2):18. https://doi.org/10.3390/proteomes4020018
Chicago/Turabian StyleBarreiro, Esther. 2016. "Role of Protein Carbonylation in Skeletal Muscle Mass Loss Associated with Chronic Conditions" Proteomes 4, no. 2: 18. https://doi.org/10.3390/proteomes4020018
APA StyleBarreiro, E. (2016). Role of Protein Carbonylation in Skeletal Muscle Mass Loss Associated with Chronic Conditions. Proteomes, 4(2), 18. https://doi.org/10.3390/proteomes4020018