
Integrated Proteomic and Transcriptomic-Based Approaches to Identifying Signature Biomarkers and Pathways for Elucidation of Daoy and UW228 Subtypes


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Preparation of Total Cell Lysate
2.3. One-Dimensional SDS-PAGE Electrophoresis and Trypsin In-Gel Digestion of Total Cellular Proteins and Analysis by LC-MS/MS
2.4. Protein Identification and Data Analysis
2.5. RNA Extraction
2.6. cDNA Library Preparation and RNA Sequencing
3. Results
3.1. Proteomic Analysis of Daoy and UW228
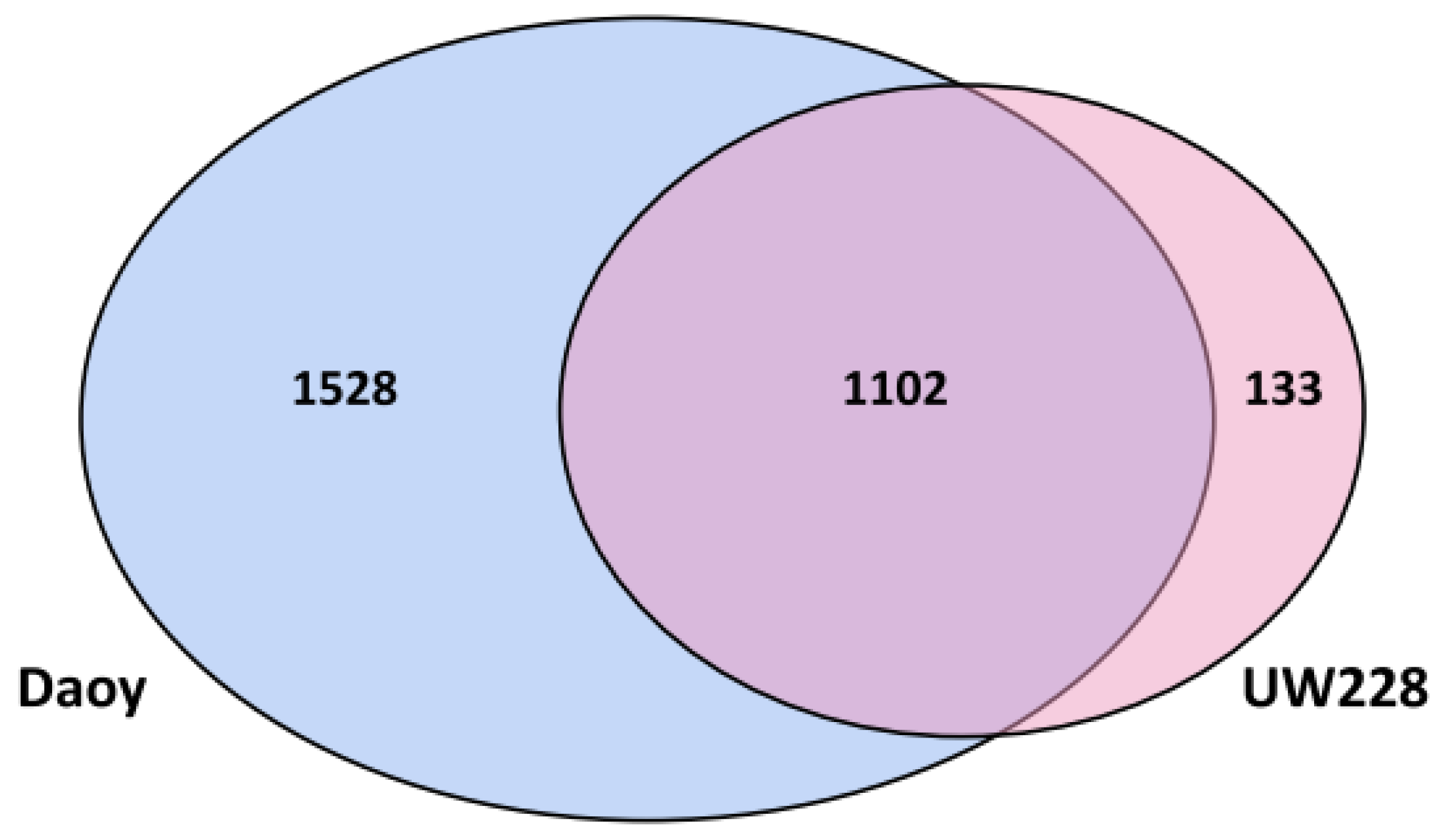
3.2. Correlation and Comparison of Proteomics and RNA-Seq Data
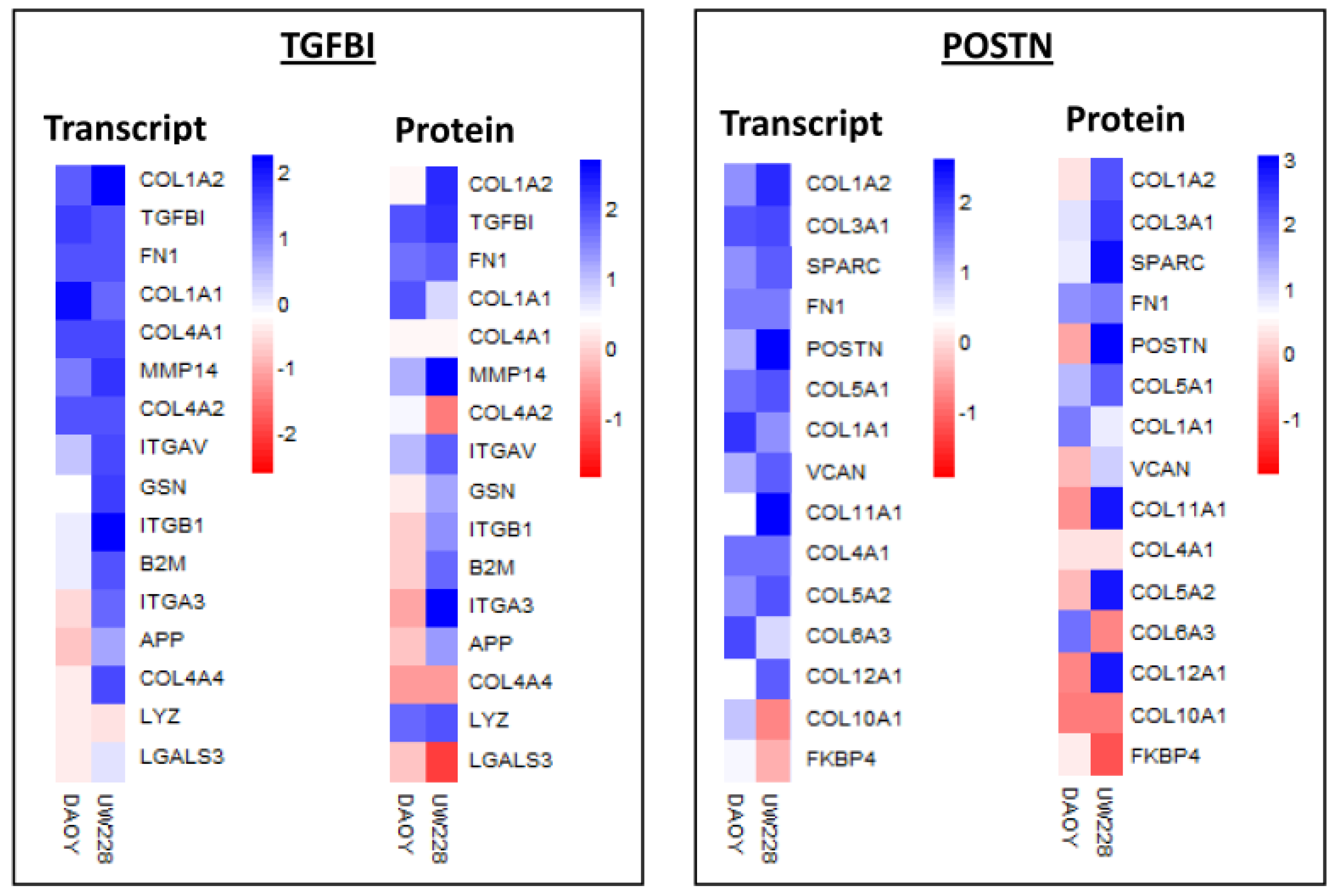
3.3. Analysis of Proteins Differentially Expressed between Daoy and UW228
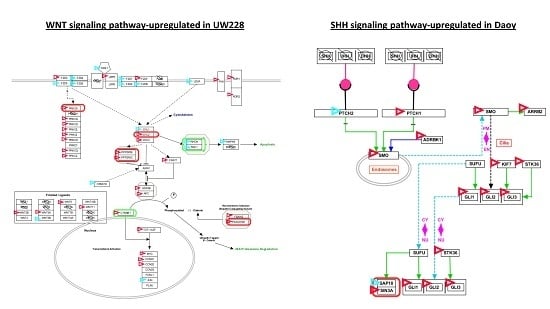
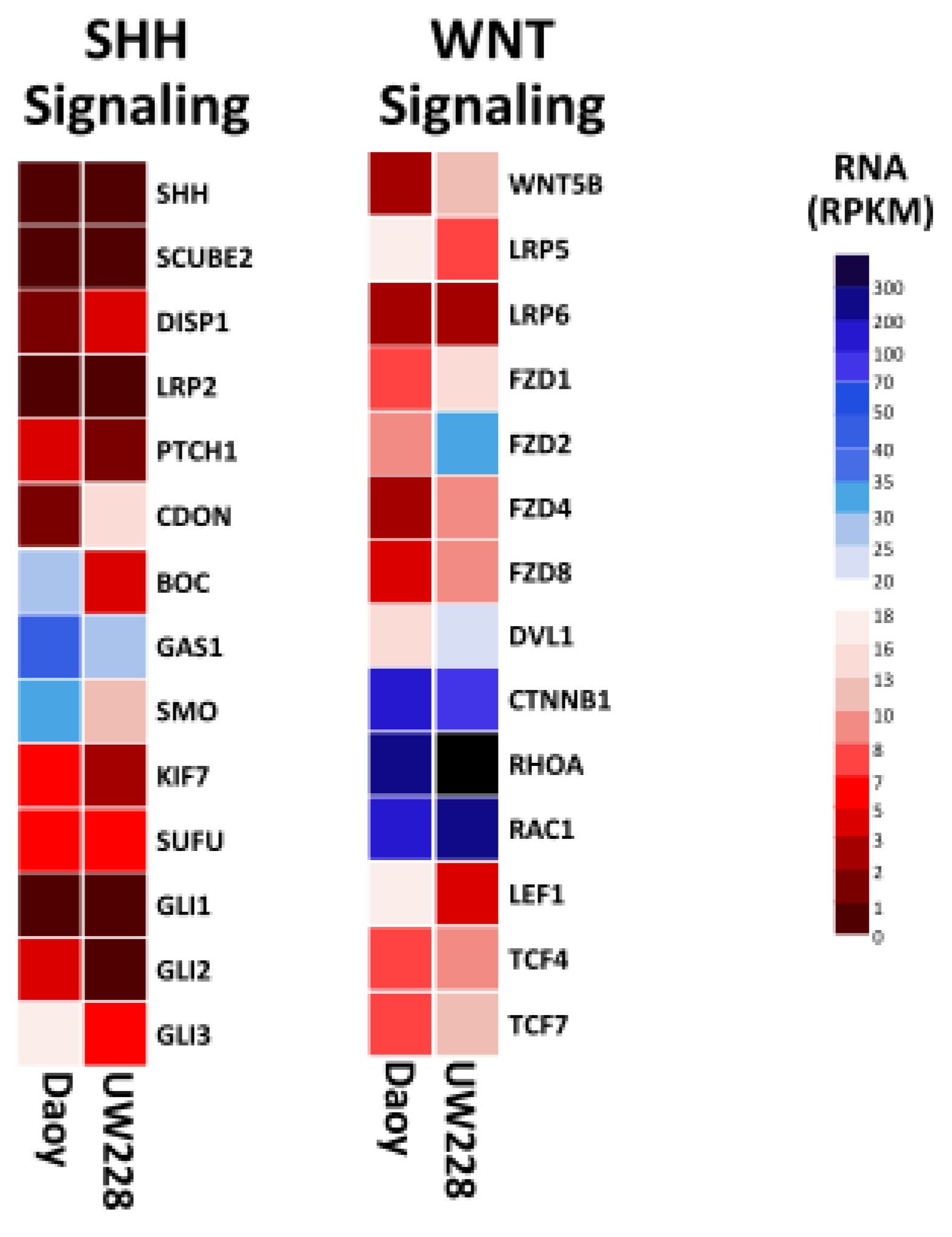
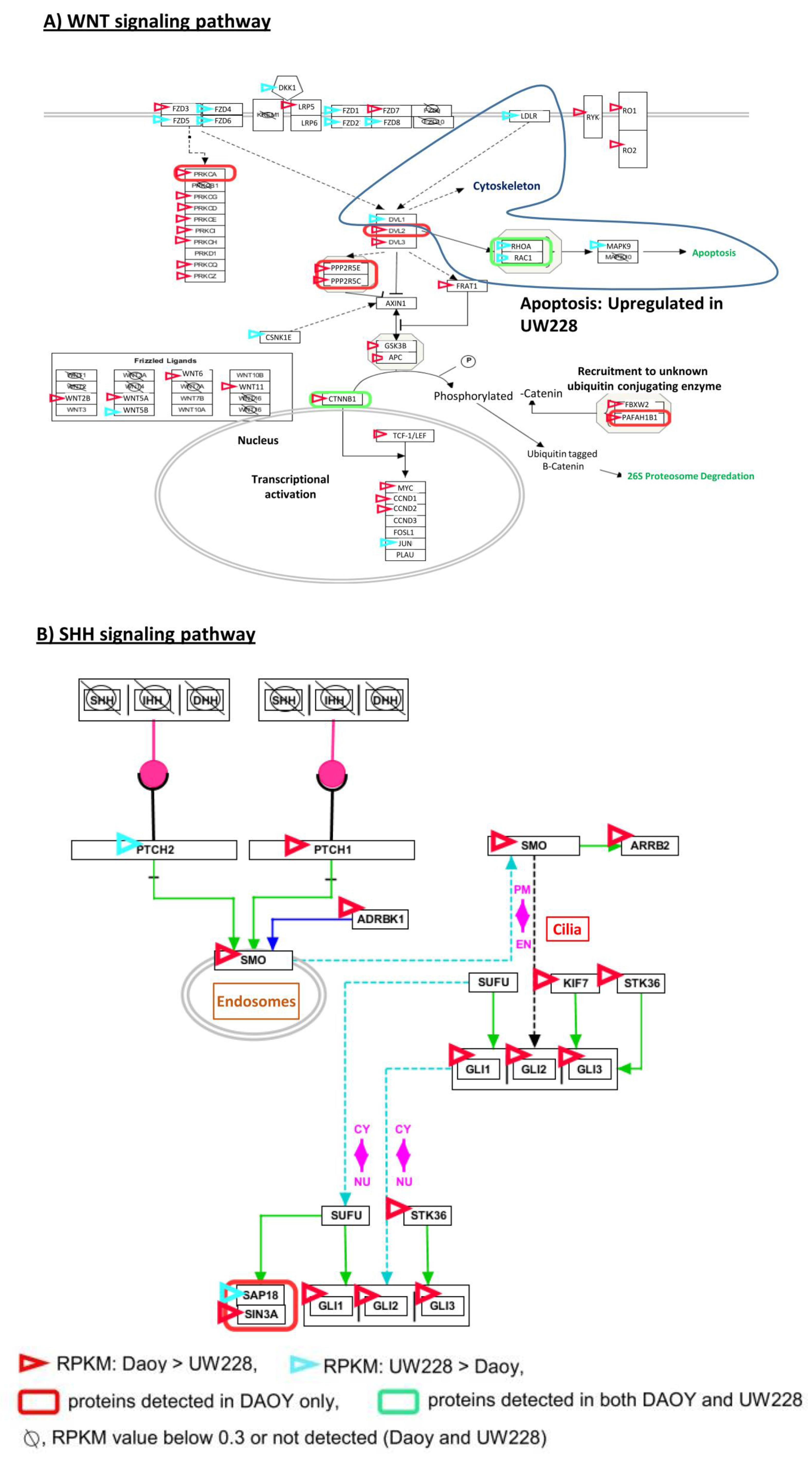
3.4. Signaling Pathways Involved in Daoy and UW228
4. Discussion and Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MB | Medulloblastoma; |
| LC | Liquid chromatography |
| MS/MS | Tandem mass spectrometry |
| RNASeq | RNA sequencing |
| SHH | Sonic hedgehog |
| D/N | Desmoplastic/nodular |
| MBEN | Medulloblastoma with extensive nodularity |
| CTNNB1 | Beta-catenin |
| DDX3X | DEAD-box RNA helicase gene |
| SMARCA4 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4 |
| TP53 | Tumor suppressor P53 |
| KMT2D | Lysine (K)-specific methyltransferase 2D |
| PTCH1 | Patched 1 |
| GLI1/GLI2 | GLI family zinc finger 1 or 2 |
| KDM6A | Lysine (K)-Specific Demethylase 6A |
| CDK6 | Cyclin-Dependent Kinase 6 |
| NPM1 | Neuplasmin |
| EZR | Ezrin |
| SET | SET nuclear oncogene |
| TGFBI | Transforming growth factor, beta induced |
| MCAM | Melanoma cell adhesion molecule |
| POSTN | Periostin, osteoblast specific factor |
| SUFU | Suppressor of fused homolog |
| SMO | Smoothened, frizzled class receptor |
| CRK | V-Crk avian sarcoma virus CT10 oncogene homolog |
| LASP1 | LIM And SH3 protein 1 |
| PCNA | Proliferating cell nuclear antigen |
| JUP | Junction plakoglobin |
| CRYAB | Crystallin Alpha B |
References
- Giordana, M.T.; Schiffer, P.; Lanotte, M.; Girardi, P.; Chio, A. Epidemiology of adultmedulloblastoma. Int. J. Cancer 1999, 80, 689–692. [Google Scholar] [CrossRef]
- Smoll, N.R.; Drummond, K.J. The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J. Clin. Neurosci. 2012, 19, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Gudrunardottir, T.; Lannering, B.; Remke, M.; Taylor, M.D.; Wells, E.M.; Keating, R.F.; Packer, R.J. Treatment developments and the unfolding of the quality of life discussion in childhood medulloblastoma: A review. Childs Nerv. Syst. 2014, 30, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.; Chintagumpala, M.; Ashley, D.; Kellie, S.; Kun, L.E.; Merchant, T.E.; Woo, S.; Wheeler, G.; Ahern, V.; Krasin, M.J.; et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): Long-term results from a prospective, multicentre trial. Lancet Oncol. 2006, 7, 813–820. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.C.; Fuller, C.; Hogg, T.L.; Dalton, J.; Finkelstein, D.; Lau, C.C.; Chintagumpala, M.; Adesina, A.; Ashley, D.M.; Kellie, S.J.; et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J. Clin. Oncol. 2006, 24, 1924–1931. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Koster, J.; Bunt, J.; Hasselt, N.E.; Lakeman, A.; van Sluis, P.; Troost, D.; Meeteren, N.S.; Caron, H.N.; Cloos, J.; et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE 2008, 3, e3088. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.J.; Tsherniak, A.; Tamayo, P.; Santagata, S.; Ligon, A.; Greulich, H.; Berhoukim, R.; Amani, V.; Goumnerova, L.; Eberhart, C.G.; et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J. Clin. Oncol. 2011, 29, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.W.; Schlanstein, M.; Northcott, P.A.; Cho, Y.-J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, K.; Gururangan, S. Molecular variants and mutations in medulloblastoma. Pharmgenom. Pers. Med. 2014, 7, 43–51. [Google Scholar]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Li, M.; Zhang, X.; Jones, S.; Leary, R.J.; Lin, J.C.; Boca, S.M.; Carter, H.; Samayoa, J.; Bettegowda, C.; et al. The genetic landscape of the childhood cancer medulloblastoma. Science 2011, 331, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Jones, D.T.W.; Jäger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Zurawel, R.H.; Chiappa, S.A.; Allen, C.; Raffel, C. Sporadic medulloblastomas contain oncogenic beta catenin mutations. Cancer Res. 1998, 58, 896–899. [Google Scholar] [PubMed]
- Ellison, D.W.; Dalton, J.; Kocak, M.; Nicholson, S.L.; Fraga, C.; Neale, G.; Kenney, A.M.; Brat, D.J.; Perry, A.; Yong, W.H.; et al. Medulloblastoma: Clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011, 121, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, E.; Remke, M.; Sturm, D.; Benner, A.; Witt, H.; Milde, T.; von Bueren, A.O.; Wittmann, A.; Schöttler, A.; Jorch, N.; et al. TP53 mutation is frequently associated with CTNNB1 mutation or MYCN amplification and is compatible with long-term survival in medulloblastoma. J. Clin. Oncol. 2010, 28, 5188–5196. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Jones, D.T.; Kool, M.; Robinson, G.W.; Gilbertson, R.J.; Cho, Y.-J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Shih, D.J.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, R.J.; Ellison, D.W. The origins of medulloblastoma subtypes. Annu. Rev. Pathol. 2008, 3, 341–365. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Nakahara, Y.; Wu, X.; Feuk, L.; Ellison, D.W.; Croul, S.; Mack, S.; Kongkham, P.N.; Peacock, J.; Dubuc, A.; et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 2009, 41, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Sengoku, T.; Yokoyama, S. Structural basis for histone H3 Lys 27 demethylation by UTX/KDM6A. Genes Dev. 2011, 25, 2266–2277. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, P.F.; Jenkyn, D.J.; Papadimitriou, J.M. Establishment of a human medulloblastoma cell line and its heterotransplantation into nude mice. J. Neuropathol. Exp. Neurol. 1985, 44, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Keles, G.E.; Berger, M.S.; Srinivasan, J.; Kolstoe, D.D.; Bobola, M.S.; Silber, J.R. Establishment and characterization of four human medulloblastoma-derived cell lines. Oncol. Res. 1995, 7, 493–503. [Google Scholar] [PubMed]
- Peyrl, A.; Krapfenbauer, K.; Slavc, I.; Yang, J.W.; Strobel, T.; Lubec, G. Protein profiles of medulloblastoma cell lines DAOY and D283: Identification of tumor-related proteins and principles. Proteomics 2003, 3, 1781–1800. [Google Scholar] [CrossRef] [PubMed]
- Martelli, C.; D’Angelo, L.; Barba, M.; Baranzini, M.; Inserra, I.; Iavarone, F.; Vincenzoni, F.; Tamburrini, G.; Massimi, L.; Di Rocco, C.; et al. Top-down proteomic characterization of DAOY medulloblastoma tumor cell line. EuPA Open Proteom. 2016, 12, 13–21. [Google Scholar] [CrossRef]
- Zanini, C.; Ercole, E.; Giorgia Mandili, G.; Salaroli, R.; Poli, A.; Renna, C.; Papa, V.; Cenacchi, G.; Forni, M. Medullospheres from DAOY, UW228 and ONS-76 Cells: Increased Stem Cell Population and Proteomic Modifications. PLoS ONE 2013, 8, e63748. [Google Scholar] [CrossRef] [PubMed]
- Pambid, M.R.; Berns, R.; Adomat, H.H.; Hu, K.; Triscott, J.; Maurer, N.; Zisman, N.; Ramaswamy, V.; Hawkins, C.E.; Taylor, M.D.; et al. Overcoming resistance to sonic hedgehog inhibition by targeting p90 ribosomal S6 kinase in pediatric medulloblastoma. Pediatr. Blood Cancer 2014, 61, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Othman, R.T.; Kimishi, I.; Bradshaw, T.D.; Storer, L.C.D.; Korshunov, A.; Pfister, S.M.; Grundy, R.G.; Kerr, I.D.; Coyle, B. Overcoming multiple drug resistance mechanisms in medulloblastoma. Acta Neuropathol. Commun. 2014, 2, 57. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wu, W.W.; Zhang, Z.; Masilamani, S.; Shen, R.-F. Decoy Methods for Assessing False Positives and False Discovery Rates in Shotgun Proteomics. Anal Chem. 2009, 81, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Fanayan, S.; Smith, J.T.; Lee, L.Y.; Yan, F.; Snyder, M.; Hancock, W.S.; Nice, E. Proteogenomic analysis of human colon carcinoma cell lines LIM1215, LIM1899, and LIM2405. J. Proteome Res. 2013, 12, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Waern, K.; Snyder, M. RNA-Seq: A method for comprehensive transcriptome analysis. Curr. Protoc. Mol. Biol. 2010. [Google Scholar] [CrossRef]
- Warden, C.D.; Yuan, Y.-C.; Wu, X. Optimal calculation of RNA-Seq fold-change values. Int. J. Comput. Bioinform. In Silico Model. 2013, 2, 285–292. [Google Scholar]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Cha, S.; Rejtar, T.; Richardson, E.A.; Karger, B.L.; Sgroi, D.C. Integrated proteomic, transcriptomic, and biological network analysis of breast carcinoma reveals molecular features of tumorigenesis and clinical relapse. Mol. Cell. Proteom. 2012, 11, M111-014910. [Google Scholar] [CrossRef] [PubMed]
- Ning, K.; Fermin, D.; Nesvizhskii, A.I. Comparative analysis of different label-free mass spectrometry based protein abundance estimates and their correlation with RNA-Seq gene expression data. J. Proteome Res. 2012, 11, 2261–2271. [Google Scholar] [CrossRef] [PubMed]
- Gygi, S.P.; Rochon, Y.; Franza, B.R.; Aebersold, R. Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 1999, 19, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Pascal, L.E.; True, L.D.; Deutsch, E.W.; Campbell, D.S.; Risk, M.; Coleman, I.M.; Eichner, L.J.; Nelson, P.S.; Liu, A.Y. Correlation of mRNA and protein levels: Cell type-specific gene expression of cluster designation antigens in the prostate. BMC Genom. 2008, 9, 246. [Google Scholar] [CrossRef] [PubMed]
- Yeung, E.S. Genome-wide correlation between mRNA and protein in a single cell. Angew. Chem. Int. Ed. Engl. 2011, 50, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Ranger, A.; McDonald, W.; Moore, E.; Delmaestro, R. The invasiveness of five medulloblastoma cell lines in collagen gels. J. Neurooncol. 2010, 96, 181–189. [Google Scholar] [CrossRef] [PubMed]
- 4Assinder, S.J.; Stanton, J.A.L.; Prasad, P.D. Transgelin: An actin-binding protein and tumour suppressor. Int. J. Biochem. Cell Biol. 2009, 41, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Shields, J.M.; Rogers-Graham, K.; Der, C.J. Loss of transgelin in breast and colon tumors and in RIE-1 cells by Ras deregulation of gene expression through Raf-independent pathways. J. Biol. Chem. 2002, 277, 9790–9799. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.D.; Stanton, J.A.; Assinder, S.J. Expression of the actin-associated protein transgelin (SM22) is decreased in prostate cancer. Cell Tissue Res. 2010, 339, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, H.; Deng, Y.J.; Wang, S.; Liu, C.; Jin, H.; Ding, Y.Q. Transgelin as a suppressor is associated with poor prognosis in colorectal carcinoma patients. Mod. Pathol. 2009, 22, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Wen, G.; Partridge, M.A.; Li, B.; Hong, M.; Liao, W.; Cheng, S.K.; Zhao, Y.; Calaf, G.M.; Liu, T.; Zhou, J.; et al. TGFBI expression reduces in vitro and in vivo metastatic potential of lung and breast tumor cells. Cancer Lett. 2011, 308, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Kyutoku, M.; Taniyama, Y.; Katsuragi, N.; Shimizu, H.; Kunugiza, Y.; Iekushi, K.; Koibuchi, N.; Sanada, F.; Oshita, Y.; Morishita, R. Role of periostin in cancer progression and metastasis: Inhibition of breast cancer progression and metastasis by anti-periostin antibody in a murine model. Int. J. Mol. Med. 2011, 28, 181–186. [Google Scholar] [PubMed]
- Buhren, J.; Christoph, A.H.; Buslei, R.; Albrecht, S.; Wiestler, O.D.; Pietsch, T. Expression of the neurotrophin receptor p75NTR in medulloblastomas is correlated with distinct histological and clinical features: Evidence for a medulloblastoma subtype derived from the external granule cell layer. J. Neuropathol. Exp. Neurol. 2000, 59, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Park, P.C.; Taylor, M.D.; Mainprize, T.G.; Becker, L.E.; Ho, M.; Dura, W.T.; Squire, J.; Rutka, J.T. Transcriptional profiling of medulloblastoma in children. J. Neurosurg. 2003, 99, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Yokota, N.; Mainprize, T.G.; Taylor, M.D.; Kohata, T.; Loreto, M.; Ueda, S.; Dura, W.; Grajkowska, W.; Kuo, J.S.; Rutka, J.T. Identification of differentially expressed and developmentally regulated genes in medulloblastoma using suppression subtraction hybridization. Oncogene 2004, 23, 3444–3453. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhuang, L.; Szatmary, P.; Wen, L.; Sun, H.; Lu, Y.; Xu, Q.; Chen, X. Upregulation of heat shock proteins (HSPA12A, HSP90B1, HSPA4, HSPA5 and HSPA6) in tumour tissues is associated with poor outcomes from HBV-related early-stage hepatocellular carcinoma. Int. J. Med. Sci. 2015, 12, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Chang, J.T.; Geradts, J.; Neckers, L.M.; Haystead, T.; Spector, N.L.; Lyerly, H.K. Amplification and high-level expression of heat shock protein 90 marks aggressive phenotypes of human epidermal growth factor receptor 2 negative breast cancer. Breast Cancer Res. 2012, 14, R62. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Aiken, C.; McClelland, R.; Morrison, L.C.; Tatari, N.; Remke, M.; Ramaswamy, V.; Issaivanan, M.; Ryken, T.; Del Bigio, M.R.; et al. Characterization of novel biomarkers in selecting for subtype specific medulloblastoma phenotypes. Oncotarget 2015, 6, 38881–38900. [Google Scholar] [PubMed]
- Reardon, D.A.; Michalkiewicz, E.; Boyett, J.M.; Sublett, J.E.; Entrekin, R.E.; Ragsdale, S.T.; Valentine, M.B.; Behm, F.G.; Li, H.; Heideman, R.L.; et al. Extensive genomic abnormalities in childhood medulloblastoma by comparative genomic hybridization. Cancer Res. 1997, 57, 4042–4047. [Google Scholar] [PubMed]
- Ellison, D. Classifying the medulloblastoma: Insights from morphology and molecular genetics. Neuropathol. Appl. Neurobiol. 2002, 28, 257–282. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zhou, L.; Killela, P.; Rasheed, A.B.; Di, C.; Poe, W.E.; McLendon, R.E.; Bigner, D.D.; Nicchitta, C.; Yan, H. Glioblastoma proto-oncogene SEC61gamma is required for tumor cell survival and response to endoplasmic reticulum stress. Cancer Res. 2009, 69, 9105–9111. [Google Scholar] [CrossRef] [PubMed]
- Eiriksdottir, G.; Barkardottir, R.B.; Agnarsson, B.A.; Johannesdottir, G.; Olafsdottir, K.; Egilsson, V.; Ingvarsson, S. High incidence of loss of heterozygosity at chromosome 17p13 in breast tumours from BRCA2 mutation carriers. Oncogene 1998, 16, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Mano, M.S.; Rosa, D.D.; De Azambuja, E.; Ismael, G.F.; Durbecq, V. The 17q12-q21 amplicon: Her2 and topoisomerase-IIα and their importance to the biology of solid tumours. Cancer Treat. Rev. 2007, 33, 64–77. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subtype | WNT | SHH | Group 3 | Group 4 |
|---|---|---|---|---|
| Prevalence | 10% | 30% | 25% | 35% |
| Patients | Common in children and adults, uncommon in infants. | Common in infants and adults, less common in children. | Common in infants and children, uncommon in adults. | Most common in children, less common in infants and adults. |
| Male = Female | Male = Female | Males > Females | Males > Females | |
| Prognosis (5 year survival) | Very good (95%) | Infants good others intermediate (75%) | Poor (50%) | Intermediate (75%) |
| Histology | Classic, rare large cell/anaplastic | Classic, large cell/anaplastic, desmoplastic, nodular | Classic, large cell anaplastic | Classic, large cell anaplastic |
| Metastasis | Rare (5%–10%) | Uncommon (15%–20%) | Very frequent (40%–45%) | Frequent (35%–40%) |
| Genetic characteristics | CTNNB1 mutations | PTCH1, SMO, SUFU mutations; GLI1, GLI2, MYCN amplification | MYC amplification | CDK6, MYCN amplification |
| WNT signaling | ||||
| MYC over-expressed |
| Accession | Gene | Description | Chromosome | Daoy | UW228 | ||
|---|---|---|---|---|---|---|---|
| RNA (RPKM) | # Unique Peptides | RNA (RPKM) | # Unique Peptides | ||||
| Overexpressed in Daoy, relative to UW228 | |||||||
| P07900 | HSP90AA1 | Heat Shock Protein 90kDa Alpha Family Class A Member 1 | 14q32 | 421.65 | 18.00 | 167.03 | 7.00 |
| P08238 | HSP90AB1 | Heat Shock Protein 90kDa Alpha Family Class B Member 1 | 6p21 | 775.45 | 19.00 | 362.14 | 9.00 |
| P06748 | NPM1 | Nucleophosmin (Nucleolar Phosphoprotein B23, Numatrin) | 5q35 | 668.25 | 5.33 | 239.64 | 3.00 |
| Q15582 | TGFBI | Transforming Growth Factor Beta Induced | 5q31 | 3135.54 | 17.00 | 1249.52 | 7.00 |
| Q00325 | DSP | Desmoplakin | 6q24 | 22.83 | 10.00 | 0.03 | 2.00 |
| P24821 | TNC | Tenascin C | 9q33 | 241.59 | 8.00 | 37.95 | 1.00 |
| P00533 | EGFR | Epidermal Growth Factor Receptor | 7p11 | 21.24 | 8.00 | 13.13 | 2.00 |
| Q9NS86 | LANCL2 | LanC Lantibiotic Synthetase Component C-Like 2 (Bacterial)1 | 7p11 | 22.28 | 3.00 | 6.52 | 1.00 |
| P62993 | GRB2 | Growth Factor Receptor-Bound Protein 2 | 17q24 | 44.9 | 5.00 | 30.56 | 1.00 |
| P11388 | TOP2A | Topoisomerase (DNA) II Alpha | 17q21 | 129.62 | 8.00 | 45.76 | 0.00 |
| P14923 | JUP | Junction Plakoglobin | 17q21 | 56.97 | 7.00 | 19.84 | 1.00 |
| P46108 | CRK | V-Crk Avian Sarcoma Virus CT10 Oncogene Homolog | 17p13 | 50.20 | 8.00 | 17.16 | 1.00 |
| P04637 | TP53 | Tumor Protein P53 | 17p13 | 235.10 | 5.00 | 36.74 | ND |
| P62826 | RAN | RAN, Member RAS Oncogene Family | 12q24 | 479.63 | 6.00 | 240.29 | 3.00 |
| P43246 | MSH2 | MutS Homolog 2 | 2p21 | 18.07 | 6.00 | 10.87 | ND |
| P52701 | MSH6 | MutS Homolog 6 | 2p16 | 25.35 | 8.00 | 19.98 | 1.00 |
| Overexpressed in UW228, relative to Daoy | |||||||
| P02511 | CRYAB | Crystallin Alpha B | 11q23 | 0.74 | ND | 438.60 | 4.67 |
| Q14847 | LASP1 | LIM And SH3 Protein 1 | 17q12 | 59.55 | 10.00 | 124.73 | 18.00 |
| Q15063 | POSTN | Periostin | 13q13 | 6.53 | ND | 1023.51 | 6.00 |
| P43121 | MCAM | Melanoma Cell Adhesion Molecule | 11q23 | 36.97 | ND | 61.89 | 10.33 |
| P32004 | L1CAM | L1 Cell Adhesion Molecule | Xq28 | 1.15 | ND | 324.27 | 10.33 |
| P46939 | UTRN | Utrophin | 6q24 | 3.7 | 1.00 | 9.86 | 12.00 |
| Q07954 | LRP1 | Low-density lipoprotein receptor-related protein 1 | 12q13 | 6.73 | 2.00 | 79.98 | 13.67 |
| P80723 | BASP1 | Brain Abundant Membrane Attached Signal Protein 1 | 5p15 | 63.7 | 5.00 | 270.37 | 9.00 |
| P98082 | DAB2 | DAB2, Clathrin Adaptor Protein | 5p13 | 7.8 | ND | 68.7 | 2.00 |
| Q6UVK1 | CSPG4 | Chondroitin Sulfate Proteoglycan 4 | 15q24 | 6.8 | 3.00 | 17.8 | 10.00 |
| Q01995 | TAGLN | Transgelin | 11q23 | 27.27 | 8.00 | 353.95 | 12.00 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Higdon, R.; Kala, J.; Wilkins, D.; Yan, J.F.; Sethi, M.K.; Lin, L.; Liu, S.; Montague, E.; Janko, I.; Choiniere, J.; et al. Integrated Proteomic and Transcriptomic-Based Approaches to Identifying Signature Biomarkers and Pathways for Elucidation of Daoy and UW228 Subtypes. Proteomes 2017, 5, 5. https://doi.org/10.3390/proteomes5010005
Higdon R, Kala J, Wilkins D, Yan JF, Sethi MK, Lin L, Liu S, Montague E, Janko I, Choiniere J, et al. Integrated Proteomic and Transcriptomic-Based Approaches to Identifying Signature Biomarkers and Pathways for Elucidation of Daoy and UW228 Subtypes. Proteomes. 2017; 5(1):5. https://doi.org/10.3390/proteomes5010005
Chicago/Turabian StyleHigdon, Roger, Jessie Kala, Devan Wilkins, Julia Fangfei Yan, Manveen K. Sethi, Liang Lin, Siqi Liu, Elizabeth Montague, Imre Janko, John Choiniere, and et al. 2017. "Integrated Proteomic and Transcriptomic-Based Approaches to Identifying Signature Biomarkers and Pathways for Elucidation of Daoy and UW228 Subtypes" Proteomes 5, no. 1: 5. https://doi.org/10.3390/proteomes5010005
APA StyleHigdon, R., Kala, J., Wilkins, D., Yan, J. F., Sethi, M. K., Lin, L., Liu, S., Montague, E., Janko, I., Choiniere, J., Kolker, N., Hancock, W. S., Kolker, E., & Fanayan, S. (2017). Integrated Proteomic and Transcriptomic-Based Approaches to Identifying Signature Biomarkers and Pathways for Elucidation of Daoy and UW228 Subtypes. Proteomes, 5(1), 5. https://doi.org/10.3390/proteomes5010005