A Novel Analysis of the Peptide Terminome Characterizes Dynamics of Proteolytic Regulation in Vertebrate Skeletal Muscle Under Severe Stress

Abstract
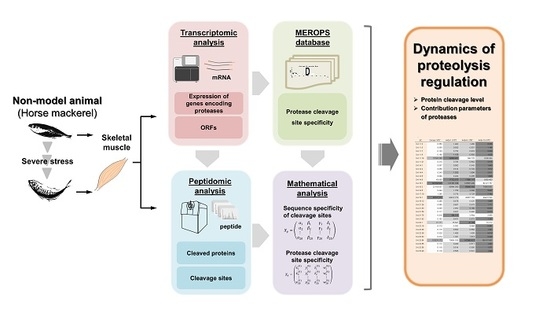
:
1. Introduction
2. Material and Methods
2.1. Reagents
2.2. Fish Samples
2.3. Transcriptomic Analysis
2.3.1. RNA Extraction
2.3.2. cDNA Library Construction and Sequencing
2.3.3. Sequence Data Processing and De Novo Assembly
2.3.4. Gene Expression Analysis of Protease
2.4. Peptidomic Analysis
2.4.1. Free Peptide Extraction
2.4.2. Liquid Chromatography
2.4.3. Shotgun Analysis
2.4.4. SWATH Analysis
2.4.5. Database Search
2.4.6. SWATH Data Processing
2.4.7. Multiple Linear Regression Model
3. Results
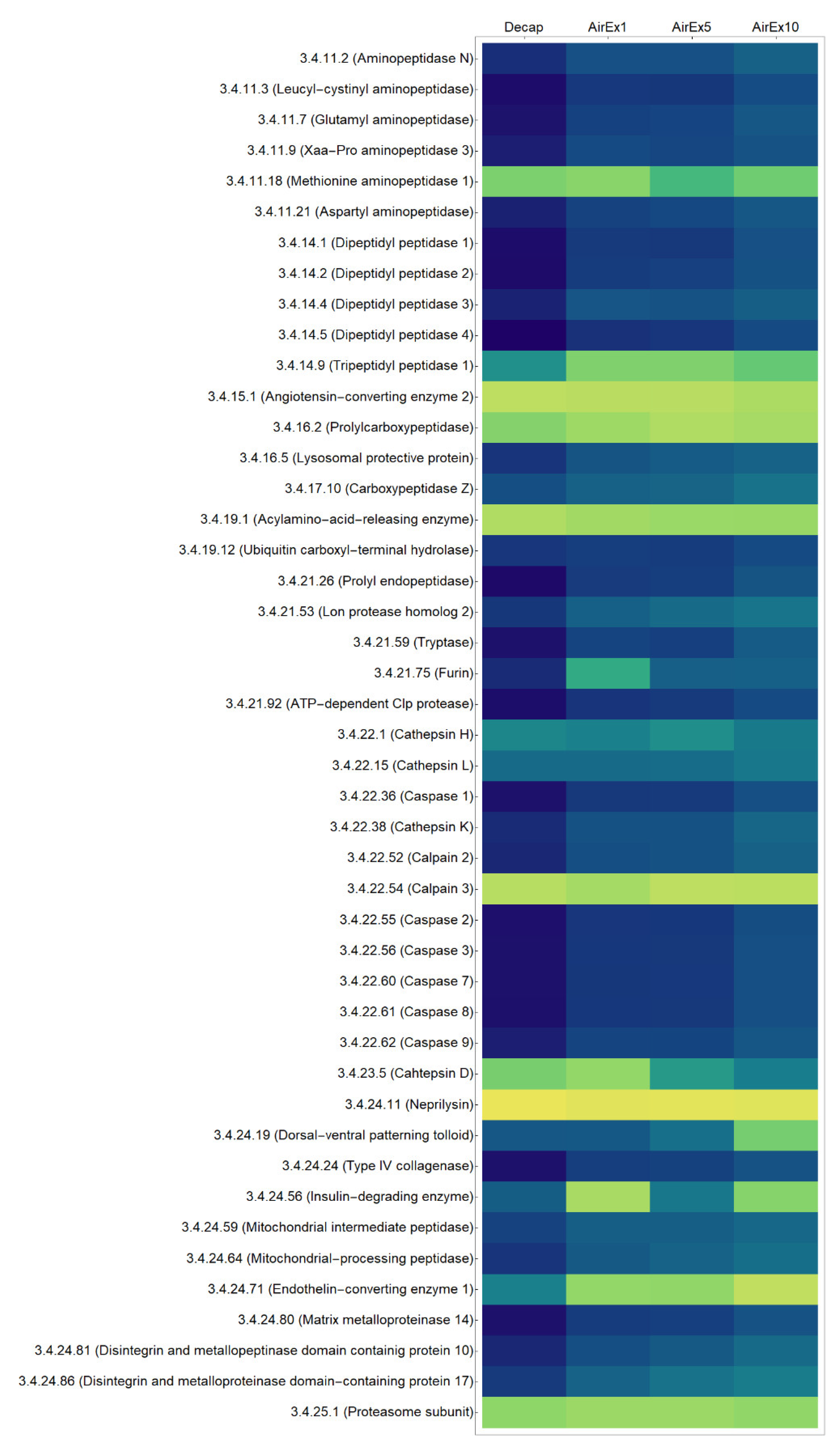
3.1. Expression of Genes Encoding Proteases
3.2. Protein cleavage levels
3.3. Dynamics of Proteolytic Regulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Urbe, S.; Clague, M.J. Selective protein degradation in cell signalling. Semin. Cell Dev. Biol. 2012, 23, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Flick, K.; Kaiser, P. Protein degradation and the stress response. Semin. Cell Dev. Biol. 2012, 23, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Ichijo, H. Mitochondrial proteolysis: Its emerging roles in stress responses. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Lopez-Soto, A.; Kumar, S.; Kroemer, G. Caspases connect cell-death signaling to organismal homeostasis. Immunity 2016, 44, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Repnik, U.; Stoka, V.; Turk, V.; Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2012, 1824, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Mahrus, S.; Trinidad, J.C.; Barkan, D.T.; Sali, A.; Burlingame, A.L.; Wells, J.A. Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 2008, 134, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Baggerman, G.; Verleyen, P.; Clynen, E.; Huybrechts, J.; De Loof, A.; Schoofs, L. Peptidomics. J. Chromatogr. B 2004, 803, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Knappe, P.; Schrader, M.; Zucht, H.D. The peptidomics concept. Comb. Chem. High Throughput Screen 2005, 8, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Gelman, J.S.; Sironi, J.; Castro, L.M.; Ferro, E.S.; Fricker, L.D. Peptidomic analysis of human cell lines. J. Proteome Res. 2011, 10, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Schulz-Knappe, P. Peptidomics technologies for human body fluids. Trends Biotechnol. 2001, 19, S55–S60. [Google Scholar] [CrossRef]
- Lone, A.M.; Kim, Y.G.; Saghatelian, A. Peptidomics methods for the identification of peptidase-substrate interactions. Curr. Opin. Chem. Biol. 2013, 17, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Dallas, D.C.; Guerrero, A.; Parker, E.A.; Robinson, R.C.; Gan, J.; German, J.B.; Barile, D.; Lebrilla, C.B. Current peptidomics: Applications, purification, identification, quantification, and functional analysis. Proteomics 2015, 15, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Luge, T.; Sauer, S. Generating sample-specific databases for mass spectrometry-based proteomic analysis by using RNA sequencing. In Proteomics in Systems Biology: Methods and Protocols; Reinders, J., Ed.; Humana Press: New York, NY, USA, 2016; pp. 219–232. [Google Scholar]
- Evans, V.C.; Barker, G.; Heesom, K.J.; Fan, J.; Bessant, C.; Matthews, D.A. De novo derivation of proteomes from transcriptomes for transcript and protein identification. Nat. Methods 2012, 9, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Slebos, R.J.; Wang, D.; Halvey, P.J.; Tabb, D.L.; Liebler, D.C.; Zhang, B. Protein identification using customized protein sequence databases derived from RNA-Seq data. J. Proteome Res. 2012, 11, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Yabu, T.; Imamura, S.; Mizusawa, N.; Touhata, K.; Yamashita, M. Induction of autophagy by amino acid starvation in fish cells. Mar. Biotechnol. 2012, 14, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Sugita, T.; Yamashita, M. Stress response in fish at whole body and cellular levels. In Quality Improvement of Coastal Fish and Marine Invertebrates-Achievement by Short-Term Rearing and aSsociated Systems for Transportation and Marketing; Fukuda, Y., Watabe, S., Eds.; Koseisha Koseikaku: Tokyo, Japan, 2012; pp. 9–24. (In Japanese) [Google Scholar]
- Yamashita, M. Stress response of fish during capture. In Quality Control of Tuna Meat through Optimization of Fishing and Handling; Konno, K., Ochiai, Y., Fukuda, Y., Eds.; Kouseisha Kouseikaku: Tokyo, Japan, 2010. (In Japanese) [Google Scholar]
- Davis, M.W.; Parker, S.J. Fish size and exposure to air: Potential effects on behavioral impairment and mortality rates in discarded sablefish. N. Am. J. Fish. Manag. 2004, 24, 518–524. [Google Scholar] [CrossRef]
- Poli, B.; Parisi, G.; Scappini, F.; Zampacavallo, G. Fish welfare and quality as affected by pre-slaughter and slaughter management. Aquac. Int. 2005, 13, 29–49. [Google Scholar] [CrossRef]
- Addis, M.F.; Pisanu, S.; Preziosa, E.; Bernardini, G.; Pagnozzi, D.; Roggio, T.; Uzzau, S.; Saroglia, M.; Terova, G. 2D DIGE/MS to investigate the impact of slaughtering techniques on postmortem integrity of fish filet proteins. J. Proteom. 2012, 75, 3654–3664. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Joka, M.; Mochizuki, S.; Satoh, K.I.; Tsukamasa, Y.; Makinodan, Y. Influence of death struggle on the structural changes in chub mackerel muscle during chilled storage. Fish. Sci. 2001, 67, 744–751. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Betancur, R.R.; Broughton, R.E.; Wiley, E.O.; Carpenter, K.; Lopez, J.A.; Li, C.; Holcroft, N.I.; Arcila, D.; Sanciangco, M.; Cureton Ii, J.C.; et al. The tree of life and a new classification of bony fishes. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Ishii, K.; Kozaki, T.; Ogiwara, I.; Kanekatsu, M.; Yamada, T. Removal of redundant contigs from de novo RNA-Seq assemblies via homology search improves accurate detection of differentially expressed genes. BMC Genom. 2015, 16, 1031. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Pachter, L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods 2013, 10, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2017, 46, D624–D632. [Google Scholar] [CrossRef] [PubMed]
- Moon, T.W. Glucose intolerance in teleost fish: Fact or fiction? Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2001, 129, 243–249. [Google Scholar] [CrossRef]
- Polakof, S.; Panserat, S.; Soengas, J.L.; Moon, T.W. Glucose metabolism in fish: A review. J. Comp. Physiol. B 2012, 182, 1015–1045. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Feature | Number |
|---|---|
| Number of total reads | 30,554,904 |
| Number of assembled contigs | 279,308 |
| Number of contigs after removal of redundancy | 17,214 |
| N50 | 1793 bp |
| Minimum contig length | 201 bp |
| Maximum contig length | 27,079 bp |
| Total length | 20,694,310 bp |
| Number of contigs classified to EC 3.4 | 165 |
| Number of contigs annotated as protease | 156 |
| Feature | Number | ||
|---|---|---|---|
| AirEx1 | AirEx5 | AirEx10 | |
| Total proteins | 19 | 20 | 14 |
| Sarcomeric proteins | 8 | 12 | 4 |
| Cytosolic proteins | 7 | 2 | 2 |
| Other proteins | 4 | 6 | 8 |
| Cleaved Protein * | Fold Change ** | ||
|---|---|---|---|
| AirEx1/Decap | AirEx5/Decap | AirEx10/Decap | |
| PREDICTED: nuclear factor NF-kappa-B p105 subunit [Nothobranchius furzeri] | 0.48 | 0.79 | 0.80 |
| PREDICTED: phosphoglycerate kinase 1 [Nothobranchius furzeri] | 2.65 | 1.57 | 1.93 |
| PREDICTED: keratin, type II cytoskeletal cochleal-like [Nothobranchius furzeri] | 1.70 | 3.64 | 1.90 |
| PREDICTED: cofilin-2 [Nothobranchius furzeri] | 1.01 | 1.49 | 0.87 |
| PREDICTED: elongation factor 1-alpha [Nothobranchius furzeri] | 0.79 | 0.49 | 0.75 |
| Nebulin [Fundulus heteroclitus] | 1.03 | 0.72 | 0.62 |
| 40S ribosomal protein S19 [Fundulus heteroclitus] | 0.37 | 0.53 | 0.69 |
| Nebulin [Fundulus heteroclitus] | 1.53 | 1.76 | 1.69 |
| Skeletal muscle fast troponin T isoform 2 [Hippoglossus hippoglossus] | 2.83 | 2.80 | 2.88 |
| Skeletal muscle fast troponin T isoform 2 [Hippoglossus hippoglossus] | 1.62 | 1.09 | 0.71 |
| Hemoglobin subunit beta [Decapterus maruadsi] | 1.15 | 0.34 | 0.26 |
| Muscle actin OlMA1 [Oryzias latipes] | 0.51 | 0.96 | 1.89 |
| Lactate dehydrogenase-A [Sphyraena idiastes] | 4.59 | 1.00 | 1.24 |
| Troponin I [Siniperca chuatsi] | 0.94 | 0.89 | 0.40 |
| PDZ and LIM domain 7 [Epinephelus coioides] | 1.75 | 1.58 | 1.47 |
| Glycerol-3-phosphate dehydrogenase [Rachycentron canadum] | 1.15 | 0.28 | 0.25 |
| Glyceraldehyde-3-phosphate dehydrogenase [Rachycentron canadum] | 11.96 | 1.37 | 0.79 |
| Parvalbumin 3 [Siniperca chuatsi] | 0.49 | 0.33 | 0.85 |
| Troponin I [Epinephelus coioides] | 1.88 | 1.16 | 1.34 |
| Ubiquitin [Siniperca chuatsi] | 0.46 | 0.70 | 1.11 |
| Keratin 8, partial [Oreochromis mossambicus] | 3.03 | 19.68 | 0.93 |
| Tropomyosin [Siniperca chuatsi] | 2.61 | 0.77 | 0.60 |
| PREDICTED: desmin-like [Oreochromis niloticus] | 1.42 | 0.87 | 0.60 |
| PREDICTED: nudC domain-containing protein 1 [Oreochromis niloticus] | 1.37 | 2.49 | 1.84 |
| PREDICTED: keratin, type II cytoskeletal 8-like [Oreochromis niloticus] | 1.98 | 7.34 | 1.63 |
| PREDICTED: 28S ribosomal protein S36, mitochondrial [Oreochromis niloticus] | 1.35 | 0.91 | 0.89 |
| PREDICTED: protein deglycase DJ-1 [Oreochromis niloticus] | 0.99 | 0.78 | 0.94 |
| PREDICTED: apoptosis-inducing factor 1, mitochondrial [Oreochromis niloticus] | 0.75 | 0.31 | 0.34 |
| Parvalbumin 1 [Siniperca chuatsi] | 1.40 | 0.58 | 0.60 |
| PREDICTED: early endosome antigen 1-like [Oryzias latipes] | 3.59 | 3.59 | 0.38 |
| PREDICTED: myozenin-2 [Maylandia zebra] | 0.16 | 0.92 | 2.10 |
| PREDICTED: troponin I, fast skeletal muscle-like [Maylandia zebra] | 0.13 | 0.07 | 0.05 |
| PREDICTED: myozenin-1-like [Oreochromis niloticus] | 0.95 | 1.08 | 0.92 |
| PREDICTED: ankyrin repeat domain-containing protein 1 [Pundamilia nyererei] | 5.85 | 2.22 | 2.84 |
| PREDICTED: myosin heavy chain, fast skeletal muscle-like [Pundamilia nyererei] | 1.77 | 1.58 | 1.40 |
| PREDICTED: keratin, type I cytoskeletal 18-like [Xiphophorus maculatus] | 1.83 | 2.84 | 1.06 |
| PREDICTED: aldose reductase-like [Haplochromis burtoni] | 0.58 | 1.93 | 1.03 |
| PREDICTED: creatine kinase M-type [Haplochromis burtoni] | 1.09 | 1.13 | 1.22 |
| Type 1 collagen alpha 2 [Paralichthys olivaceus] | 0.86 | 0.21 | 0.33 |
| SET and MYND domain containing protein 1a [Siniperca chuatsi] | 1.46 | 2.92 | 1.95 |
| PREDICTED: collagen alpha-2(I) chain-like isoform X1 [Neolamprologus brichardi] | 0.98 | 0.94 | 0.46 |
| PREDICTED: myc box-dependent-interacting protein 1-like isoform X1 [Neolamprologus brichardi] | 1.12 | 1.75 | 2.18 |
| PREDICTED: ATP synthase subunit beta, mitochondrial-like [Neolamprologus brichardi] | 0.74 | 0.73 | 1.52 |
| PREDICTED: putative peptidyl-tRNA hydrolase PTRHD1-like [Neolamprologus brichardi] | 1.06 | 1.37 | 0.84 |
| Cytochrome c oxidase subunit VIa precursor [Thunnus obesus] | 0.59 | 0.26 | 0.54 |
| PREDICTED: 60S ribosomal protein L3-like [Poecilia formosa] | 0.70 | 1.98 | 2.78 |
| Alpha hemoglobin A [Seriola quinqueradiata] | 1.60 | 1.52 | 0.17 |
| PREDICTED: myosin-binding protein H-like isoform X1 [Stegastes partitus] | 0.31 | 0.92 | 0.72 |
| PREDICTED: dnaJ homolog subfamily C member 8 [Stegastes partitus] | 1.79 | 0.84 | 0.48 |
| PREDICTED: proteasome subunit alpha type-1 [Stegastes partitus] | 0.19 | 0.38 | 0.63 |
| PREDICTED: 40S ribosomal protein S25 [Stegastes partitus] | 0.66 | 1.04 | 0.78 |
| PREDICTED: malate dehydrogenase, mitochondrial [Stegastes partitus] | 0.61 | 0.32 | 0.34 |
| PREDICTED: adenylate kinase isoenzyme 1 [Stegastes partitus] | 0.26 | 0.07 | 0.16 |
| PREDICTED: transcription factor BTF3 homolog 4 isoform X2 [Stegastes partitus] | 0.75 | 0.92 | 1.17 |
| PREDICTED: unconventional myosin-XVIIIb isoform X1 [Stegastes partitus] | 0.38 | 0.67 | 0.20 |
| PREDICTED: nascent polypeptide-associated complex subunit alpha isoform X1 [Stegastes partitus] | 0.79 | 1.54 | 1.60 |
| PREDICTED: poly(rC)-binding protein 2-like isoform X2 [Stegastes partitus] | 1.17 | 1.28 | 0.92 |
| PREDICTED: 26S proteasome non-ATPase regulatory subunit 14 [Stegastes partitus] | 1.47 | 4.82 | 3.08 |
| PREDICTED: voltage-dependent anion-selective channel protein 3 isoform X2 [Stegastes partitus] | 1.44 | 0.41 | 0.84 |
| PREDICTED: reticulon-3-like isoform X1 [Stegastes partitus] | 0.60 | 0.43 | 0.51 |
| PREDICTED: myomesin-1-like isoform X3 [Stegastes partitus] | 1.78 | 3.11 | 1.48 |
| PREDICTED: dehydrogenase/reductase SDR family member 7C-A-like [Stegastes partitus] | 2.23 | 0.47 | 0.78 |
| PREDICTED: ATP-binding cassette sub-family A member 1-like isoform X2 [Stegastes partitus] | 1.86 | 2.57 | 0.46 |
| PREDICTED: transcription factor E2-alpha-like isoform X1 [Stegastes partitus] | 0.81 | 1.26 | 1.19 |
| PREDICTED: creatine kinase M-type [Stegastes partitus] | 1.10 | 0.93 | 1.03 |
| PREDICTED: titin-like isoform X11 [Stegastes partitus] | 4.95 | 1.22 | 1.72 |
| PREDICTED: titin-like isoform X20 [Stegastes partitus] | 1.69 | 2.06 | 1.51 |
| PREDICTED: peptidyl-prolyl cis-trans isomerase A-like [Stegastes partitus] | 1.03 | 1.16 | 0.69 |
| PREDICTED: glutamate dehydrogenase, mitochondrial [Stegastes partitus] | 0.65 | 1.00 | 0.99 |
| PREDICTED: retinal dehydrogenase 2 [Stegastes partitus] | 1.71 | 2.74 | 8.28 |
| PREDICTED: creatine kinase S-type, mitochondrial [Stegastes partitus] | 0.93 | 0.57 | 0.56 |
| PREDICTED: CDK5 regulatory subunit-associated protein 2 isoform X3 [Stegastes partitus] | 2.13 | 2.19 | 1.18 |
| PREDICTED: sarcoplasmic/endoplasmic reticulum calcium ATPase 1 isoform X1 [Stegastes partitus] | 0.94 | 0.91 | 0.89 |
| PREDICTED: calpain-3-like isoform X1 [Stegastes partitus] | 0.57 | 0.62 | 1.07 |
| PREDICTED: creatine kinase M-type-like [Stegastes partitus] | 0.88 | 1.20 | 1.59 |
| PREDICTED: 40S ribosomal protein S11 [Stegastes partitus] | 0.85 | 0.66 | 0.77 |
| PREDICTED: mucin-5AC-like [Cynoglossus semilaevis] | 1.29 | 1.27 | 0.77 |
| PREDICTED: NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 8 [Poecilia reticulata] | 1.23 | 1.33 | 1.23 |
| PREDICTED: phosphate carrier protein, mitochondrial isoform X1 [Poecilia reticulata] | 0.74 | 0.72 | 0.51 |
| PREDICTED: lactoylglutathione lyase-like [Larimichthys crocea] | 0.99 | 0.60 | 0.38 |
| PREDICTED: myelin basic protein-like isoform X2 [Larimichthys crocea] | 1.47 | 0.95 | 1.04 |
| PREDICTED: 14 kDa phosphohistidine phosphatase-like [Larimichthys crocea] | 0.40 | 0.63 | 0.24 |
| PREDICTED: LOW QUALITY PROTEIN: myosin heavy chain, fast skeletal muscle-like [Larimichthys crocea] | 1.13 | 1.13 | 0.82 |
| PREDICTED: AMP deaminase 1 isoform X1 [Larimichthys crocea] | 0.92 | 0.66 | 0.41 |
| PREDICTED: myosin-binding protein C, fast-type-like isoform X5 [Larimichthys crocea] | 0.40 | 0.95 | 0.40 |
| PREDICTED: glucose-6-phosphate isomerase [Larimichthys crocea] | 1.24 | 0.66 | 0.81 |
| PREDICTED: SH3 domain-binding glutamic acid-rich protein isoform X2 [Larimichthys crocea] | 0.60 | 1.60 | 1.79 |
| PREDICTED: peroxisome proliferator-activated receptor gamma coactivator-related protein 1-like [Larimichthys crocea] | 0.93 | 1.01 | 1.38 |
| PREDICTED: 60S ribosomal protein L4-B isoform X1 [Larimichthys crocea] | 0.39 | 0.89 | 1.19 |
| PREDICTED: NADH dehydrogenase [ubiquinone] iron-sulfur protein 6, mitochondrial [Larimichthys crocea] | 0.48 | 0.31 | 0.56 |
| PREDICTED: alpha-2-macroglobulin-like isoform X1 [Larimichthys crocea] | 2.96 | 2.61 | 1.81 |
| PREDICTED: protocadherin-18-like isoform X2 [Larimichthys crocea] | 0.48 | 0.17 | 0.44 |
| PREDICTED: guanidinoacetate N-methyltransferase [Larimichthys crocea] | 0.60 | 1.15 | 1.28 |
| PREDICTED: calpastatin isoform X10 [Larimichthys crocea] | 1.33 | 1.97 | 1.14 |
| PREDICTED: fructose-bisphosphate aldolase A-like [Larimichthys crocea] | 1.23 | 1.01 | 1.19 |
| PREDICTED: proteoglycan 4-like [Larimichthys crocea] | 1.32 | 0.80 | 0.57 |
| PREDICTED: plasminogen activator inhibitor 1 RNA-binding protein-like isoform X1 [Larimichthys crocea] | 0.82 | 0.71 | 1.83 |
| PREDICTED: alpha-actinin-3 [Larimichthys crocea] | 0.79 | 0.93 | 0.28 |
| PREDICTED: protein S100-A14 [Larimichthys crocea] | 1.74 | 0.59 | 0.49 |
| PREDICTED: methionine aminopeptidase 2 [Larimichthys crocea] | 0.81 | 1.03 | 0.82 |
| PREDICTED: muscle-related coiled-coil protein-like [Larimichthys crocea] | 0.96 | 1.10 | 0.71 |
| PREDICTED: asparagine--tRNA ligase, cytoplasmic [Larimichthys crocea] | 0.43 | 0.28 | 0.35 |
| PREDICTED: patellin-2-like [Notothenia coriiceps] | 2.04 | 5.15 | 4.29 |
| PREDICTED: collagen alpha-1(I) chain-like [Notothenia coriiceps] | 1.18 | 0.05 | 0.03 |
| Pyruvate kinase [Trachinotus ovatus] | 9.68 | 1.71 | 1.41 |
| PREDICTED: 60S ribosomal protein L9 isoform X1 [Oryzias latipes] | 0.36 | 1.58 | 2.13 |
| PREDICTED: rho guanine nucleotide exchange factor 9 isoform X3 [Oryzias latipes] | 0.46 | 1.71 | 2.13 |
| Myosin light chain 1 [Trachurus trachurus] | 1.93 | 0.49 | 0.32 |
| Myosin light chain 3 [Trachurus trachurus] | 1.48 | 0.79 | 0.74 |
| Myosin light chain 2 [Trachurus trachurus] | 1.20 | 0.70 | 0.51 |
| White muscle parvalbumin [Trachurus japonicus] | 1.41 | 0.63 | 0.52 |
| Peroxiredoxin 5 [Oplegnathus fasciatus] | 0.47 | 0.46 | 0.30 |
| Nebulin [Larimichthys crocea] | 1.14 | 1.05 | 1.04 |
| Heat shock cognate protein [Larimichthys crocea] | 0.75 | 1.25 | 0.93 |
| Ral GTPase-activating protein subunit beta [Larimichthys crocea] | 1.01 | 0.64 | 0.98 |
| Myosin heavy chain, fast skeletal muscle [Larimichthys crocea] | 2.93 | 3.09 | 2.74 |
| PREDICTED: ATP-dependent 6-phosphofructokinase, muscle type-like [Fundulus heteroclitus] | 0.63 | 0.25 | 0.18 |
| PREDICTED: hematological and neurological expressed 1-like protein [Fundulus heteroclitus] | 2.07 | 1.28 | 0.74 |
| PREDICTED: LIM domain-binding protein 3-like isoform X3 [Oreochromis niloticus] | 0.61 | 0.45 | 0.39 |
| PREDICTED: PDZ and LIM domain protein 5 isoform X4 [Oreochromis niloticus] | 4.74 | 0.69 | 0.51 |
| PREDICTED: phosphorylase b kinase gamma catalytic chain, skeletal muscle/heart isoform [Oreochromis niloticus] | 0.54 | 0.68 | 1.99 |
| PREDICTED: neurofilament heavy polypeptide-like isoform X5 [Pundamilia nyererei] | 1.63 | 0.79 | 0.53 |
| PREDICTED: dysferlin [Pundamilia nyererei] | 1.54 | 0.72 | 0.43 |
| PREDICTED: fibrous sheath CABYR-binding protein-like [Haplochromis burtoni] | 3.52 | 4.93 | 1.77 |
| PREDICTED: glycogen phosphorylase, muscle form isoform X1 [Xiphophorus maculatus] | 0.83 | 1.06 | 1.14 |
| Aldolase A, partial [Poeciliopsis prolifica] | 0.90 | 0.91 | 0.84 |
| PREDICTED: 40S ribosomal protein S2 [Xiphophorus maculatus] | 0.59 | 1.18 | 2.10 |
| PREDICTED: triosephosphate isomerase [Poecilia latipinna] | 0.76 | 0.74 | 0.76 |
| PREDICTED: keratin, type I cytoskeletal 18 [Poecilia latipinna] | 2.05 | 4.94 | 1.06 |
| PREDICTED: guanine nucleotide-binding protein subunit beta-2-like 1 [Poecilia mexicana] | 0.52 | 0.84 | 1.43 |
| PREDICTED: phosphoglycerate mutase 2 [Cyprinodon variegatus] | 0.68 | 1.21 | 1.80 |
| PREDICTED: cold-inducible RNA-binding protein isoform X4 [Cyprinodon variegatus] | 1.57 | 2.13 | 1.68 |
| PREDICTED: glycogen [starch] synthase, muscle-like [Cyprinodon variegatus] | 0.54 | 1.21 | 2.55 |
| PREDICTED: ATP synthase subunit delta, mitochondrial [Cyprinodon variegatus] | 0.95 | 1.79 | 1.88 |
| PREDICTED: beta-enolase [Cyprinodon variegatus] | 1.15 | 1.37 | 1.51 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kominami, Y.; Hayashi, T.; Tokihiro, T.; Ushio, H. A Novel Analysis of the Peptide Terminome Characterizes Dynamics of Proteolytic Regulation in Vertebrate Skeletal Muscle Under Severe Stress. Proteomes 2019, 7, 6. https://doi.org/10.3390/proteomes7010006
Kominami Y, Hayashi T, Tokihiro T, Ushio H. A Novel Analysis of the Peptide Terminome Characterizes Dynamics of Proteolytic Regulation in Vertebrate Skeletal Muscle Under Severe Stress. Proteomes. 2019; 7(1):6. https://doi.org/10.3390/proteomes7010006
Chicago/Turabian StyleKominami, Yuri, Tatsuya Hayashi, Tetsuji Tokihiro, and Hideki Ushio. 2019. "A Novel Analysis of the Peptide Terminome Characterizes Dynamics of Proteolytic Regulation in Vertebrate Skeletal Muscle Under Severe Stress" Proteomes 7, no. 1: 6. https://doi.org/10.3390/proteomes7010006
APA StyleKominami, Y., Hayashi, T., Tokihiro, T., & Ushio, H. (2019). A Novel Analysis of the Peptide Terminome Characterizes Dynamics of Proteolytic Regulation in Vertebrate Skeletal Muscle Under Severe Stress. Proteomes, 7(1), 6. https://doi.org/10.3390/proteomes7010006