
Comparative Proteomics of Root Apex and Root Elongation Zones Provides Insights into Molecular Mechanisms for Drought Stress and Recovery Adjustment in Switchgrass

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Construction of a “Sandwich” Drought Treatment System
2.2. Preparation of Plant Materials
2.3. Root Tissue Harvest and Processing
2.4. Physiological Measurement
2.5. Preparation of Protein Samples
2.6. Tandem Mass Tags (TMT) Labeling and Mass Spectrometry Analysis
2.7. Protein Identification, Quantification, and Statistics Analysis
3. Results
3.1. Physiological Changes under Different Levels of Drought Condition
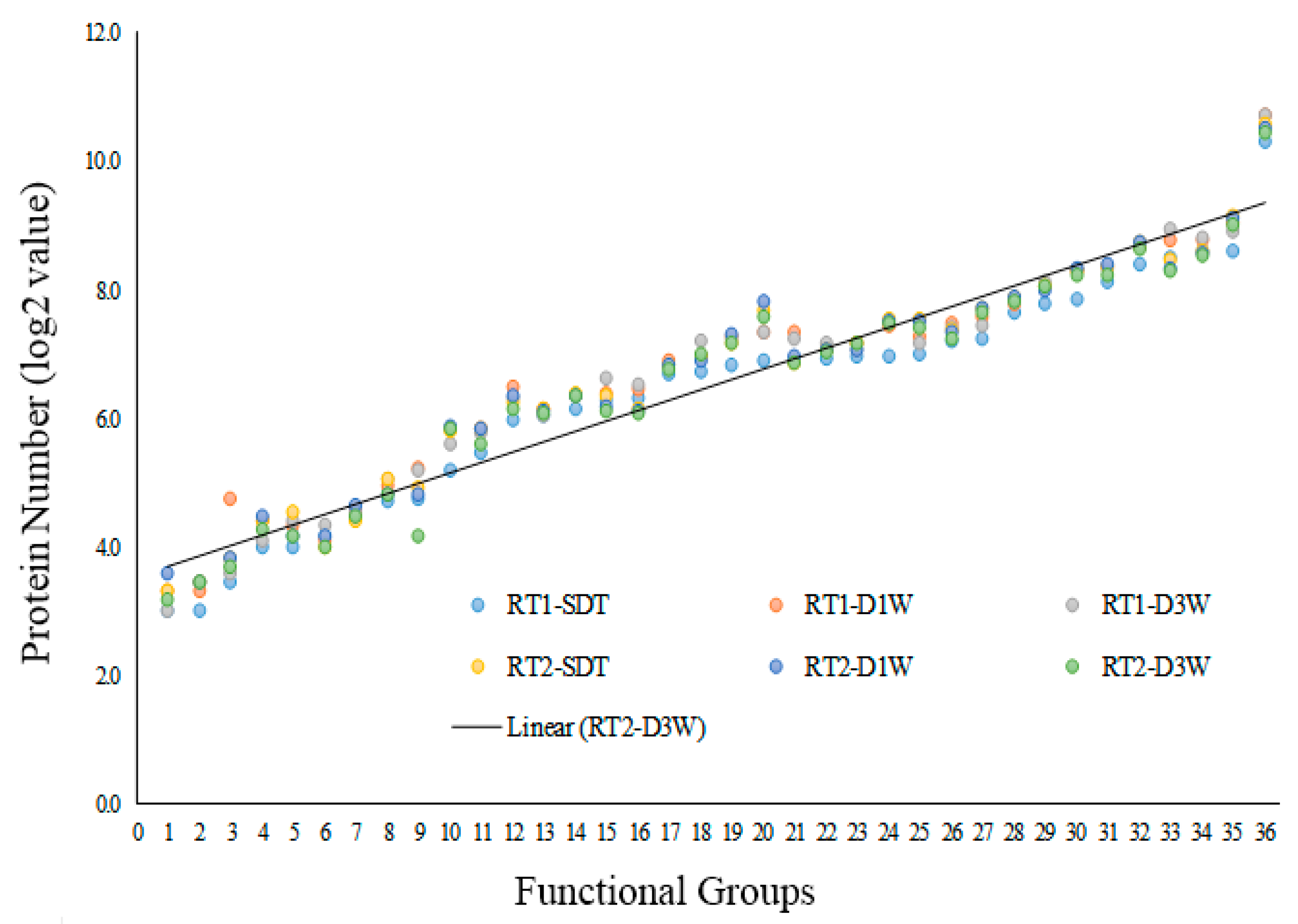
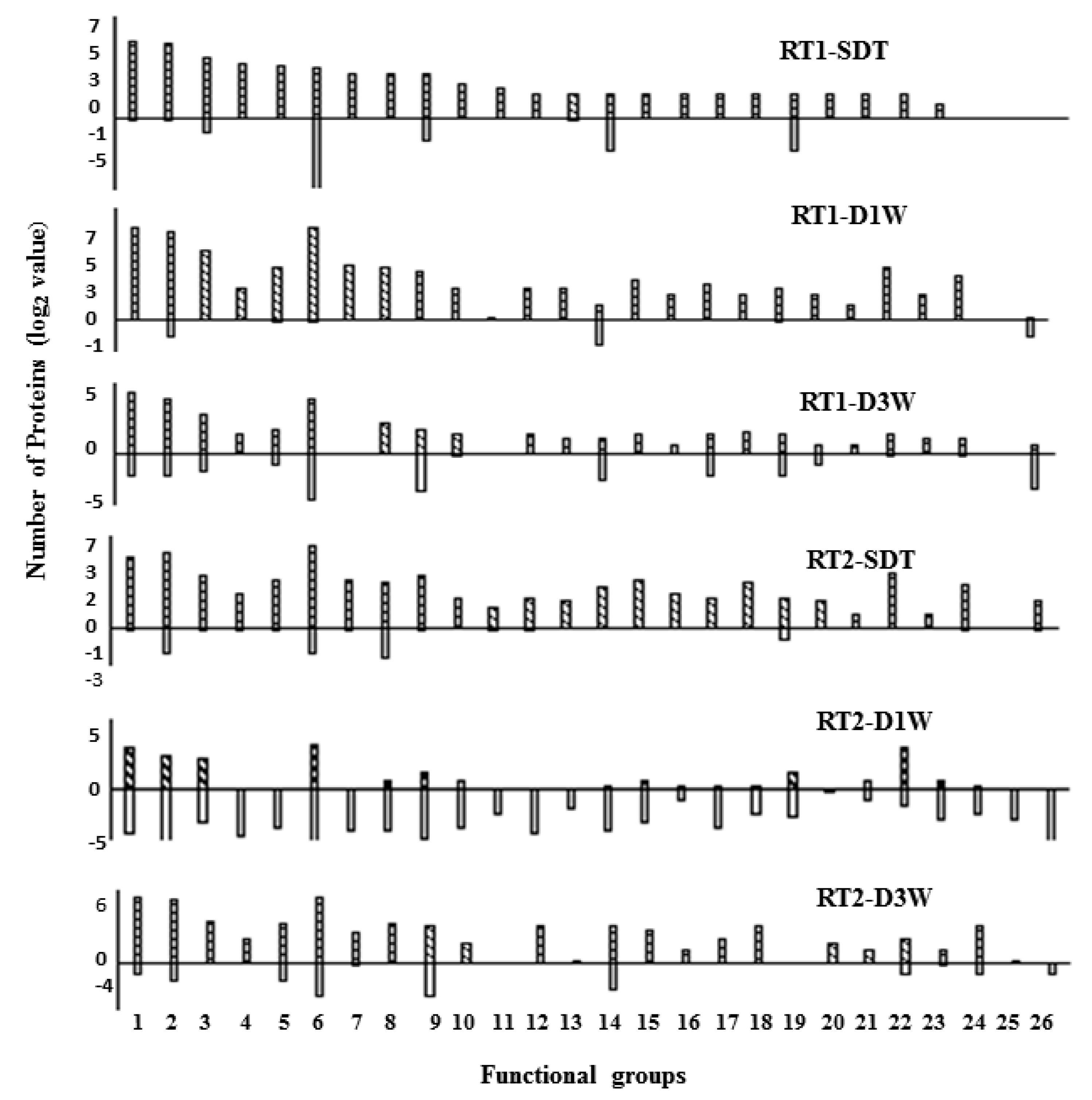
3.2. The Drought-Induced Proteins and Functional Classifications
3.3. Drought-Induced Differentially Accumulated Proteins Involved in Phytohormone Perception and Signaling Network
3.4. The Drought-Induced Differentially Accumulated Proteins (DAPs) Involved in Cellular Activities and Organogenesis
3.5. The Drought-Induced Differentially Accumulated Proteins (DAPs) Involved in Tolerance/Resistance to Physiological Stresses in Roots
3.6. The Drought-Induced Differentially Accumulated Proteins (DAPs) Involved in Gene Transcription and Protein Translation Activities
3.7. The Drought-Induced Differentially Accumulated Proteins (DAPs) Involved in Metabolic Pathways
3.8. Access to Mass Spectrometry Proteomics Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parrisha, D.J.; Fike, J.H. The biology and agronomy of switchgrass for biofuels. CRC Crit. Rev. Plant Sci. 2005, 24, 423–459. [Google Scholar] [CrossRef]
- Wright, L.L. Historical Perspective on How and Why Switchgrass Was Selected as a ‘Model’ High-Potential Energy Crop. Available online: https://info.ornl.gov/sites/publications/files/Pub7047.pdf (accessed on 16 February 2020).
- Keyser, P.; Harper, C.; Bates, G.; Waller, J.; Doxon, E. Native Warm-Season Grasses for Mid-South Forage Production. UT Ext. SP731—A. Available online: https://extension.tennessee.edu/publications/Documents/SP731-A.pdf (accessed on 16 February 2020).
- Sanderson, M.A.; Reed, R.L. Switchgrass growth and development: Water, nitrogen, and plant density effects. J. Range Manag. 2000, 53, 221–227. [Google Scholar] [CrossRef]
- Vogel, K.P.; Sarath, G.; Saathoff, A.J.; Mitchell, R.B. Switchgrass in Energy Crops. In RSC Energy and Environment Series No. 3, Energy Crops; Halford, N., Karp, A., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2011; pp. 341–380. [Google Scholar]
- Jones, M.; Turner, N.; Osmond, C. Mechanisms of drought resistance. In The Physiology and Biochemistry of Drought Resistance in Plants; Paleg, L.G., Aspinall, D., Eds.; Academic Press: Sydney, Australia, 1981; pp. 15–35. [Google Scholar]
- Jones, H. What is water use efficiency. In Water Use Efficiency in Plant Biology; Bacon, M., Ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2004; pp. 27–41. [Google Scholar]
- Turner, N.C.; Kramer, P.J. Adaptation of Plants to Water and High Temperature Stress; John Wiley and Sons: Hoboken, NJ, USA, 1980; pp. 97–131. [Google Scholar]
- Sharp, R.E.; Poroyko, V.; Hejlek, L.G.; Spollen, W.G.; Springer, G.K.; Bohnert, H.J.; Nguyen, H.T. Root growth maintenance during water deficits: Physiology to functional genomics. J. Exp. Bot. 2004, 55, 2343–2351. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, N.J.; Urwin, P.E. The interaction of plant biotic and abiotic stresses: From genes to the field. J. Exp. Bot. 2012, 63, 3523–3544. [Google Scholar] [CrossRef]
- Ghosh, D.; Xu, J. Abiotic stress responses in plant roots: A proteomics perspective. Front. Plant. Sci. 2014, 5, 1–13. [Google Scholar] [CrossRef]
- Gowda, V.R.P.; Henry, A.; Yamauchi, A.; Shashidhar, H.E.; Serraj, R. Root biology and genetic improvement for drought avoidance in rice. Field Crop. Res. 2011, 122, 1–13. [Google Scholar] [CrossRef]
- Fraser, T.E.; Silk, W.K.; Rost, T.L. Effects of low water potential on cortical cell length in growing regions of maize roots. Plant Physiol. 1990, 93, 648–651. [Google Scholar] [CrossRef]
- Tomos, D.; Pritchard, J. Biophysical and biochemical control of cell expansion in roots and leaves. J. Exp. Bot. 1994, 45, 1721–1731. [Google Scholar] [CrossRef]
- Xiong, L.; Wang, R.G.; Mao, G.; Koczan, J.M. Identification of drought tolerance determinants by genetic analysis of root response to drought stress and abscisic acid. Plant Physiol. 2006, 142, 1065–1074. [Google Scholar] [CrossRef]
- O’Toole, J.C.; Bland, W.L. Genotypic variation in crop plant root systems. Adv. Agron. 1987, 41, 91–145. [Google Scholar] [CrossRef]
- Sponchiadoa, B.N.; Whitea, J.W.; Castilloa, J.A.; Jonesa, P.G. Root growth of four common bean cultivars in relation to drought tolerance in environments with contrasting soil types. Exp. Agric. 1989, 25, 249–257. [Google Scholar] [CrossRef]
- Akinci, Ş.; Lösel, D.M. Plant water-stress response mechanisms. In Water Stress; Ismail, M.D., Rahman, M., Eds.; InTechOpen: London, UK, 2012; pp. 149–172. [Google Scholar]
- Izanloo, A.; Condon, A.G.; Langridge, P.; Tester, M.; Schnurbusch, T. Different mechanisms of adaptation to cyclic water stress in two South Australian bread wheat cultivars. J. Exp. Bot. 2008, 59, 3327–3346. [Google Scholar] [CrossRef]
- Bogeat-Triboulot, M.B.; Brosché, M.; Renaut, J.; Jouve, L.; Thiec, D.L.; Fayyaz, P.; Vinocur, B.; Witters, E.; Laukens, K.; Teichmann, T.; et al. Gradual soil water depletion results in reversible changes of gene expression, protein profiles, ecophysiology, and growth performance in Populus euphratica, a poplar growing in arid regions. Plant Physiol. 2007, 143, 876–892. [Google Scholar] [CrossRef]
- Bonhomme, L.; Monclus, R.; Vincent, D.; Carpin, S.; Lomenech, A.M.; Plomion, C.; Brignolas, F.; Morabito, D. Leaf proteome analysis of eight Populus x euramericana genotypes: Genetic variation in drought response and in water-use efficiency involves photosynthesis-related proteins. Proteomics 2009, 9, 4121–4142. [Google Scholar] [CrossRef]
- Moumeni, A.; Satoh, K.; Kondoh, H.; Asano, T.; Hosaka, A.; Venuprasad, R.; Serraj, R.; Kumar, A.; Leung, H.; Kikuchi, S. Comparative analysis of root transcriptome profiles of two pairs of drought-tolerant and susceptible rice near-isogenic lines under different drought stress. BMC Plant Biol. 2011, 11, 174. [Google Scholar] [CrossRef]
- Ye, Z.; Sangireddy, S.; Okekeogbu, I.; Zhou, S.; Yu, C.L.; Hui, D.; Howe, K.J.; Fish, T.; Thannhauser, T.W. Drought-induced leaf proteome changes in switchgrass seedlings. Int. J. Mol. Sci. 2016, 17, 1251. [Google Scholar] [CrossRef]
- Zhang, M.H.; Li, G.W.; Huang, W.; Bi, T.; Chen, G.Y.; Tang, Z.C.; Su, W.A.; Sun, W.N. Proteomic study of Carissa spinarum in response to combined heat and drought stress. Proteomics 2010, 10, 3117–3129. [Google Scholar] [CrossRef]
- Ghaffari, M.; Toorchi, M.; Valizadeh, M.; Komatsu, S. Differential response of root proteome to drought stress in drought sensitive and tolerant sunflower inbred lines. Funct. Plant Biol. 2013, 40, 609–617. [Google Scholar] [CrossRef]
- Paul, S.; Gayen, D.; Datta, S.K.; Datta, K. Dissecting root proteome of transgenic rice cultivars unravels metabolic alterations and accumulation of novel stress responsive proteins under drought stress. Plant Sci. 2015, 234, 133–143. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Valliyodan, B.; Zhang, J.; Lenoble, M.E.; Yu, O.; Rogers, E.E.; Nguyen, H.T.; Sharp, R.E. Regulation of growth response to water stress in the soybean primary root. I. Proteomic analysis reveals region-specific regulation of phenylpropanoid metabolism and control of free iron in the elongation zone. Plant Cell Environ. 2010, 33, 223–243. [Google Scholar] [CrossRef]
- Zhou, S.P.; Palmer, M.; Zhou, J.; Bhatti, S.; Howe, K.J.; Fish, T.; Thannhauser, T.W. Differential root proteome expression in tomato genotypes with contrasting drought tolerance exposed to dehydration. J. Am. Soc. Hortic. Sci. 2013, 138, 131–141. [Google Scholar] [CrossRef]
- Gilar, M.; Olivova, P.; Daly, A.E.; Gebler, J.C. Orthogonality of separation in two-dimensional liquid chromatography. Anal. Chem. 2005, 77, 6426–6434. [Google Scholar] [CrossRef] [PubMed]
- Gilar, M.; Olivova, P.; Daly, A.E.; Gebler, J.C. Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J. Sep. Sci. 2005, 28, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, F.; Gritsenko, M.A.; Clauss, T.; Liu, T.; Shen, Y.; Monroe, M.E.; Lopez-Ferrer, D.; Reno, T.; Moore, R.J.; et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 2011, 11, 2019–2026. [Google Scholar] [CrossRef] [PubMed]
- Rangu, M.; Ye, Z.; Bhatti, S.; Zhou, S.; Yang, Y.; Fish, T.; Thannhauser, T.W. Association of proteomics changes with Al-sensitive root zones in switchgrass. Proteomes 2018, 22, 6. [Google Scholar] [CrossRef]
- Zhou, S.P.; Sangireddy, S.; Ye, Z.; Li, H.; Bhatti, S.; Hui, D.; McDonald, D.; Yang, Y.; Giri, S.; Howe, K.; et al. Proteome modification in tomato plants upon long-term aluminum treatment. J. Proteome Res. 2016, 15, 1670–1684. [Google Scholar] [CrossRef]
- Thannhauser, T.; Shen, M.; Sherwood, R.; Howe, K.; Fish, T.; Yang, Y.; Chen, W.; Zhang, S. A workflow for large-scale empirical identification of cell wall N-linked glycoproteins of tomato (Solanum lycopersicum). Electrophoresis 2013, 34, 2417–2431. [Google Scholar] [CrossRef]
- Yang, Q.S.; Wu, J.H.; Li, C.Y.; Wei, Y.R.; Sheng, O. Quantitative proteomic analysis reveals that antioxidation mechanisms contribute to cold tolerance in plantain (Musa paradisiaca L.; ABB Group) seedlings. Mol. Cell Proteom. 2012, 11, 1853–1869. [Google Scholar] [CrossRef]
- Yang, Y.; Qiang, X.; Owsiany, K.; Zhang, S.; Thannhauser, T.W.; Li, L. Evaluation of different multidimensional LC-MS/MS pipelines for iTRAQ-based proteomic analysis of potato tubers in response to cold storage. J. Proteome Res. 2011, 10, 4647–4660. [Google Scholar] [CrossRef]
- Okekeogbu, I.; Ye, Z.J.; Sangireddy, S.R.; Li, H.; Bhatti, S.; Hui, D.F.; Zhou, S.P.; Howe, K.J.; Fish, T.; Yang, Y.; et al. Effect of aluminum treatment on proteomes of radicles of seeds derived from Al-treated tomato plants. Proteomes 2014, 2, 169–190. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Usadel, B.; Poree, F.; Nagel, A.; Lohse, M.; Czedik-Eysenberg, A.; Stitt, M. A guide to using MapMan to visualize and compare Omics data in plants: A case study in the crop species, Maize. Plant. Cell Environ. 2009, 32, 1211–1229. [Google Scholar] [CrossRef] [PubMed]
- Lyon, D.; Castillejo, M.A.; Mehmeti-Tershani, V.; Staudinger, C.; Kleemaier, C.; Wienkoop, S. Drought and recovery: InDAPendently regulated processes highlighting the importance of protein turnover dynamics and translational regulation in Medicago truncatula. Mol. Cell Proteom. 2016, 15, 1921–1937. [Google Scholar] [CrossRef] [PubMed]
- Royer, M.; Cohen, D.; Aubry, N.; Vendramin, V.; Scalabrin, S.; Cattonaro, F.; Bogeat- Triboulot, M.B.; Hummel, I. The build-up of osmotic stress responses within the growing root apex using kinematics and RNA-sequencing. J. Exp. Bot. 2016, 67, 5961–5973. [Google Scholar] [CrossRef] [PubMed]
- Chakrabortee, S.; Boschetti, C.; Walton, L.J.; Sarkar, S.; Rubinsztein, D.C.; Tunnacliffe, A. Hydrophilic protein associated with desiccation tolerance exhibits broad protein stabilization function. Proc. Natl. Acad. Sci. USA 2007, 104, 18073–18078. [Google Scholar] [CrossRef]
- Li, N.; Zhang, S.; Liang, Y.; Qi, Y.; Chen, J.; Zhu, W.; Zhang, L. Label-free quantitative proteomic analysis of drought stress-responsive late embryogenesis abundant proteins in the seedling leaves of two wheat (Triticum aestivum L.) genotypes. J. Proteom. 2018, 172, 122–142. [Google Scholar] [CrossRef]
- Carvalho, M.H.C. Drought stress and reactive oxygen species Production, scavenging and signaling. Plant Signal. Behav. 2008, 3, 156–165. [Google Scholar] [CrossRef]
- Kar, R.K. Plant responses to water stress: Role of reactive oxygen species. Plant Signal. Behav. 2011, 6, 1741–1745. [Google Scholar] [CrossRef]
- Yu, L.; Chen, X.; Wang, Z.; Wang, S.; Wang, Y.; Zhu, Q.; Li, S.; Xiang, C. Arabidopsis enhanced drought tolerance1/HOMEODOMAIN GLABROUS11 confers drought tolerance in transgenic rice without yield penalty. Plant Physiol. 2013, 162, 1378–1391. [Google Scholar] [CrossRef]
- Han, Y.; Chen, Y.; Yin, S.; Zhang, M.; Wang, W. Over-expression of TaEXPB23, a wheat expansin gene, improves oxidative stress tolerance in transgenic tobacco plants. J. Plant. Physiol. 2015, 173, 62–71. [Google Scholar] [CrossRef]
- Tenhaken, R. Cell wall remodeling under abiotic stress. Front. Plant Sci. 2015, 5, 1–9. [Google Scholar] [CrossRef]
- Schuetz, M.; Benske, A.; Smith, R.A.; Watanabe, Y.; Tobimatsu, Y.; Ralph, J.; Demura, T.; Ellis, B.; Samuels, A.L. Laccases direct lignification in the discrete secondary cell wall domains of protoxylem. Plant Physiol. 2014, 166, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.L.; Cassin, A.M.; Lonsdale, A.; Bacic, A.; Doblin, M.S.; Schultz, C.J. Pipeline to identify hydroxyproline-rich glycoproteins. Plant Physiol. 2017, 174, 886–903. [Google Scholar] [CrossRef] [PubMed]
- Tseng, I.C.; Hong, C.Y.; Yu, S.M.; Ho, T.H.D. Abscisic acid- and stress-induced highly proline-rich glycoproteins regulate root growth in rice. Plant Physiol. 2013, 163, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Qian, S.B. Translational reprogramming in cellular stress response. Wiley Interdiscip. Rev. RNA 2014, 5, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Evrard, J.L.; Pieuchot, L.; Vos, J.W.; Vernos, I.; Schmit, A.C. Plant TPX2 and related proteins. Plant Signal. Behav. 2009, 4, 69–72. [Google Scholar] [CrossRef]
- Vos, J.W.; Pieuchot, L.; Evrard, J.-L.; Janski, N.; Bergdoll, M. The plant TPX2 protein regulates prospindle assembly before nuclear envelope breakdown. Plant Cell 2008, 20, 2783–2797. [Google Scholar] [CrossRef]
- Bengough, A.G.; McKenzie, B.M.; Hallett, P.D.; Valentine, T.A. Root elongation, water stress, and mechanical impedance: A review of limiting stresses and beneficial root tip traits. J. Exp. Bot. 2011, 62, 59–68. [Google Scholar] [CrossRef]
- Triplett, J.K.; Wang, Y.; Zhong, J.; Kellogg, E.A. Five nuclear loci resolve the polyploid history of switchgrass (Panicum virgatum L.) and relatives. PLoS ONE 2012, 7, e38702. [Google Scholar] [CrossRef]
- Grabowski, P.P.; Morris, G.P.; Casler, M.D.; Borevitz, J.O. Population genomic variation reveals roles of history, adaptation and ploidy in switchgrass. Mol. Ecol. 2014, 23, 4059–4073. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Levels of Drought Condition | Treatment | Leaf Photosynthetic Rate (μmol CO2 /m2/s) | Stomatal Conductance (mol H2O /m2/s) | Transpiration (mmol H2O/m2/s) | Water Use Efficiency (μmol CO2 /mmol H2O) |
|---|---|---|---|---|---|
| Level 1 (Before drought treatment) | Control | 23.31 ± 4.59 A | 0.14 ± 0.03 A | 4.44 ± 1.13 A | 5.40 ± 0.99 A |
| Drought | 22.54 ± 3.97 A | 0.14 ± 0.03 A | 4.25 ± 1.03 A | 5.46 ± 0.99 A | |
| Level 2 (28 days drought) | Control | 22.02 ± 4.06 A | 0.15 ± 0.03 A | 5.38 ± 1.23 A | 4.16 ± 0.56 A |
| Drought | 16.62 ± 5.82 B | 0.12 ± 0.05 B | 4.51 ± 1.54 B | 3.77 ± 0.75 B | |
| Level 3 (28 days drought + 1 day re-watering) | Control | 18.98 ± 4.93 A | 0.12 ± 0.03 A | 3.64 ± 0.87 A | 5.21 ± 0.64 A |
| Drought | 15.10 ± 4.99 B | 0.10 ± 0.03 B | 2.67 ± 0.93 B | 5.83 ± 1.33 B | |
| Level 4 (28 days drought + 3 days re-watering) | Control | 12.48 ± 2.91 A | 0.18 ± 0.06 A | 2.21 ± 0.64 A | 5.80 ± 0.82 A |
| Drought | 11.54 ± 4.2 A | 0.15 ± 0.05 B | 2.0 ± 0.67 A | 5.80 ± 0.86 A |
| Biological Process | Protein Accessions b | Description | Fold Change (Drought Treated/Control) c | ||
|---|---|---|---|---|---|
| SDT d | D1W e | D3W f | |||
| Phytohormones ABA | Pavir.Ab00822.1 Pavir.Da00781.1 | PYR1-like 2 (ABA RECEPTOR1) | 0.27–0.40 | ||
| Pavir.Ab01425.1 | Malectin/receptor-like protein kinase family protein (negative regulation of abscisic acid-activated signaling pathway) | 0.33 | |||
| Pavir.Ba03030.1 | RNA binding;abscisic acid binding (ABA receptor) | 2.69 | |||
| Pavir.Db00930.1 | ABA- and stress-inducible osmotic stress tolerance gene (HVA22 homologue) | 0.26 | |||
| Pavir.Aa00702.1 | Cullin 3 | 2.61 | |||
| Pavir.Gb00126.1 | IQ-domain 32 | 0.39 | |||
| Pavir.Ab01039.1 | REGULATORY COMPONENTS OF ABA RECEPTOR3 (RCAR3, PYL8) | 3.58 | |||
| Pavir.J34085.1 | ABA-RESPONSIVE ELEMENT BINDING PROTEIN 3, AREB3 | 2.51 | |||
| Pavir.Ia02723.1 | Nodulin-related protein 1 | 2.17 | |||
| Pavir.Ba01419.1; Pavir.Gb00423.1 | Protein phosphatase 2C family protein | 3.25–3.98 | |||
| Auxin | Pavir.Fa00409.1 | Auxin-responsive family protein | 0.33 | ||
| Pavir.Cb00986.1 | IBA response protein | 3.54 | |||
| Pavir.Fb00898.1 | Auxin-responsive family protein | 6.58 | |||
| Pavir.Ia02169.1 | Flavin-binding monooxygenase family protein (Auxin biosynthesis) | 0.40 | |||
| Ethylene | Pavir.Fb01856.2; Pavir.Ba01308.1; | S-adenosyl-L-methionine-dependent methyltransferase | 2.86–3.78 | ||
| Pavir.Fa00353.1 | 0.35 | ||||
| Pavir.J10341.1; | S-adenosylmethionine synthetase | 3.31 | |||
| Pavir.J19251.1 | 0.36 | ||||
| Pavir.Ea01573.1 | ACC oxidase 1 | 2.88 | 0.29 | ||
| Pavir.J10323.1 | 0.31 | 0.45 | |||
| Pavir.Ib00921.1 | Ethylene response regulator 2 | 0.21 | |||
| Pavir.Ib03775.1 | Dormancy-associated protein (Ethylene inducible TF (ERF114) down-regulated cell proliferation | 2.80 | |||
| Pavir.J19751.1 | 3.86 | ||||
| JA | Pavir.Ca00561.1 | 12-oxophytodienoate reductase 1 | 0.31 | ||
| Pavir.Da01421.1 | 12-oxophytodienoate reductase 1 | 0.37 | |||
| Lateral root initiation | Pavir.Bb03551.1 | Transducin/WD40 repeat-like superfamily protein | 2.54 | ||
| Pavir.Bb01815.1 | 2.66 | ||||
| Pavir.Ba00100.1 | 2.98 | ||||
| Pavir.J08344.1 | 3.39 | ||||
| Pavir.Cb02019.1 | 3.41 | ||||
| Pavir.Fa01415.1 | 4.04 | ||||
| Pavir.J02298.1 | 3.13 | ||||
| Aquaporin associated root meristem cell division | Pavir.Gb01084.1 | plasma membrane intrinsic protein | 0.39 | ||
| Pavir.Bb01320.2 | 0.38 | ||||
| Pavir.J11885.1 | 0.38 | 0.47 | |||
| Cell elongation growth | Pavir.Ca00886.1 | dynamin-like protein | 3.65 | ||
| Pavir.Da01210.1 | dynamin-like protein 6 | 4.43 | |||
| Pavir.Ab03037.1 | varicose-related | 8.67 | |||
| Cell wall | 67 proteins | Expansin; Pectin, Xylan, Cellulose, Fasciclin, Chitinase, Arabinogalactan, Lignin, hydroxyproline-rich glycoprotein, Other cell wall components | 2.86–9.68 (14 proteins) 0.33–0.39 (3 proteins) | 2.21–4.71 (19 proteins); 0.20–0.39 (6 proteins) | 2.01–5.01(22 proteins); 0.40–0.46 (2 proteins) |
| Water stress proteins (Dehydrins, LEA, Osmotic) | Pavir.J35962.1 | Dehydrin | 0.39 | 4.11 | |
| Pavir.Gb00029.1 | Early-responsive to dehydration stress protein (ERD4) | 5.31 | |||
| Pavir.Gb00029.1 | Responsive to dehydration 21 | 2.69 | |||
| 16 proteins | LEA | 3.54–11.07 | 2.11–9.25 | ||
| 6 proteins | Osmotin | 2.2–3.18 | 4.02–6.66 | ||
| Pavir.J37770.1 | spermidine synthase 1 | ||||
| Oxidative stress | Pavir.J22922.1 | monodehydroascorbate reductase 1 | 0.38 | 2.19 | |
| 16 | peroxidase | 2.62–17.67 (5) | 0.26–7.17 (11) | 2.07–3.55 (6) | |
| Three proteins | Plant L-ascorbate oxidase | 13.73 | 2.15 | 2.33–2.41 | |
| Pavir.Ib02577.1 Pavir.Hb01571.1 | Plant thionin | 2.29–3.87 | 2.79–4.62 | ||
| Pavir.Bb00430.1; Pavir.J05150.1; Pavir.J05150.1 | thioredoxin H-type 1 | 2.61 | 2.28 | 2.06 | |
| Detoxification | 26 proteins | glutathione peroxidase 1; glutathione S-transferase; glyoxalase; metallothionein; glutathione reductase | 2.71–3.98 | 2.28–6.10 | 2.01–4.03 |
| Transcriptional Regulation (TFs) | Pavir.J05134.1 | TCP family transcription factor (TF) | 8.12 | ||
| Pavir.J01608.1 | TBP-associated factor II 15 (TF) | 3.10 | |||
| Pavir.Fa01472.1; Pavir.Fb01387.1 | multiprotein bridging factor 1A (Transcriptional coactivator) | 3.82–3.61 | |||
| Pavir.Fa00854.1; Pavir.J16866.1; Pavir.Aa01534.1 | general regulatory factor 7 (TF for fatty acid synthase genes) | 0.35–0.36 | 0.43–0.45 | ||
| Pavir.Ib02226.1 | transcription regulators | 3.90 | |||
| Pavir.Bb01701.1 | nuclear factor Y, subunit B11 | 0.24 | |||
| Pavir.Ia04404.1 | transcription activator | 6.31 | |||
| Pavir.Ha01744.1 | Transcription elongation factor (TFIIS) family protein | 0.50 | |||
| Protein translation | Pavir.Ba00102.1 | Ribosomal protein L10 | 3.03 | ||
| Pavir.Cb01702.1 | Ribosomal protein L13 | 2.91 | |||
| Pavir.Ea00990.1 | Ribosomal L29e | 4.63 | |||
| Pavir.Ea02098.1 | Ribosomal protein L25 | 2.51 | |||
| Pavir.Ea02335.1 | ribosomal protein L12-A | 5.12 | |||
| Pavir.Fa00399.1 | Ribosomal protein L32e | 2.65 | |||
| Pavir.Fb00702.1 | Ribosomal protein L7Ae | 2.94 | |||
| Pavir.Gb02168.1 | Ribosomal protein L10 | 4.07 | |||
| Pavir.Ib01816.1 | Ribosomal protein S4 | 0.38 | |||
| Pavir.J00318.1 | ribosomal protein L24 | 0.38 | |||
| Pavir.J04126.1 | Ribosomal protein S3 | 6.41 | |||
| Pavir.J14971.1 | Ribosomal protein S21e | 2.52 | |||
| Pavir.J21269.1 | Ribosomal protein L6 family | 4.85 | |||
| Pavir.J33201.1 | Ribosomal protein S4 (RPS4A) | 0.40 | |||
| Pavir.J39391.1 | Ribosomal protein L31e | 4.85 | |||
| Pavir.Eb01973.1 | CASC3/Barentsz eIF4AIII binding | 0.26 | |||
| Pavir.Ca01872.1 | 8.26 | ||||
| Pavir.Bb02918.1 | Eukaryotic translation initiation factor | 2.78 | |||
| Pavir.Da00112.1 | Translation machinery associated TMA7 | 0.30 | |||
| Pavir.Eb02299.1 | Translation protein SH3-like family protein | 6.58 | |||
| Pavir.J11476.1 Pavir.J16816.1 | Eukaryotic translation initiation factor 3 subunit 7 (eIF-3) | 0.48–0.49 | |||
| Pavir.J01318.1 | Eukaryotic translation initiation factor 4A1 | 0.39 | |||
| Amino acids | Pavir.J17001.1 | S-methyl-5-thioribose kinase (Methionine synthesis) | 0.29 | ||
| Pavir.Ca00973.1 | Ketol-acid reductoisomerase (valine, leucine and isoleucine) | 0.38 | |||
| Pavir.J20971.1 | Imidazoleglycerol-phosphate dehydratase (histidine) | 2.76 | |||
| Pavir.Ib01852.1 | Class-II DAHP synthetase family protein (aromatic amino acids) | 2.88 | |||
| Pavir.Da02432.1 | Glutaminyl cyclase | 3.02 | |||
| Pavir.Fb01711.1 | Glutamate decarboxylase (GABA shunt) | 3.71 | |||
| Pavir.Ib02399.1 | Branched-chain amino acid transaminase 5 (BCAT5) | 4.68 | |||
| Pavir.Ia00863.1 | Glutamine synthase clone F11 | 2.29 | |||
| Pavir.J14758.1 | D-3-phosphoglycerate dehydrogenase (L-serine) | 0.45 | |||
| Pavir.J13435.1 | O-acetylserine (thiol) lyase (OAS-TL) isoform A1 (cysteine) | 0.48 | |||
| Pavir.Ab00560.1 | Chorismate mutase 2 (aromatic AA) | 2.06 | |||
| Carbohydrate metabolism | Pavir.J20347.1 | Neutral invertase | 0.25 | ||
| Pavir.Ia03415.1 | Sucrose synthase 3 | 2.29 | |||
| Pavir.Ib01813.1 | Sucrose synthase 4 | 0.36 | |||
| Pavir.J19702.1 | Sucrose synthase 6 | 2.60 | |||
| Pavir.Bb00094.1 | Sugar transporter 1 | 9.72 | |||
| Pavir.Ba01978.1 | Phosphoglucose isomerase | 9.72 | |||
| Pavir.J01199.1 | Starch branching enzyme | 3.88 | 2.76 | ||
| Pavir.J06227.1; Pavir.Ca02850.1 | Pyruvate dehydrogenase E1 alpha | 0.18–0.35 | |||
| Pavir.Ia03418.1 | Fumarase 1 | 0.39 | |||
| Pavir.J02207.1; Pavir.J15849.1 | Pyruvate orthophosphate dikinase | 0.38 | 0.23–0.29 | ||
| Pavir.Ga01969.1; Pavir.Gb01988.1; Pavir.J33969.1 | Phosphoenolpyruvate carboxylase | 0.38 | 0.45–0.48 | ||
| Pavir.Ib01540.1 | Phosphoglycerate mutase | 4.46 | |||
| Pavir.Fa02320.1; Pavir.J02390.1 | glyceraldehyde-3-phosphate dehydrogenase C subunit 1 | 3.25–4.13 | |||
| Pavir.Ga01407.1 | Phosphofructokinase 2 | 3.08 | |||
| Pavir.Ib01540.1 | Phosphoglycerate mutase | 6.11 | |||
| Pavir.Db00351.1 | Aldolase-type TIM barrel family protein | 2.8 | 2.85 | 2.6 | |
| Pavir.Ba00222.1 | Melibiase family protein | 2.72 | |||
| Biological Process | Protein Accessions b | Description | Fold Change (Drought Treated/Control) c | ||
|---|---|---|---|---|---|
| SDT d | D1W e | D3W f | |||
| Phytohormones | Pavir.Ia03517.1 | PYR1-like 6 | 2.17 | ||
| Pavir.Ib01400.1 | 2.23 | ||||
| Pavir.J39695.1 | 2.06 | ||||
| Pavir.Gb00126.1 | IQ-domain 32 | 0.33 | |||
| Pavir.Ia01238.1 | ABI3-interacting protein 3 (negative regulator of ABA signaling, PP2C) | 0.51 | |||
| Pavir.Ca02694.1 | ABI3 (AP2/B3-like) TF | 0.46 | |||
| Pavir.Ia02723.1 | Nodulin-related protein 1 (a negative regulator of the ABA signaling/synthesis) | 3.04 | 3.14 | 1.70 | |
| Pavir.Ca02736.1 | IAA-induced protein 16 | 0.38 | |||
| Pavir.J10323.1; Pavir.Ea01573.1 | ACC oxidase 1 | 0.38 | 0.38–0.39 | ||
| Pavir.J06494.1 | Gibberellin-regulated family protein | 4.15 | 4.81 | ||
| Root organogenesis (meristem cell division) | Pavir.Bb01893.1 | Annexin 5 (apoptosis) | 3.11 | 2.7 | |
| Pavir.Db00940.1 Pavir.Aa00752.1 | microtubule-associated proteins | 0.36–0.37 | |||
| Pavir.Ib03538.1 | Tetratricopeptide repeat (TPR)-like superfamily protein (anphase-promoting complex (APC) subunits cdc16, cdc23 and cdc27) | 0.34 | 0.50 | ||
| Pavir.J21702.1 | 0.32 | ||||
| Pavir.Ib03761.1 | 0.44 | ||||
| Pavir.J29419.1 | Centrin2 (Required for centriole duplication and correct spindle formation) | 0.49 | |||
| Pavir.J28355.1 | Methionine sulfoxide reductase (a central regulator of cell proliferation and differentiation) | 0.35 | |||
| Pavir.Aa01553.1 | NAP1-related protein 2 (Acts as histone H2A/H2B chaperone in nucleosome assembly, playing a critical role for the correct expression of genes involved in root proliferation and patterning) | 0.23 | 0.43 | 0.48 | |
| Pavir.J19751.1 | Dormancy-associated protein (ethylene induced-negative regulator of Cell division from TF ERF114); | 2.82 | 4.43 | ||
| Pavir.Ib03775.1 | 2.88 | ||||
| Pavir.Ga01149.1 | Aquaporin for sustaining root meristem cell division) | 2.95 | |||
| Pavir.Gb00671.1 | 20.43 | ||||
| Cell wall proteins | 21 proteins in SDT; 24 proteins in D1W; 22 proteins in D3W | Beta-1,3-glucanase; Beta-hexosaminidase; chitinase; d-arabinono-1,4-lactone oxidase; Expansin; Laccase; Hydroxyproline-rich glycoprotein | 2.68–10.34 | 20.40–6.80 | 1.98–3.91 |
| Organogenesis and differentiation | Pavir.Ba01536.1 | Root hair specific 19 | 0.37 | ||
| Pavir.J03344.1 | Root hair initiation protein root hairless 1 (RHL1) | 0.49 | 0.49 | ||
| Pavir.Eb02336.1 | Dirigent-like protein (Castrip) | ||||
| Pavir.Ha00936.1 | 4.76 | 0.47 | |||
| Pavir.Aa03031.1; Pavir.J12222.1 | TPX2 (wvd2) protein (The wvd2 gain-of-function mutant has impaired cell expansion and root waving, and changed root skewing) | 0.36–0.38 | |||
| Pavir.Ba01202.1 | Transducin family protein / WD-40 repeat family protein (LATERAL ROOT STIMULATOR 1) | 0.36 | |||
| Pavir.Bb02689.1 Pavir.Ca00095.1 Pavir.Ha01893.1 Pavir.Ia01197.1 | 0.36–0.50 | ||||
| Water stress proteins (Dehydrins, LEA, Osmotins) | Pavir.Cb00662.1 | Dehydrin | 6.39 | 4.82 | 4.68 |
| Pavir.J13075.1 | 20.51 | ||||
| Pavir.Ea02542.1 | 5.86 | ||||
| Pavir.Aa00887.1 | 0.47 | ||||
| Pavir.J04551.1 | 0.46 | ||||
| Pavir.J13075.1 | 7.88 | ||||
| Pavir.Aa02737.1 | LEA | 6.14 | 4.86 | 3.09 | |
| Pavir.Bb02409.1 | 3.71 | 3.95 | 2.95 | ||
| Pavir.Ca01780.1 | 12.15 | 5.36 | 4.90 | ||
| Pavir.Eb02512.1 | 3.23 | 3.02 | 3.19 | ||
| Pavir.Ga00596.1 | 6.30 | 5.85 | 6.82 | ||
| Pavir.Gb00543.1 | 10.76 | 7.06 | 7.80 | ||
| Pavir.Ia03678.1 | 6.29 | 4.13 | 3.46 | ||
| Pavir.J00158.1 | 21.57 | 14.23 | 6.24 | ||
| Pavir.J28600.1 | 8.29 | 9.69 | 6.55 | ||
| Pavir.J24821.1 | 7.43 | 2.43 | |||
| Three proteins | 2.63–4.27 | 2.42–4.92 | |||
| Pavir.Db00310.1 | 0.42 | ||||
| Pavir.Bb00799.1 | Osmotin34 | 6.15 | |||
| Pavir.Bb03197.1 | 6.63 | 7.83 | 6.08 | ||
| Pavir.Ea03024.1 | 6.25 | ||||
| Pavir.Eb03806.1 | 5.55 | 6.43 | 5.32 | ||
| Pavir.J40731.1 | Galactinol synthase 1 (biosynthesis of raffinose osmoprotectants) | 3.77 | |||
| Oxidative stress | Pavir.Aa01787.1; Pavir.Aa03533.1; Pavir.Ab00019.1; Pavir.Ia03296.1 | Disulfide isomerases | 0.45–0.48 | ||
| Pavir.Fb02222.1 | 2.39 | 3.098 | |||
| Pavir.Ca00381.1 | Alkenal reductase | 3.02 | 2.56 | ||
| Pavir.Ea00295.1 | 2-oxoglutarate (2OG) and Fe(II)-DAPendent oxygenase superfamily protein | 4.11 | 3.99 | 3.16 | |
| Pavir.Ea01261.1 | Peroxidase | 3.01 | 3.20 | 3.98 | |
| Pavir.Eb03999.1 | 3.27 | 2.29 | 2.37 | ||
| Pavir.Ia02811.1 | 4.41 | 2.97 | 2.83 | ||
| Pavir.J14586.1 | 2.65 | 2.94 | 2.20 | ||
| Pavir.J15786.1 | 5.24 | 3.53 | 3.55 | ||
| Pavir.J14586.1 | Calatase 1 | 2.65 | 2.94 | 2.20 | |
| Detoxification | 31 proteins | Glutathione S-transferase; Glutaredoxin; Glutathione S-transferase; Formate dehydrogenase; metallothionein 2A; Heavy metal transport/detoxification superfamily protein; Glyoxalases | 2.63–16.26 | 2.23–4.19 | 1.98–6.55 |
| Transcriptional Regulation (TFs) | Pavir.Ia00088.1; Pavir.Ba01216.1; Pavir.Ba01215.1 | Ribonucleases (post-transcriptional regulation) | 2.34–7.66 | ||
| Pavir.Fb01805.1; Pavir.Gb01520.1; Pavir.Hb00704.1; Pavir.Fa00854.1 | General regulatory factor (TF (fatty acid synthase genes) | 0.29–0.50 | |||
| Pavir.Ab02229.1; Pavir.Gb01377.1; Pavir.Bb02517.1 | Basic helix-loop-helix (bHLH) DNA-binding superfamily protein | 0.17–0.37 | |||
| Pavir.Ib01088.1 | Homeodomain-like superfamily protein | 3.05 | |||
| Pavir.Da01122.1 | 0.44 | ||||
| Pavir.Eb00895.1 | NmrA-like negative transcriptional regulator family protein | 2.92 | |||
| Pavir.Db01330.1 | PHD finger protein-related | 0.37 | |||
| Pavir.Ib01055.1 | RAD-like 1 | 0.25 | |||
| Pavir.J31192.1 | RAD-like 1 | 0.24 | |||
| Pavir.Fb01946.1 | Squamosa promoter binding protein-like 9 | 0.31 | |||
| Pavir.Ib04454.1 | Winged-helix DNA-binding transcription factor family protein | 0.29 | |||
| Pavir.J31192.1 | 0.42 | ||||
| Pavir.Bb02517.1 | Basic helix-loop-helix (bHLH) DNA-binding superfamily protein | 0.43 | |||
| Pavir.J25625.1 | Basic-leucine zipper (bZIP) transcription factor family protein | 0.40 | |||
| Pavir.Eb00253.1 | CCCH-type zinc finger family protein | 0.43 | |||
| Pavir.J10403.1 | Zinc finger (C3HC4-type RING finger) family protein | 0.27 | 0.49 | ||
| Pavir.Cb00329.1 | Zinc finger (C3HC4-type RING finger) family protein | 0.43 | |||
| Pavir.Ab00438.1 | Zinc finger C-x8-C-x5-C-x3-H type family protein | 0.46 | |||
| Pavir.Da01847.1 | Growth-regulating factor 5 (Transcription activator that plays a role in the regulation of cell expansion) | 0.31 | |||
| Pavir.Fb01387.1 | Multiprotein bridging factor 1A (Transcriptional coactivator) | 2.83 | 2.54 | ||
| Pavir.Ea03968.1 | Nuclear factor Y, subunit B5 (TF) | 0.45 | |||
| Pavir.Ia04404.1 | Transcription activator-related | 9.73 | 8.95 | ||
| Pavir.Cb02016.1 | Transcription elongation factor (TFIIS) family protein | 0.44 | |||
| Pavir.Ea00508.1 | 0.48 | ||||
| Pavir.Ia02581.1 | Transcription factor TFIIE, alpha subunit | 0.50 | |||
| Pavir.J35507.1 | Splicing factor-related | 0.35 | |||
| Protein translation | 2 proteins in SDT, 7 in D1W, 8 in D3W | Ribosomal subunits | 0.37–0.38 | 0.28–0.43 | 0.35–0.50 |
| Pavir.J20711.1; Pavir.Gb01843.1 | 2.34–2.79 | ||||
| Pavir.J16816.1; Pavir.Ga01591.1 | Eukaryotic translation initiation factors | 0.35–0.38 | |||
| Pavir.J32559.1 | Nuclear transport factor 2 (NTF2) family protein with RNA binding (RRM-RBD-RNP motifs) domain | 0.42 | 0.41 | ||
| 14 proteins | Ekaryotic release factor 1-3, eukaryotic translation initiation factors; Translation elongation factor EF1B; Translation initiation factor IF2/IF5; GTP binding Elongation factor Tu family protein; Essential protein Yae1 | 0.40–0.50 | |||
| Amino acids | Pavir.Fa00915.1 | Glutamate decarboxylase (GABA) | 4.80 | 3.69 | |
| Pavir.Cb00192.1 | 3-methylcrotonyl-CoA carboxylase 1 (MCCA) (leucine catabolism) | 2.67 | |||
| Pavir.Da02439.1 | Homogentisate 1,2-dioxygenase (break down tyrosine and phenylalanine | 3.03 | 2.84 | ||
| Pavir.Db00968.1 | Dihydrodipicolinate synthase 1(lysine biosynthesis | 2.62 | |||
| Pavir.Eb03924.1 | Pyrroline-5-carboxylate (P5C) reductase (arginine and proline metabolism) | 0.51 | |||
| Carbohydrates Sucrose/Starch | Pavir.Ab00047.1 | Sucrose invertase | 0.34 | ||
| Pavir.J01866.1; Pavir.Ia03415.1 | Sucrose synthase | 3.37 | 2.51–3.66 | 2.42–2.58 | |
| Pavir.J01199.1 | Starch branching enzyme 2.2 | 3.56 | |||
| Pavir.Ba00222.1 | Melibiase family protein | 6.02 | 4.41 | 2.93 | |
| Glycolysis | Pavir.Cb01993.1 | Fructose-bisphosphate aldolase 2 | 2.86 | ||
| Pavir.J07576.1 | Phosphoglycerate kinase | 2.71 | 2.74 | 2.00 | |
| Pavir.J15479.1 | Phosphofructokinase 2 | 3.67 | |||
| Pavir.J13866.1 | Phosphoglycerate kinase | 2.31 | |||
| Pavir.Ib01540.1 | Phosphoglycerate mutase | 5.40 | |||
| Pavir.Ga01407.1 | phosphofructokinase 2 | 2.57 | |||
| Pentose shunt | Pavir.Gb02335.1 | Transketolase | 2.78 | ||
| Pavir.Db00351.1 | Aldolase-type TIM barrel family protein | 2.23 | |||
| TCA | Pavir.Bb02852.1 | Aconitase 3 | 4.72 | ||
| Pavir.J02207.1; Pavir.J15849.1 | Pyruvate orthophosphate dikinase | 0.15–0.23 | |||
| Pavir.Gb01988.1 Pavir.J33969.1 | Phosphoenolpyruvate carboxylase | 0.43–0.41 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Z.; Sangireddy, S.R.; Yu, C.-L.; Hui, D.; Howe, K.; Fish, T.; Thannhauser, T.W.; Zhou, S. Comparative Proteomics of Root Apex and Root Elongation Zones Provides Insights into Molecular Mechanisms for Drought Stress and Recovery Adjustment in Switchgrass. Proteomes 2020, 8, 3. https://doi.org/10.3390/proteomes8010003
Ye Z, Sangireddy SR, Yu C-L, Hui D, Howe K, Fish T, Thannhauser TW, Zhou S. Comparative Proteomics of Root Apex and Root Elongation Zones Provides Insights into Molecular Mechanisms for Drought Stress and Recovery Adjustment in Switchgrass. Proteomes. 2020; 8(1):3. https://doi.org/10.3390/proteomes8010003
Chicago/Turabian StyleYe, Zhujia, Sasikiran Reddy Sangireddy, Chih-Li Yu, Dafeng Hui, Kevin Howe, Tara Fish, Theodore W. Thannhauser, and Suping Zhou. 2020. "Comparative Proteomics of Root Apex and Root Elongation Zones Provides Insights into Molecular Mechanisms for Drought Stress and Recovery Adjustment in Switchgrass" Proteomes 8, no. 1: 3. https://doi.org/10.3390/proteomes8010003
APA StyleYe, Z., Sangireddy, S. R., Yu, C.-L., Hui, D., Howe, K., Fish, T., Thannhauser, T. W., & Zhou, S. (2020). Comparative Proteomics of Root Apex and Root Elongation Zones Provides Insights into Molecular Mechanisms for Drought Stress and Recovery Adjustment in Switchgrass. Proteomes, 8(1), 3. https://doi.org/10.3390/proteomes8010003