Precursor Intensity-Based Label-Free Quantification Software Tools for Proteomic and Multi-Omic Analysis within the Galaxy Platform

 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. (A) ABRF Dataset
2.2. (B) Spiked-In UPS Benchmark Dataset
2.3. Peptide Identification
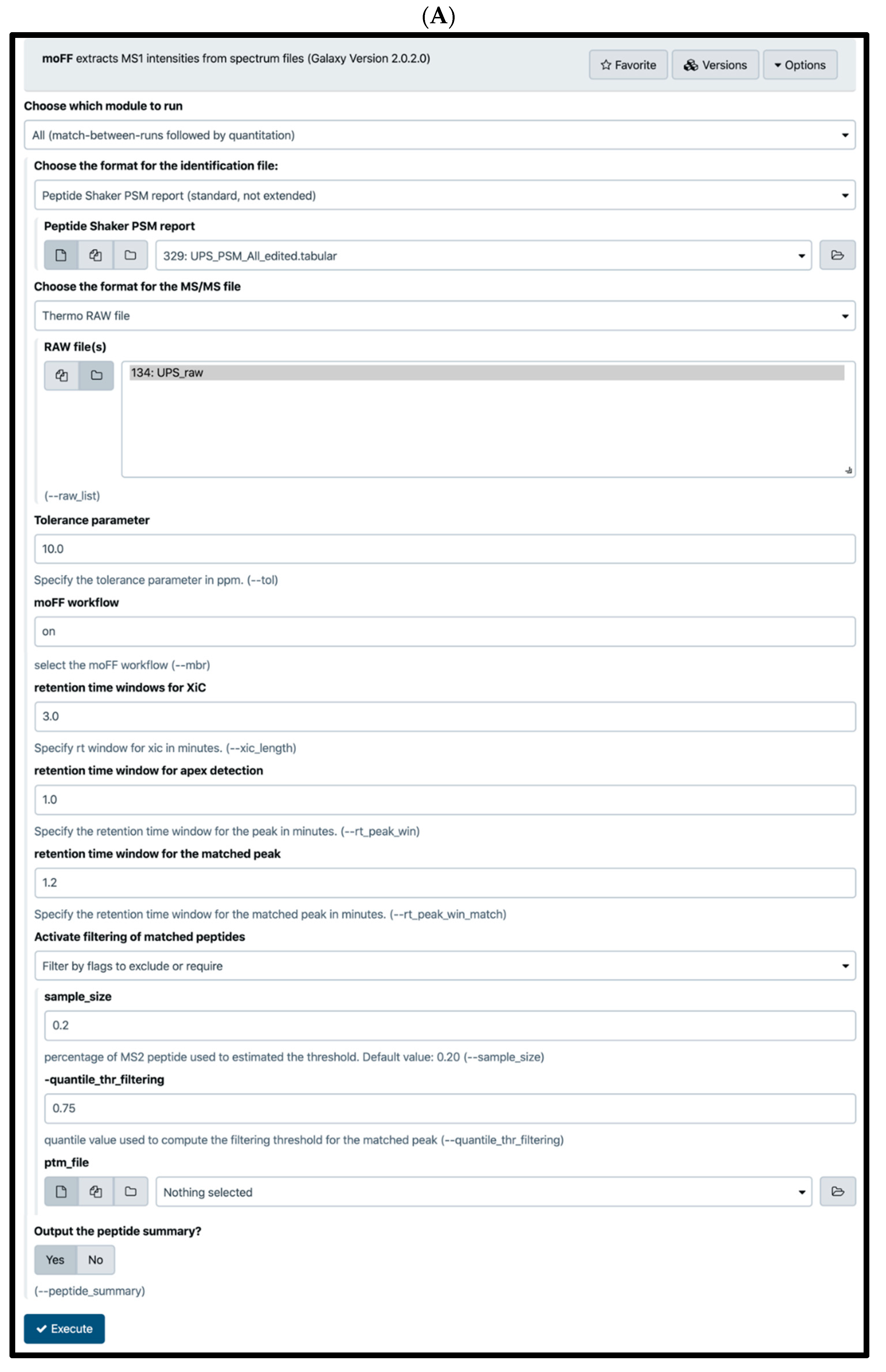
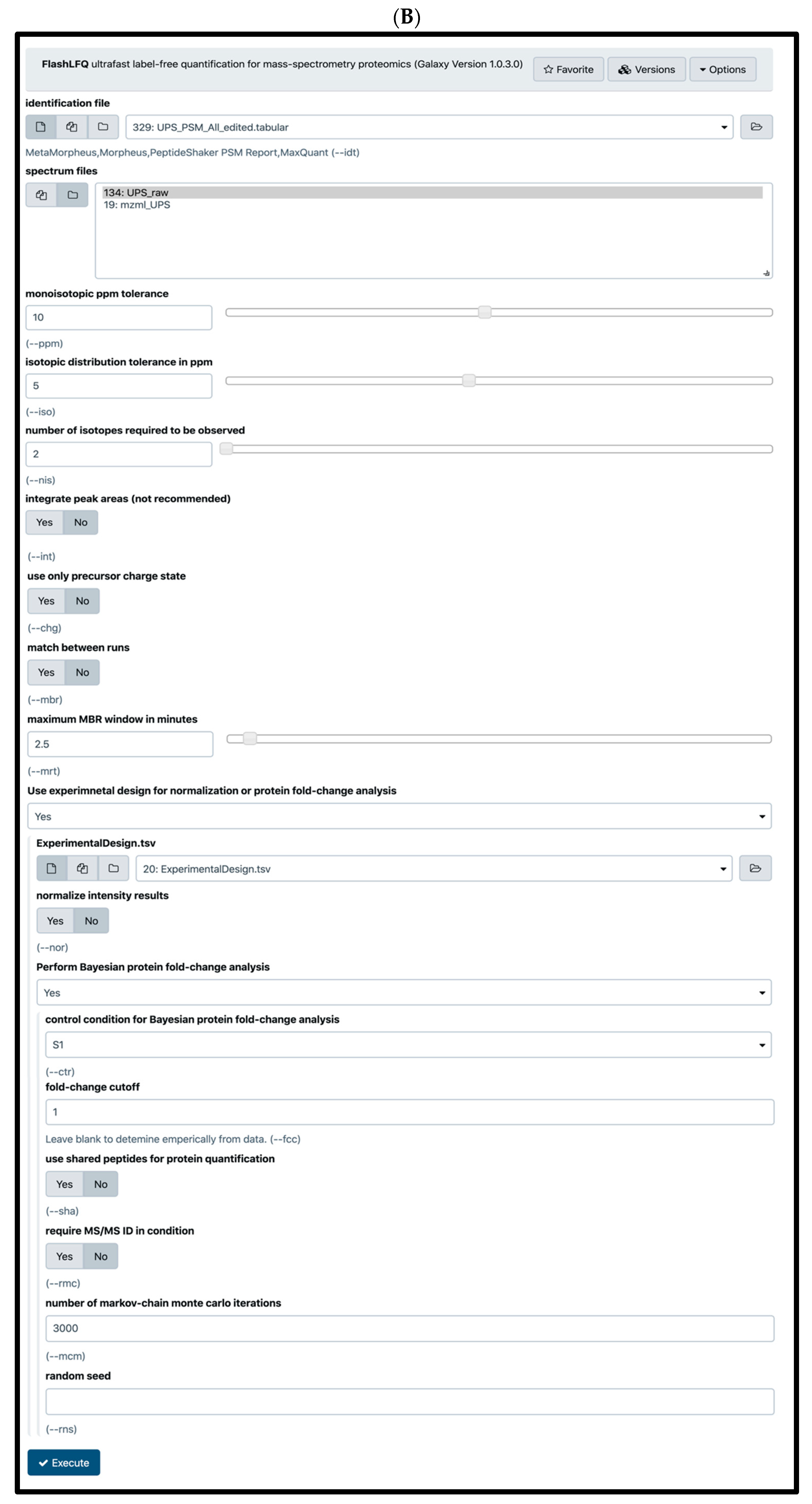
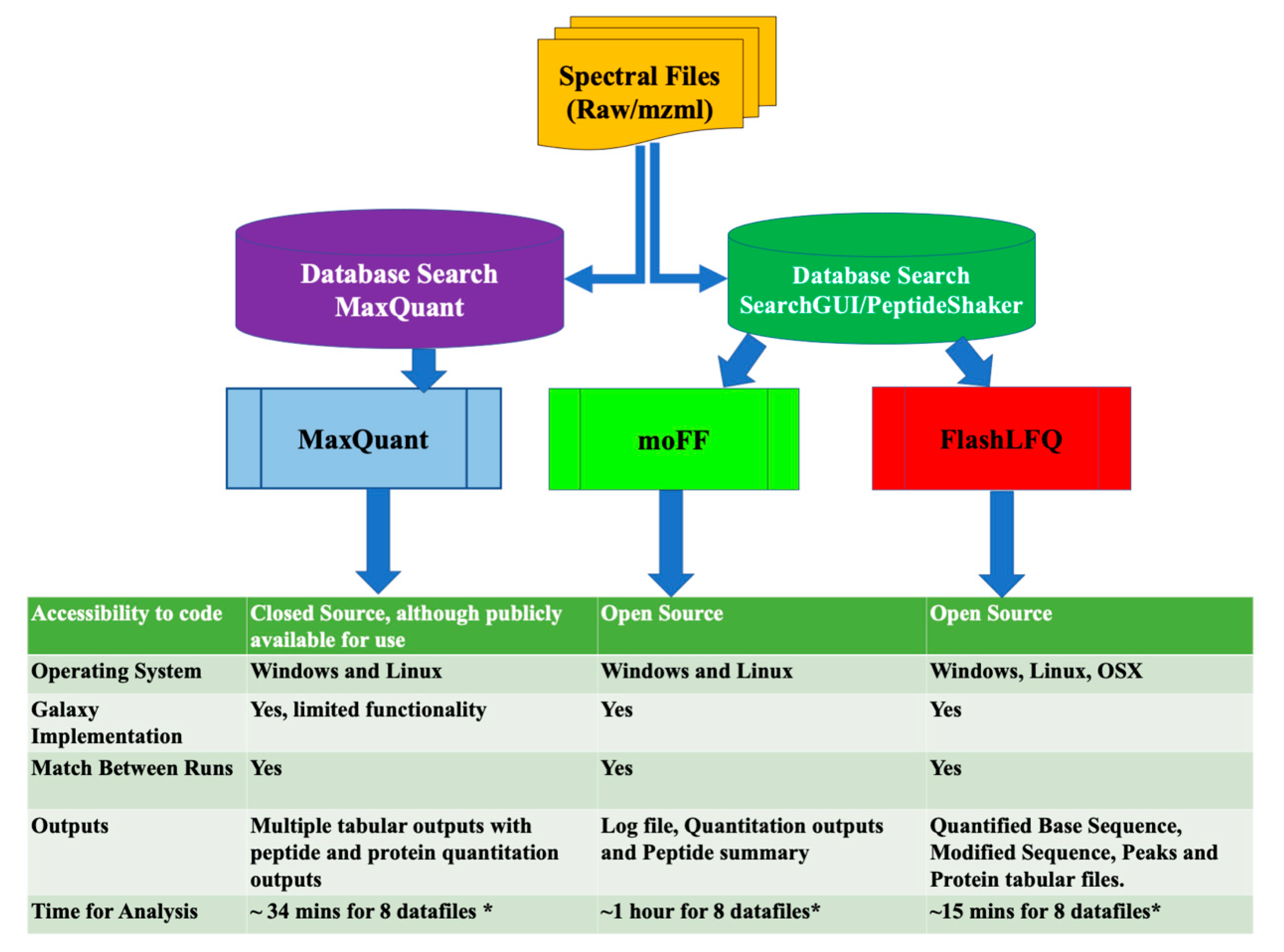
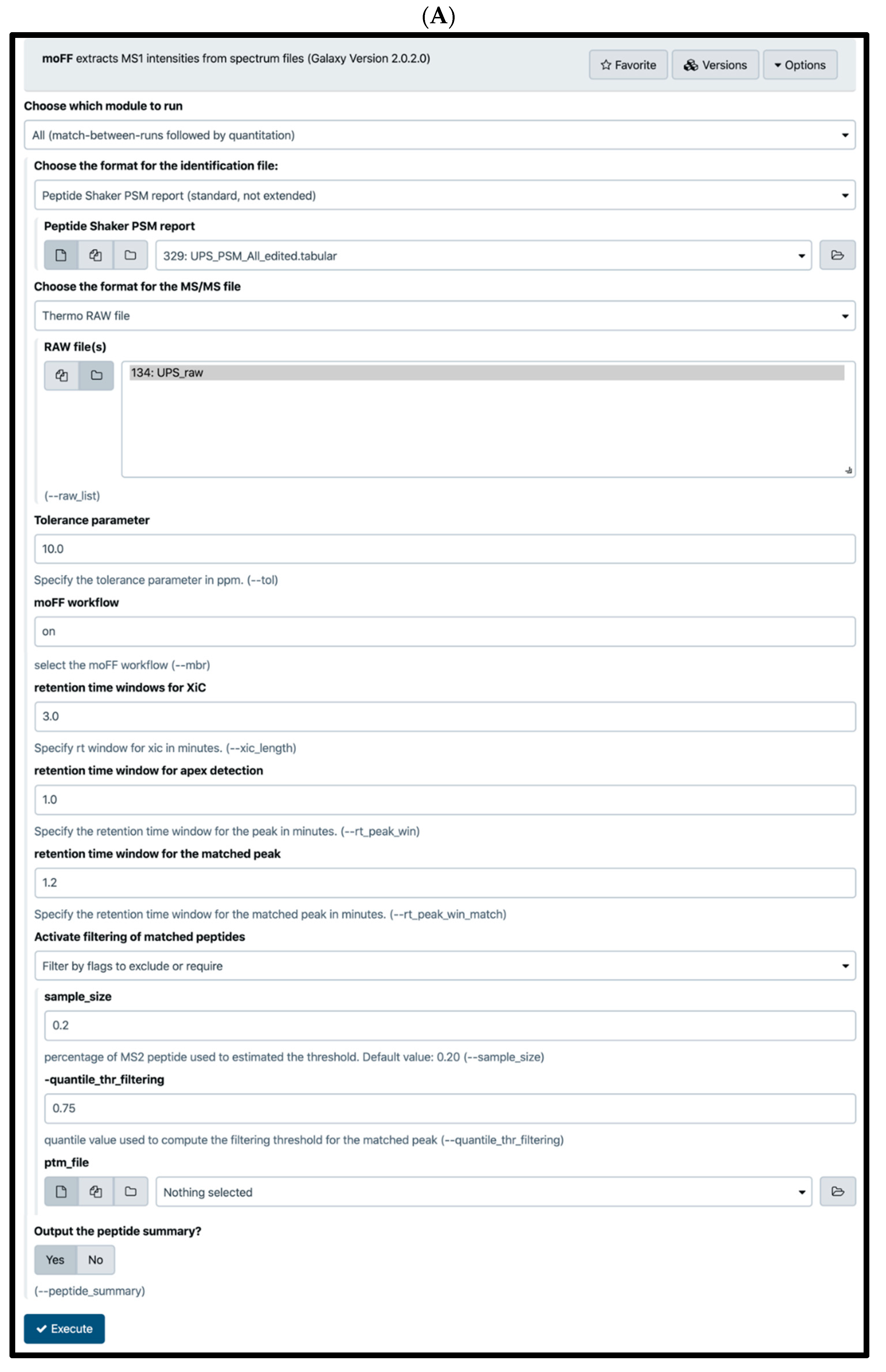
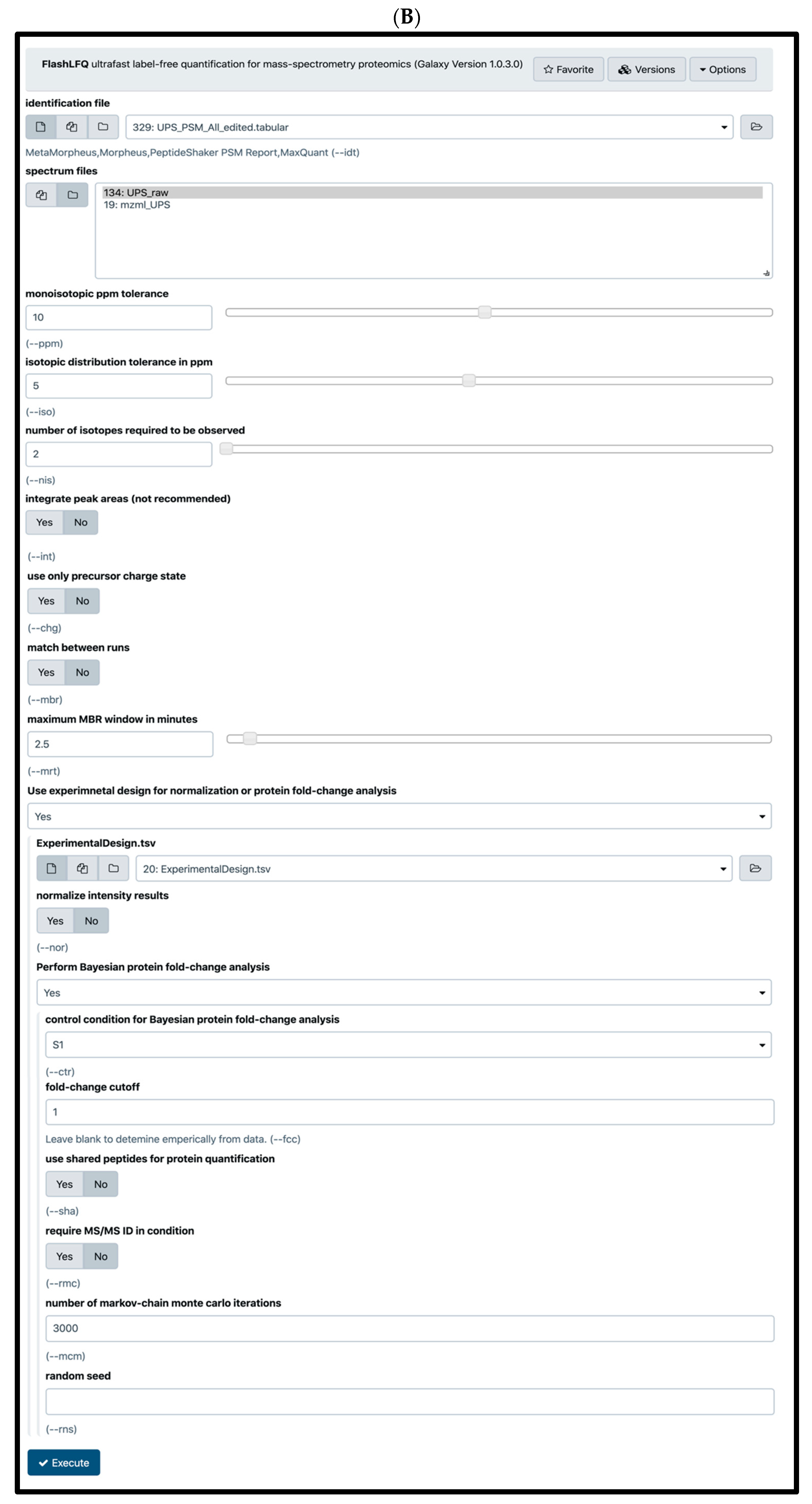
2.4. Quantification Tools
2.5. Normalization and Protein Quantification
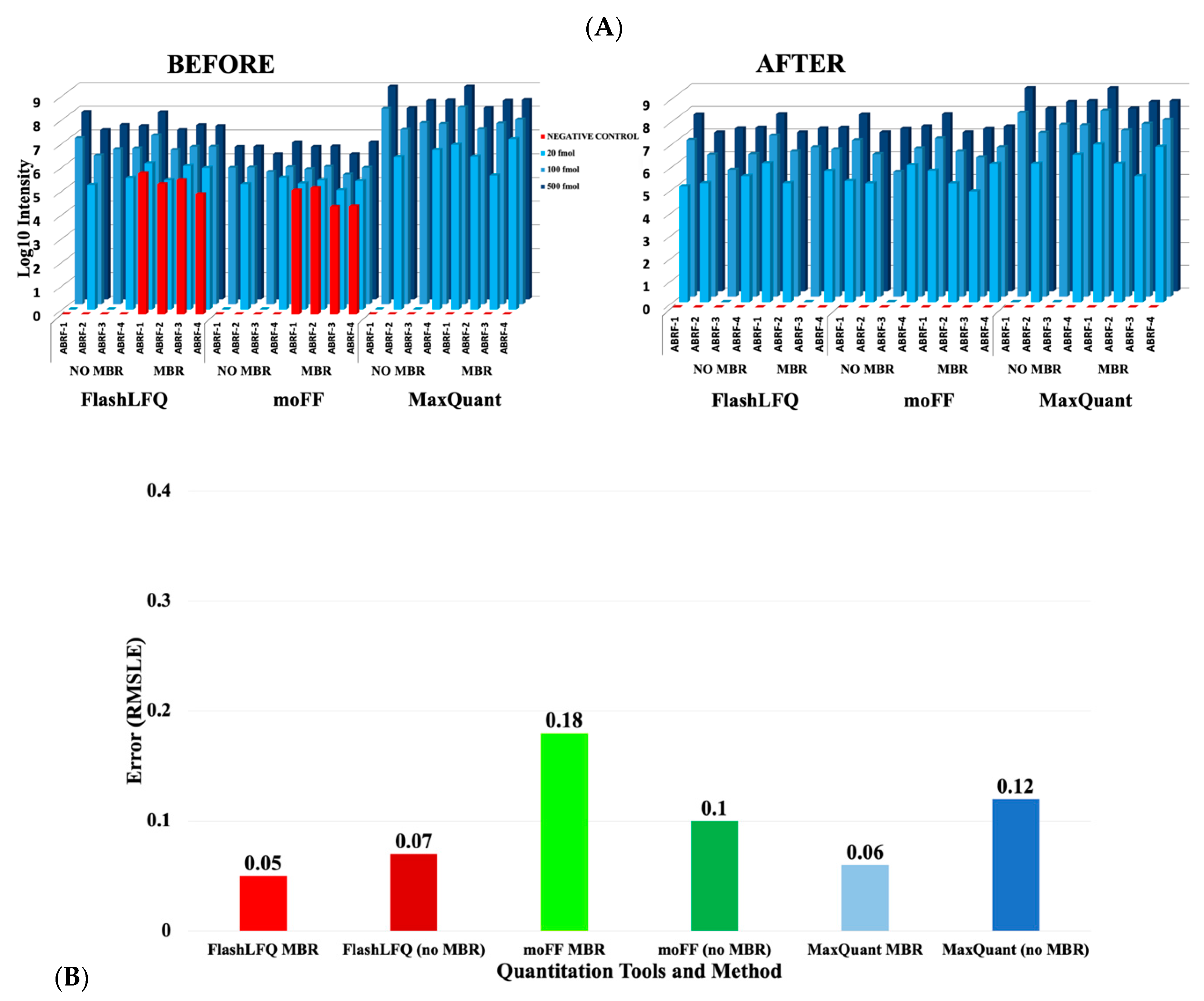
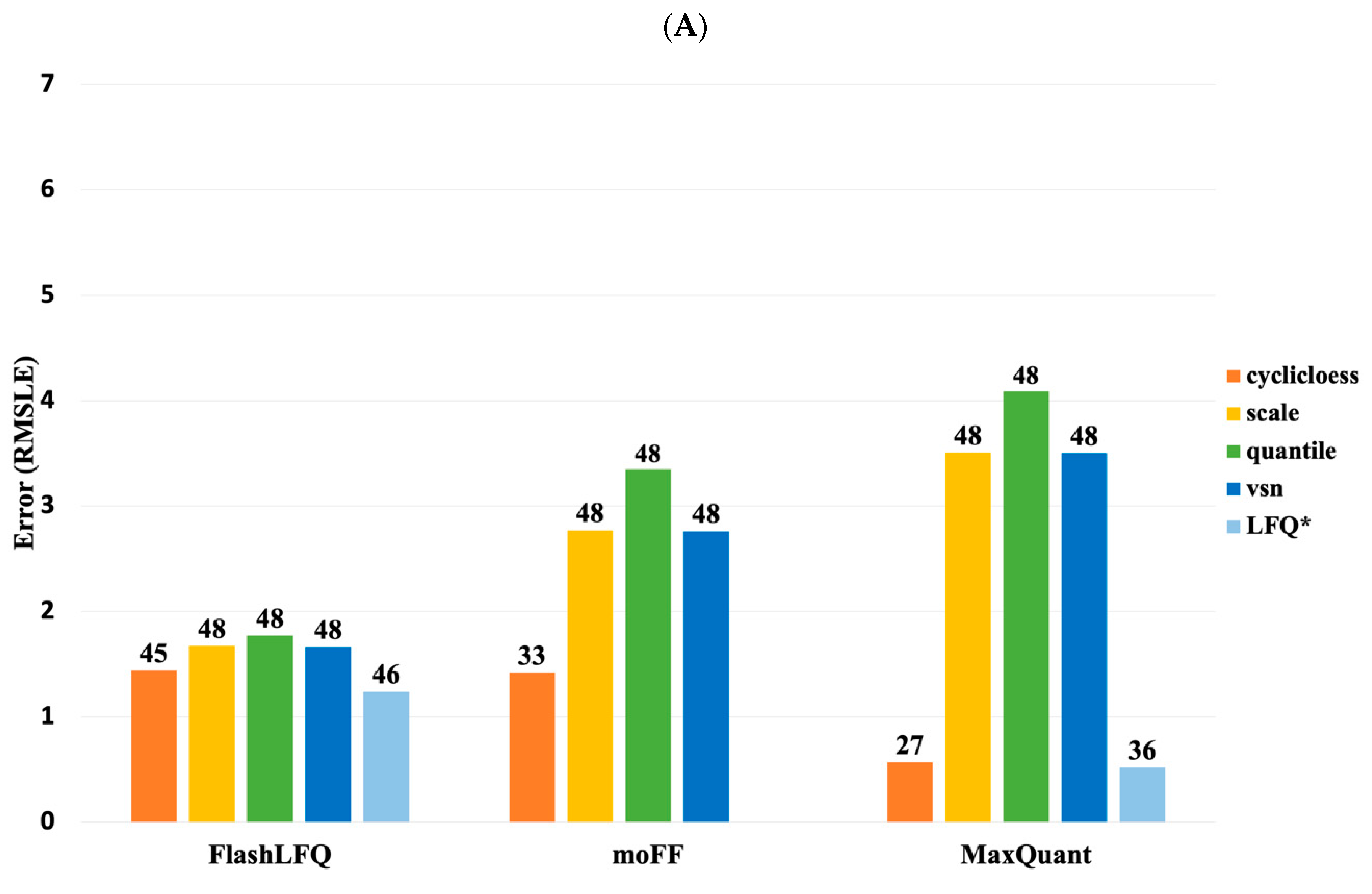
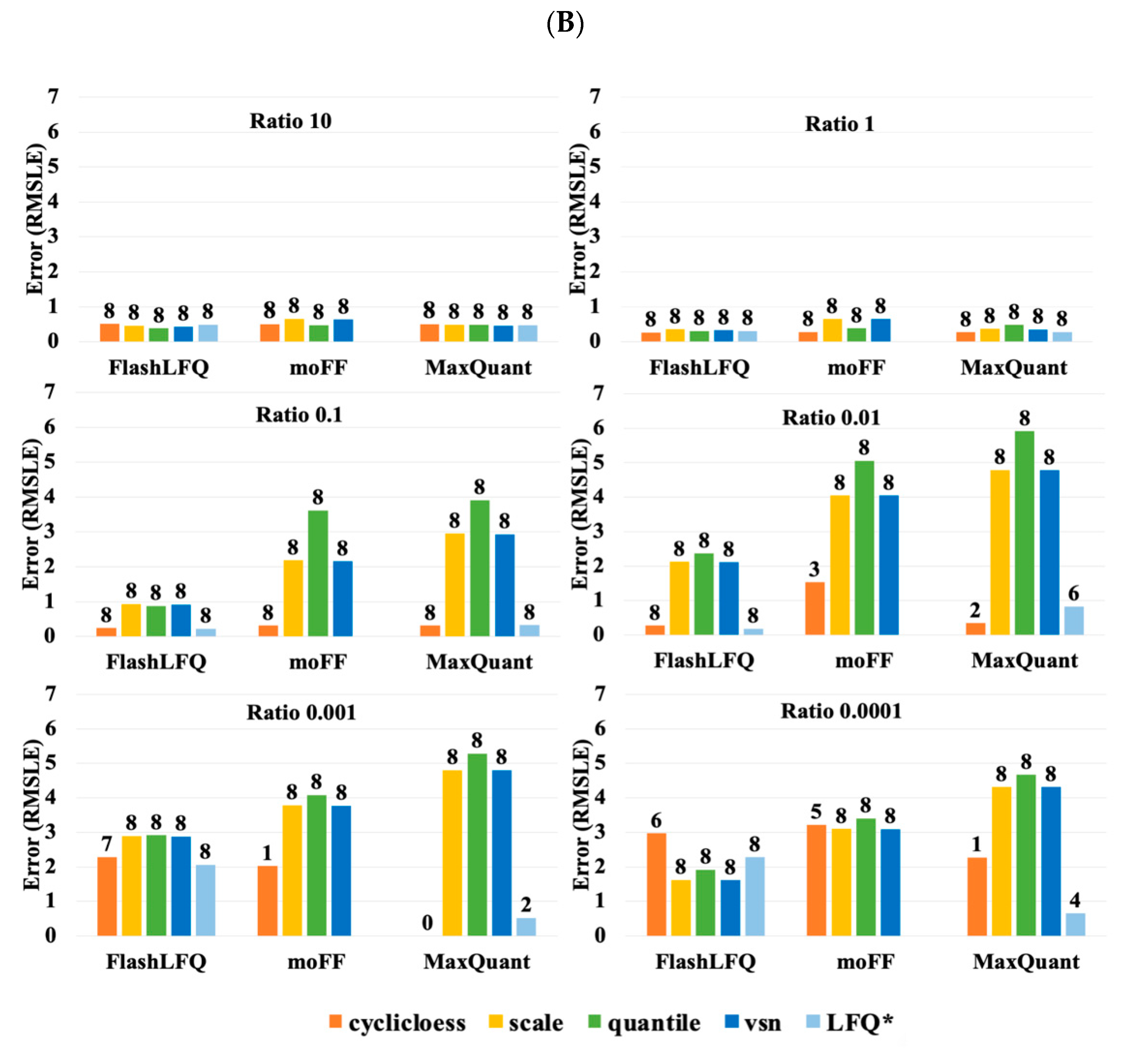
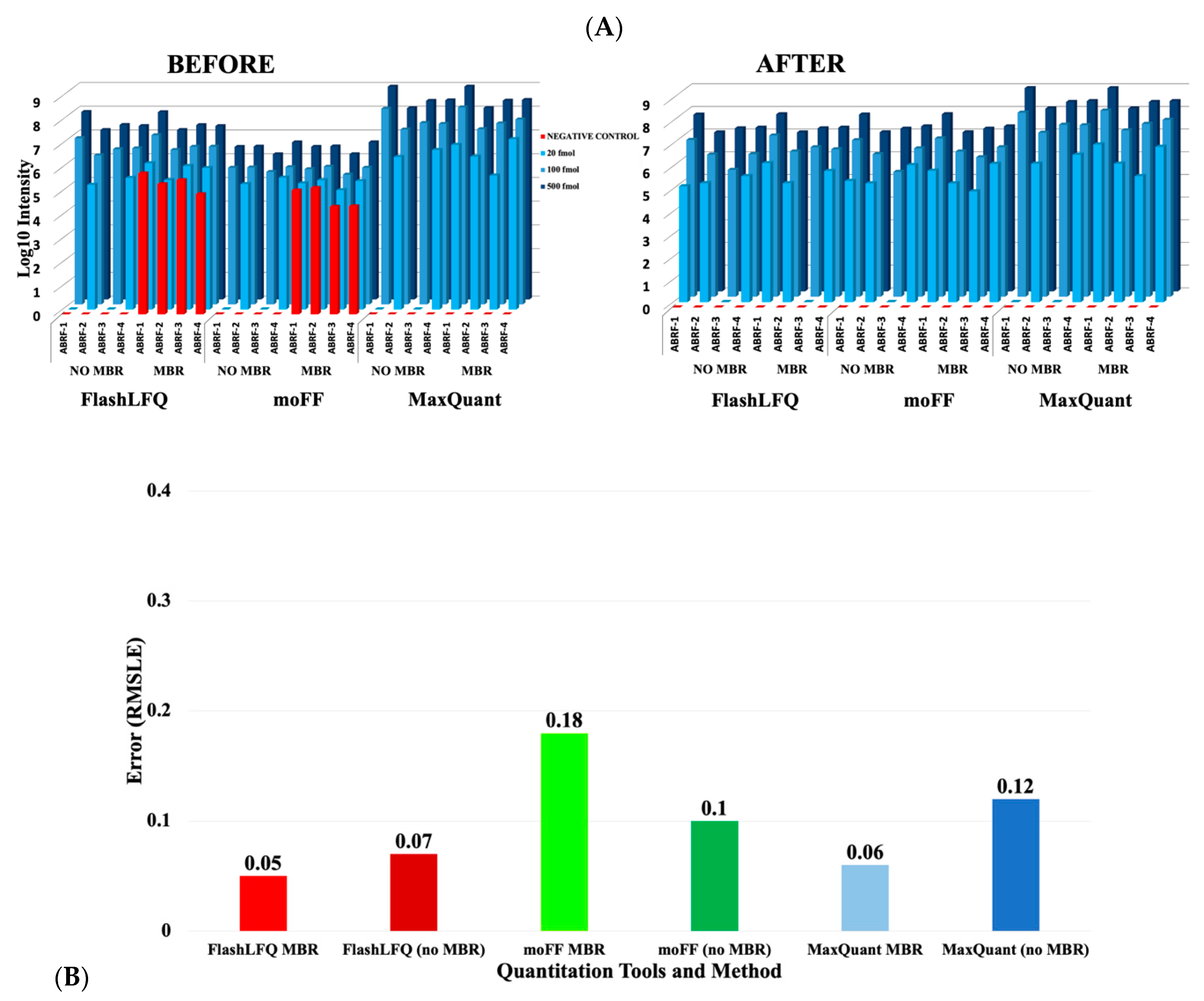
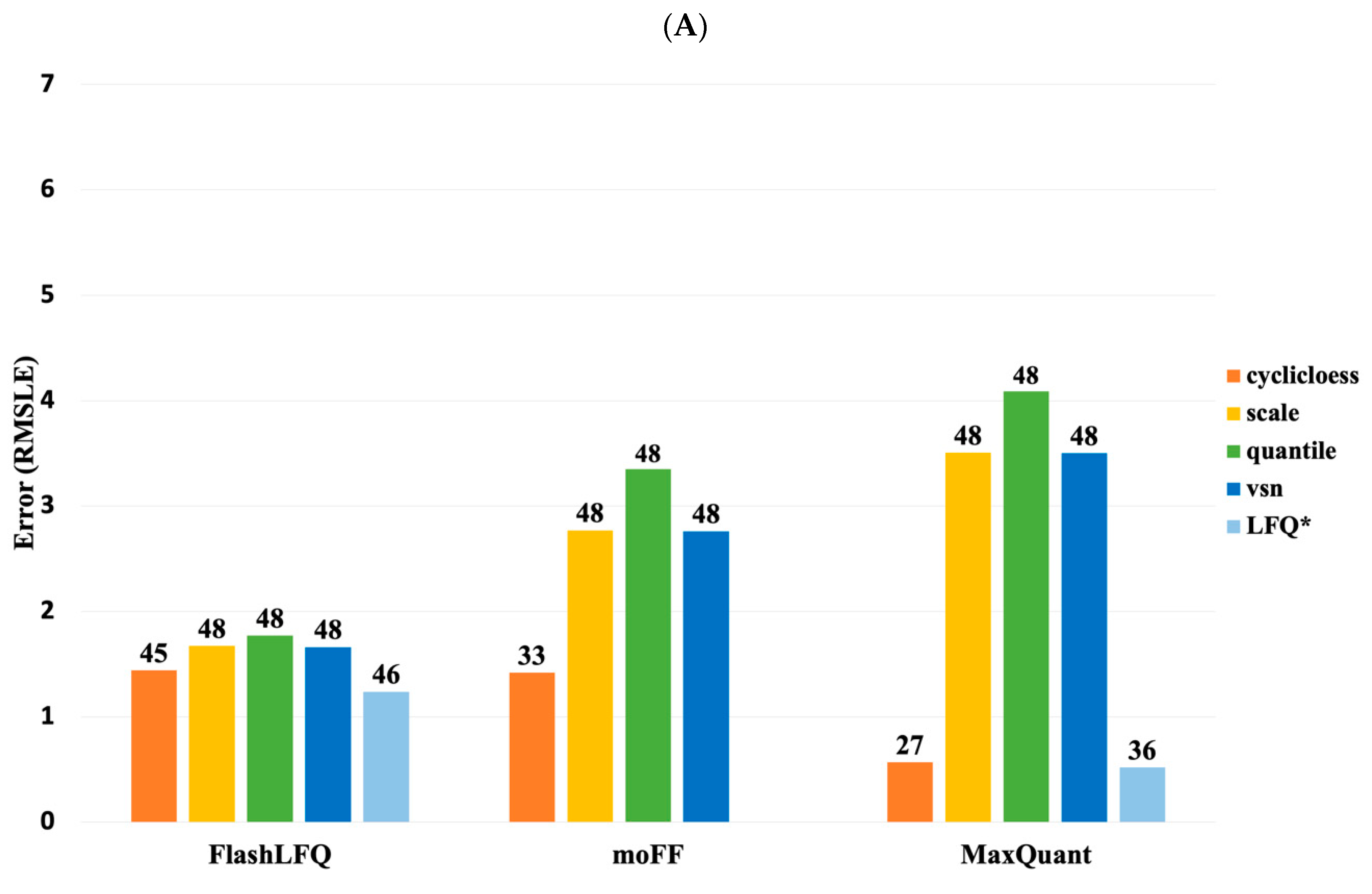
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, Y. Retrieving Quantitative Information of Histone PTMs by Mass Spectrometry, In Methods Enzymology; Academic Press Inc.: Cambridge, MA, USA, 2017; pp. 165–191. [Google Scholar] [CrossRef]
- Alfaro, J.A.; Ignatchenko, A.; Ignatchenko, V.; Sinha, A.; Boutros, P.C.; Kislinger, T. Detecting protein variants by mass spectrometry: A comprehensive study in cancer cell-lines. Genome Med. 2017, 9, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeom, J.; Kabir, M.H.; Lim, B.; Ahn, H.S.; Kim, S.Y.; Lee, C. A proteogenomic approach for protein-level evidence of genomic variants in cancer cells. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easterly, C.W.; Sajulga, R.; Mehta, S.; Johnson, J.; Kumar, P.; Hubler, S.; Mesuere, B.; Rudney, J.; Griffin, T.J.; Jagtap, P.D. metaQuantome: An Integrated, Quantitative Metaproteomics Approach Reveals Connections Between Taxonomy and Protein Function in Complex Microbiomes. Mol. Cell. Proteom. 2019, 18, S82–S91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krey, J.F.; Wilmarth, P.A.; Shin, J.B.; Klimek, J.; Sherman, N.E.; Jeffery, E.D.; Choi, D.; David, L.L.; Barr-Gillespie, P.G. Accurate label-free protein quantitation with high- and low-resolution mass spectrometers. J. Proteome Res. 2014, 13, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Välikangas, T.; Suomi, T.; Elo, L.L. A comprehensive evaluation of popular proteomics software workflows for label-free proteome quantification and imputation. Brief. Bioinform. 2017, 19, 1344–1355. [Google Scholar] [CrossRef] [Green Version]
- Chawade, A.; Sandin, M.; Teleman, J.; Malmström, J.; Levander, F. Data processing has major impact on the outcome of quantitative label-free LC-MS analysis. J. Proteome Res. 2015, 14, 676–687. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; Rardin, M.J.; MacLean, B.X.; Zawadzka, A.M.; Frewen, B.E.; Cusack, M.P.; Sorensen, D.J.; Bereman, M.S.; Jing, E.; Wu, C.C.; et al. Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: Application to protein acetylation and phosphorylation. Mol. Cell. Proteom. 2012. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Zhang, K.; Hendrie, C.; Liang, C.; Li, M.; Doherty-Kirby, A.; Lajoie, G. PEAKS: Powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2337–2342. [Google Scholar] [CrossRef]
- Progenesis QI for Proteomics: Waters. Available online: https://www.waters.com/waters/en_US/Progenesis-QI-for-Proteomics/nav.htm?locale=en_US&cid=134790665 (accessed on 8 October 2019).
- Argentini, A.; Goeminne, L.J.E.; Verheggen, K.; Hulstaert, N.; Staes, A.; Clement, L.; Martens, L. MoFF: A robust and automated approach to extract peptide ion intensities. Nat. Methods 2016, 13, 964–966. [Google Scholar] [CrossRef]
- Millikin, R.J.; Solntsev, S.K.; Shortreed, M.R.; Smith, L.M. Ultrafast Peptide Label-Free Quantification with FlashLFQ. J. Proteome Res. 2018, 17, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Van Riper, S.; Chen, E.; Remmer, H.; Chien, A.; Stemmer, P.; Wang, Y.; Jagtap, P. Identification of Low Abundance Proteins in a Highly Complex Protein Mixture. 2016. Available online: https://zenodo.org/record/3563207#.XwRcuhJ5t2E (accessed on 7 July 2020). [CrossRef]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagtap, P.D.; Johnson, J.E.; Onsongo, G.; Sadler, F.W.; Murray, K.; Wang, Y.; Shenykman, G.M.; Bandhakavi, S.; Smith, L.M.; Griffin, T.J. Flexible and accessible workflows for improved proteogenomic analysis using the galaxy framework. J. Proteome Res. 2014, 13, 5898–5908. [Google Scholar] [CrossRef]
- Anderson, K.J.; Vermillion, K.L.; Jagtap, P.; Johnson, J.E.; Griffin, T.J.; Andrews, M.T. Proteogenomic Analysis of a Hibernating Mammal Indicates Contribution of Skeletal Muscle Physiology to the Hibernation Phenotype. J. Proteome Res. 2016, 15, 1253–1261. [Google Scholar] [CrossRef]
- Blank, C.; Easterly, C.; Gruening, B.; Johnson, J.; Kolmeder, C.A.; Kumar, P.; May, D.; Mehta, S.; Mesuere, B.; Brown, Z.; et al. Disseminating metaproteomic informatics capabilities and knowledge using the galaxy-P framework. Proteomes 2018, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagtap, P.D.; Blakely, A.; Murray, K.; Stewart, S.; Kooren, J.; Johnson, J.E.; Rhodus, N.L.; Rudney, J.; Griffin, T.J. Metaproteomic analysis using the Galaxy framework. Proteomics 2015, 15, 3553–3565. [Google Scholar] [CrossRef]
- Proteomics moFF Galaxy Toolshed Repository. Available online: https://toolshed.g2.bx.psu.edu/view/galaxyp/proteomics_moff/7af419c90f5f (accessed on 7 July 2020).
- Proteomics FlashLFQ Galaxy Toolshed Repository. Available online: https://toolshed.g2.bx.psu.edu/view/galaxyp/flashlfq/908ab13490dc (accessed on 7 July 2020).
- Adusumilli, R.; Mallick, P. Data conversion with proteoWizard msConvert. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; pp. 339–368. [Google Scholar] [CrossRef]
- Barsnes, H.; Vaudel, M. SearchGUI: A Highly Adaptable Common Interface for Proteomics Search and de Novo Engines. J. Proteome Res. 2018, 17, 2552–2555. [Google Scholar] [CrossRef] [PubMed]
- Vaudel, M.; Burkhart, J.M.; Zahedi, R.P.; Oveland, E.; Berven, F.S.; Sickmann, A.; Martens, L.; Barsnes, H. PeptideShaker enables reanalysis of MS-derived proteomics data sets: To the editor. Nat. Biotechnol. 2015, 33, 22–24. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Linear Models for Microarray and RNA-Seq Data. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Suomi, T.; Corthals, G.L.; Nevalainen, O.S.; Elo, L.L. Using peptide-level proteomics data for detecting differentially expressed proteins. J. Proteome Res. 2015, 14, 4564–4570. [Google Scholar] [CrossRef] [PubMed]
- Välikangas, T.; Suomi, T.; Elo, L.L. A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief. Bioinform. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- Argentini, A.; Staes, A.; Grüning, B.; Mehta, S.; Easterly, C.; Griffin, T.J.; Jagtap, P.; Impens, F.; Martens, L. Update on the moFF Algorithm for Label-Free Quantitative Proteomics. J. Proteome Res. 2019, 18, 728–731. [Google Scholar] [CrossRef]
- Dale, R.; Grüning, B.; Sjödin, A.; Rowe, J.; Chapman, B.A.; Tomkins-Tinch, C.H.; Valieris, R.; Batut, B.; Caprez, A.; Cokelaer, T.; et al. Bioconda: Sustainable and comprehensive software distribution for the life sciences. Nat. Methods 2018, 15, 475–476. [Google Scholar] [CrossRef]
- Proteomics moFF GitHub Repository. Available online: https://github.com/compomics/moFF (accessed on 7 July 2020).
- Luersen, M.A.; le Riche, R.; Guyon, F. A constrained, globalized, and bounded Nelder-Mead method for engineering optimization. Struct. Multidiscip. Optim. 2004, 27, 43–54. [Google Scholar] [CrossRef]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific containers for mobility of compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef] [PubMed]
- Proteomics FlashLFQ GitHub Repository. Available online: https://github.com/smith-chem-wisc/FlashLFQ (accessed on 7 July 2020).
- Walther, B.A.; Moore, J.L. The concepts of bias, precision and accuracy, and their use in testing the performance of species richness estimators, with a literature review of estimator performance. Ecography 2005, 28, 815–829. [Google Scholar] [CrossRef]
- Bensimon, A.; Heck, A.J.R.; Aebersold, R. Mass Spectrometry–Based Proteomics and Network Biology. Annu. Rev. Biochem. 2012, 81, 379–405. [Google Scholar] [CrossRef]
- Vermillion, K.L.; Jagtap, P.; Johnson, J.E.; Griffin, T.J.; Andrews, M.T. Characterizing cardiac molecular mechanisms of mammalian hibernation via quantitative proteogenomics. J. Proteome Res. 2015, 14, 4792–4804. [Google Scholar] [CrossRef]
- Ori, A.; Toyama, B.H.; Harris, M.S.; Bock, T.; Iskar, M.; Bork, P.; Ingolia, N.T.; Hetzer, M.W.; Beck, M. Integrated Transcriptome and Proteome Analyses Reveal Organ-Specific Proteome Deterioration in Old Rats. Cell Syst. 2015, 1, 224–237. [Google Scholar] [CrossRef] [Green Version]
- Roumeliotis, T.I.; Williams, S.P.; Gonçalves, E.; Alsinet, C.; Velasco-Herrera, M.D.; Aben, N.; Ghavidel, F.Z.; Michaut, M.; Schubert, M.; Price, S.; et al. Genomic Determinants of Protein Abundance Variation in Colorectal Cancer Cells. Cell Rep. 2017, 20, 2201–2214. [Google Scholar] [CrossRef] [Green Version]
- Schlaffner, C.N.; Pirklbauer, G.J.; Bender, A.; Steen, J.A.J.; Choudhary, J.S. A fast and quantitative method for post-translational modification and variant enabled mapping of peptides to genomes. J. Vis. Exp. 2018. [Google Scholar] [CrossRef]
- Gans, J.; Wolinsky, M.; Dunbar, J. Microbiology: Computational improvements reveal great bacterial diversity and high toxicity in soil. Science 2005, 309, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; LeBlanc, J.; Truong, A.; Vuthoori, R.; Chen, S.S.; Lustgarten, J.L.; Roth, B.; Allard, J.; Ippoliti, A.; Presley, L.L.; et al. A metaproteomic approach to study human-microbial ecosystems at the mucosal luminal interface. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; Ning, Z.; Zhang, X.; Li, L.; Liao, B.; Mayne, J.; Stintzi, A.; Figeys, D. MetaLab: An automated pipeline for metaproteomic data analysis. Microbiome 2017, 5, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, M.C.; Jagtap, P.D.; Johnson, J.E.; McGowan, T.; Kumar, P.; Onsongo, G.; Guerrero, C.R.; Barsnes, H.; Vaudel, M.; Martens, L.; et al. An Accessible Proteogenomics Informatics Resource for Cancer Researchers. Cancer Res. 2017, 77, e43–e46. [Google Scholar] [CrossRef]
- Mayer, G.; Montecchi-Palazzi, L.; Ovelleiro, D.; Jones, A.R.; Binz, P.A.; Deutsch, E.W.; Chambers, M.; Kallhardt, M.; Levander, F.; Shofstahl, J.; et al. The HUPO proteomics standards initiativemass spectrometry controlled vocabulary. Database 2013. [Google Scholar] [CrossRef] [PubMed]
- Smith, R. Conversations with 100 Scientists in the Field Reveal a Bifurcated Perception of the State of Mass Spectrometry Software. J. Proteome Res. 2018, 17, 1335–1339. [Google Scholar] [CrossRef]
- Proteomics MaxQuant Galaxy Toolshed Repository. Available online: https://toolshed.g2.bx.psu.edu/view/galaxyp/maxquant/175e062b6a17 (accessed on 7 July 2020).
- Proteomics MaxQuant GitHub Repository. Available online: https://github.com/galaxyproteomics/tools-galaxyp/tree/master/tools/maxquant (accessed on 7 July 2020).


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehta, S.; Easterly, C.W.; Sajulga, R.; Millikin, R.J.; Argentini, A.; Eguinoa, I.; Martens, L.; Shortreed, M.R.; Smith, L.M.; McGowan, T.; et al. Precursor Intensity-Based Label-Free Quantification Software Tools for Proteomic and Multi-Omic Analysis within the Galaxy Platform. Proteomes 2020, 8, 15. https://doi.org/10.3390/proteomes8030015
Mehta S, Easterly CW, Sajulga R, Millikin RJ, Argentini A, Eguinoa I, Martens L, Shortreed MR, Smith LM, McGowan T, et al. Precursor Intensity-Based Label-Free Quantification Software Tools for Proteomic and Multi-Omic Analysis within the Galaxy Platform. Proteomes. 2020; 8(3):15. https://doi.org/10.3390/proteomes8030015
Chicago/Turabian StyleMehta, Subina, Caleb W. Easterly, Ray Sajulga, Robert J. Millikin, Andrea Argentini, Ignacio Eguinoa, Lennart Martens, Michael R. Shortreed, Lloyd M. Smith, Thomas McGowan, and et al. 2020. "Precursor Intensity-Based Label-Free Quantification Software Tools for Proteomic and Multi-Omic Analysis within the Galaxy Platform" Proteomes 8, no. 3: 15. https://doi.org/10.3390/proteomes8030015
APA StyleMehta, S., Easterly, C. W., Sajulga, R., Millikin, R. J., Argentini, A., Eguinoa, I., Martens, L., Shortreed, M. R., Smith, L. M., McGowan, T., Kumar, P., Johnson, J. E., Griffin, T. J., & Jagtap, P. D. (2020). Precursor Intensity-Based Label-Free Quantification Software Tools for Proteomic and Multi-Omic Analysis within the Galaxy Platform. Proteomes, 8(3), 15. https://doi.org/10.3390/proteomes8030015