
Protamine Characterization by Top-Down Proteomics: Boosting Proteoform Identification with DBSCAN

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Purification of Human Sperm Protamine
2.3. Protamine Quantification
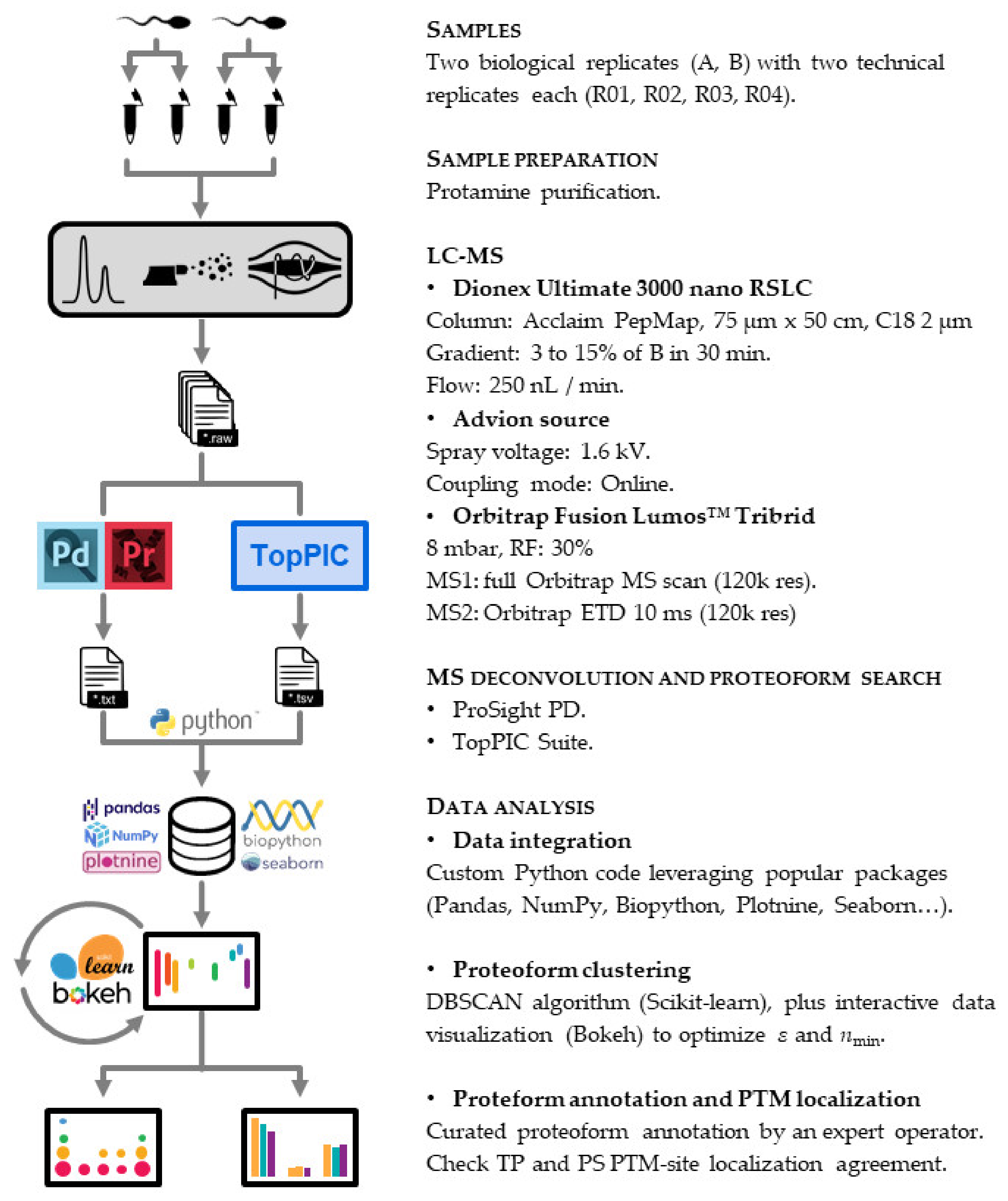
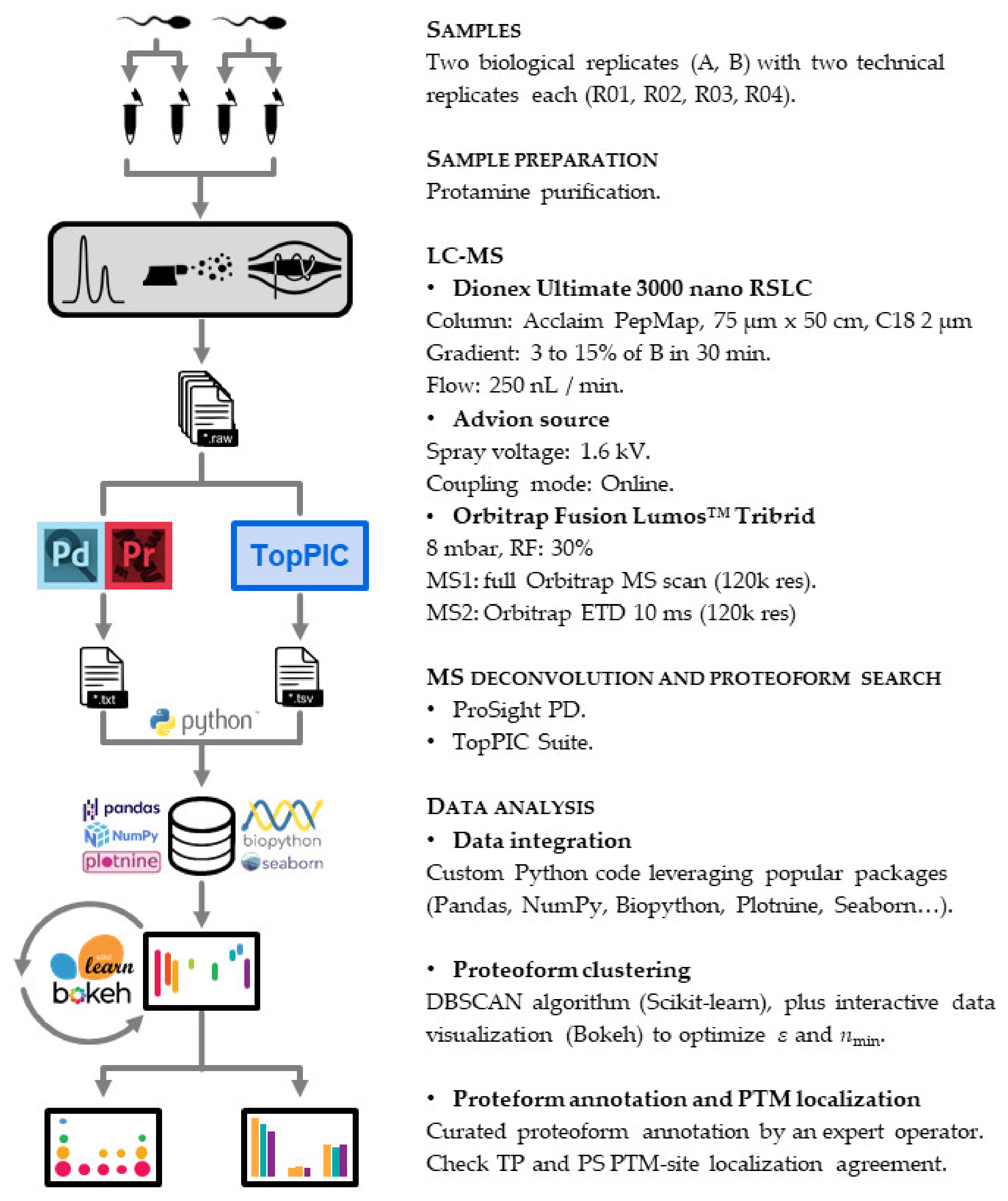
2.4. Top-Down Proteomics
2.4.1. LC-MS
2.4.2. Proteoform Search
2.4.3. Data Analysis
2.4.4. Proteoform Annotation
3. Results and Discussion
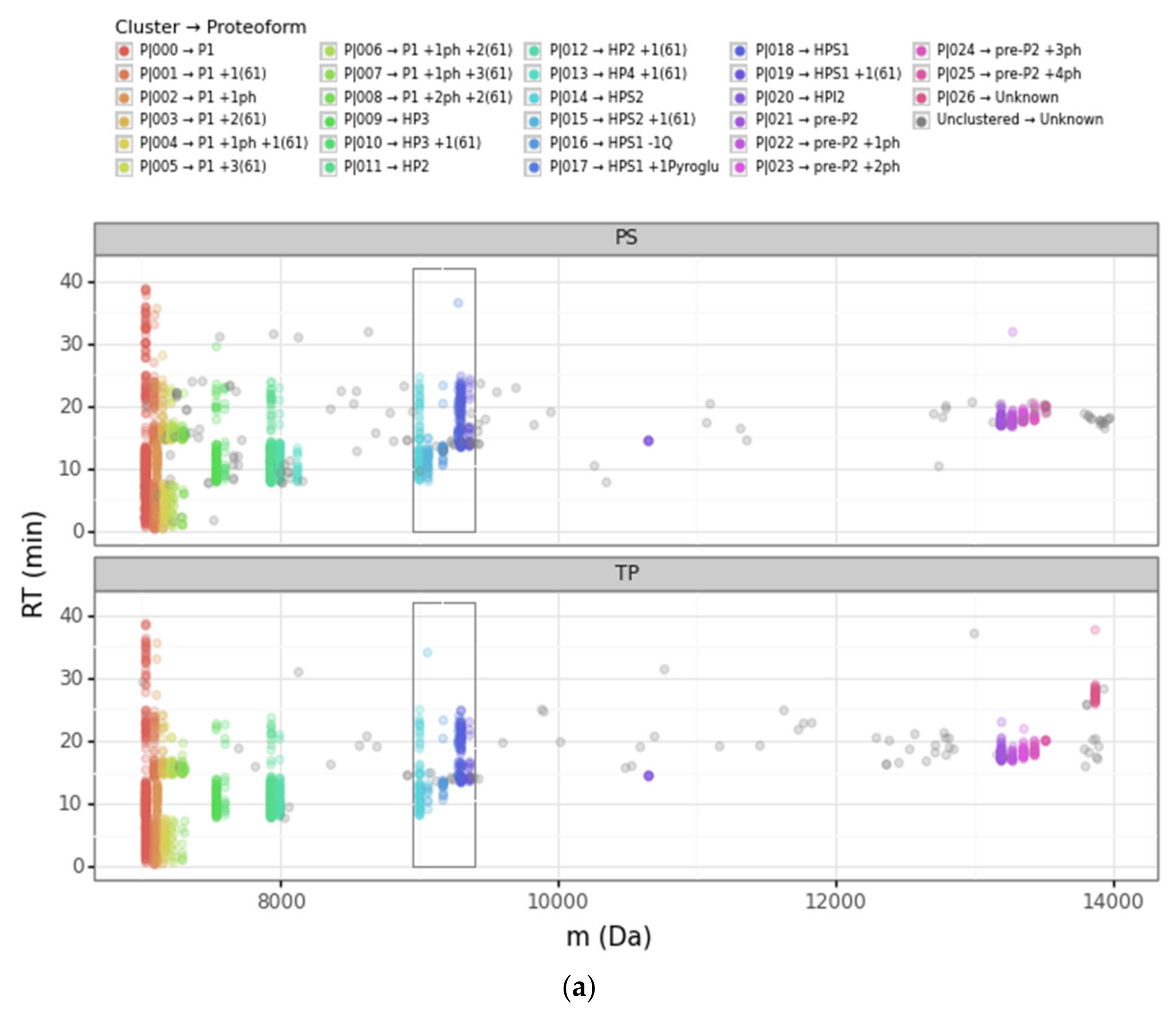
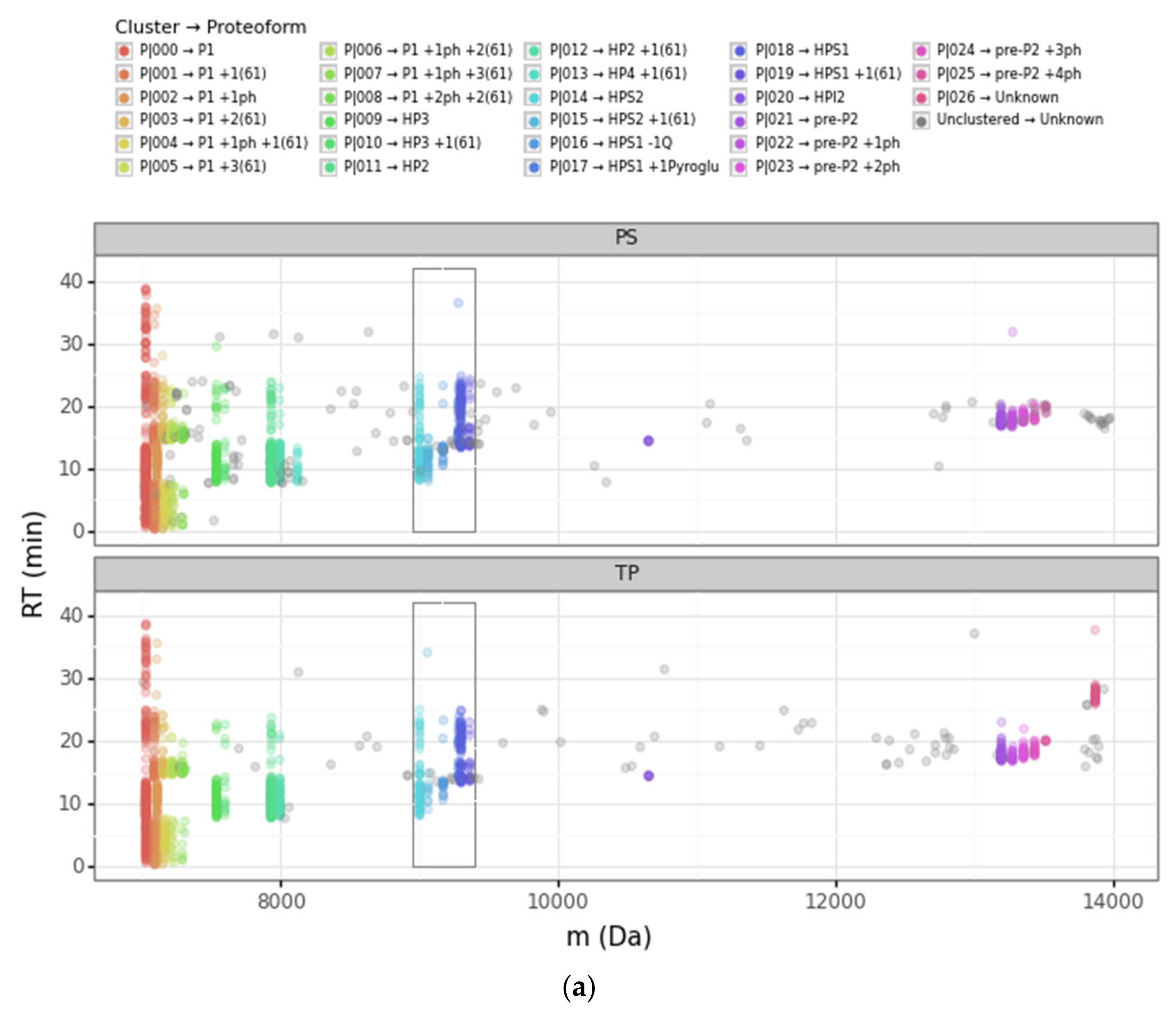
3.1. Overview of the DBSCAN Approach
3.2. Advantages of the DBSCAN Approach vs. PS and TP
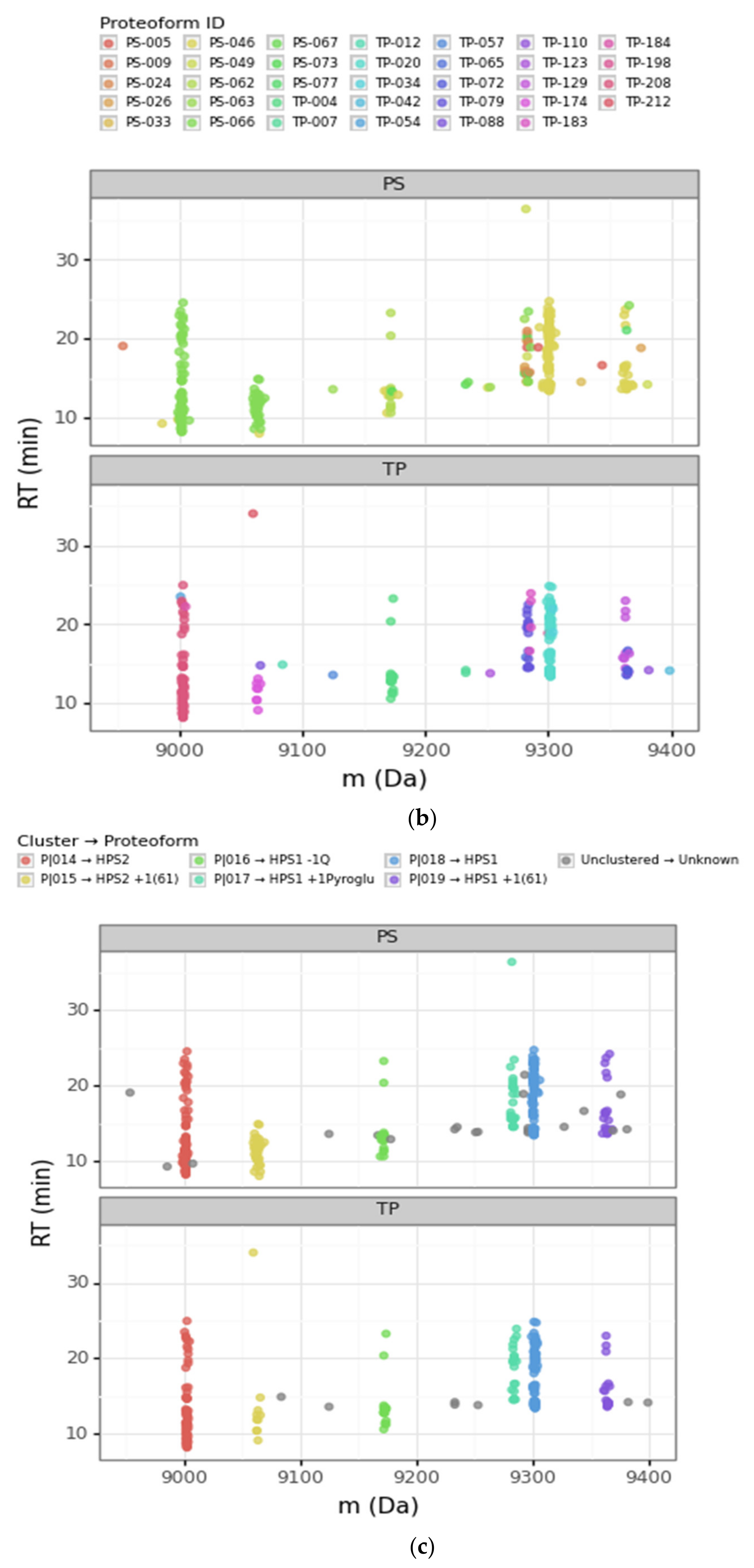
3.2.1. Converging and Diverging Misassignment of PrSMs
3.2.2. DBSCAN Approach Limitations
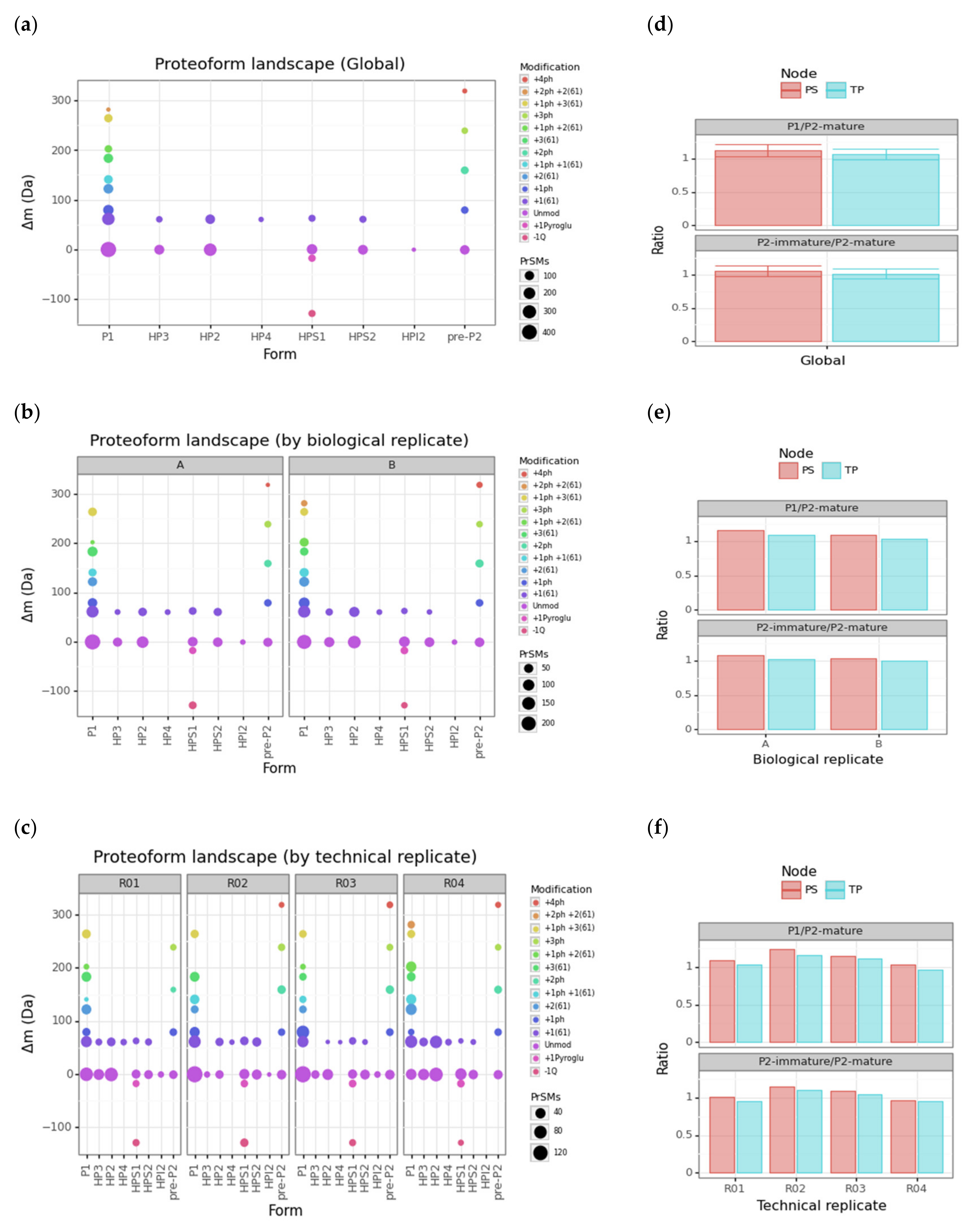
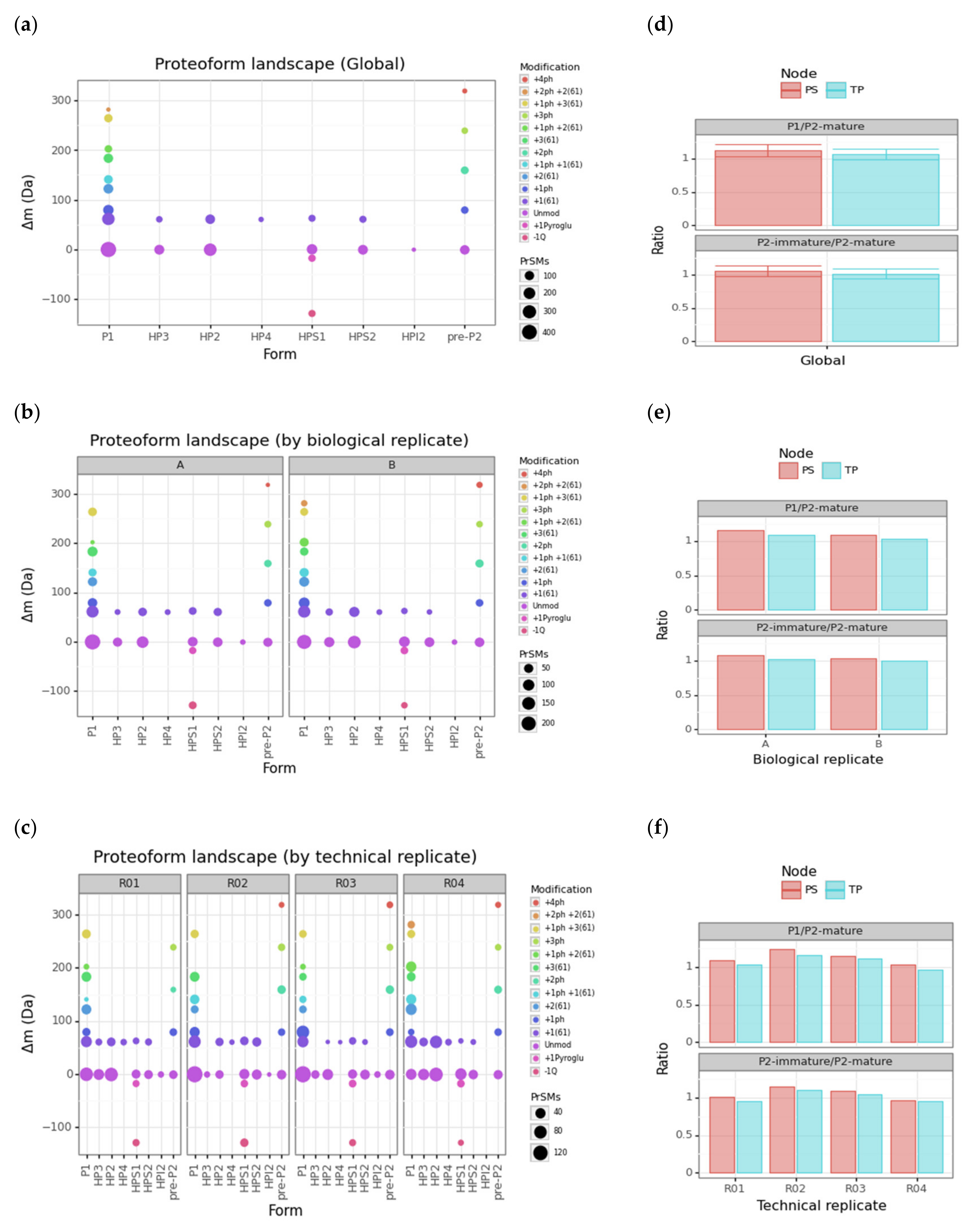
3.3. Protamine Proteoform Landscape and Protamine Ratios
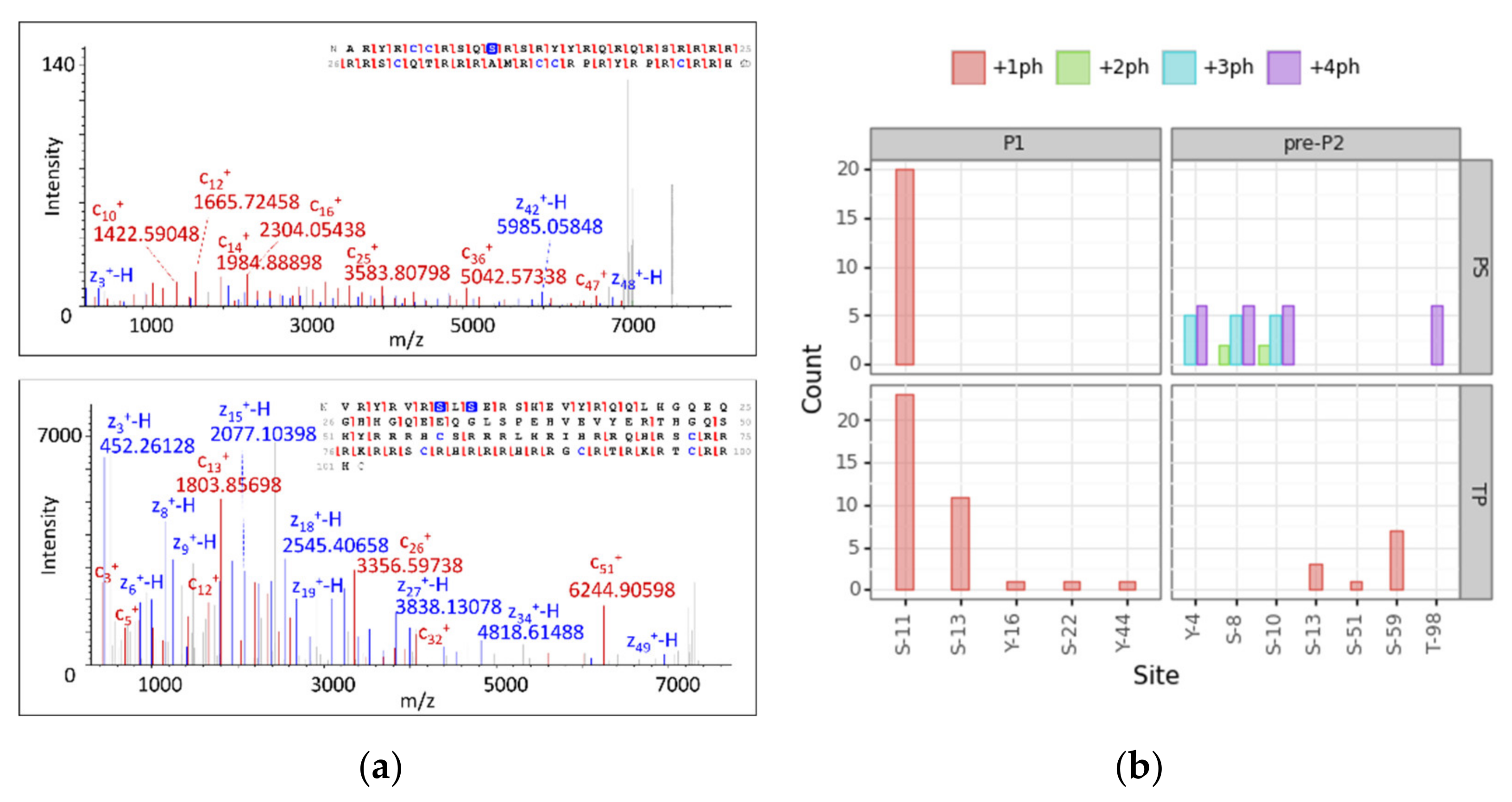
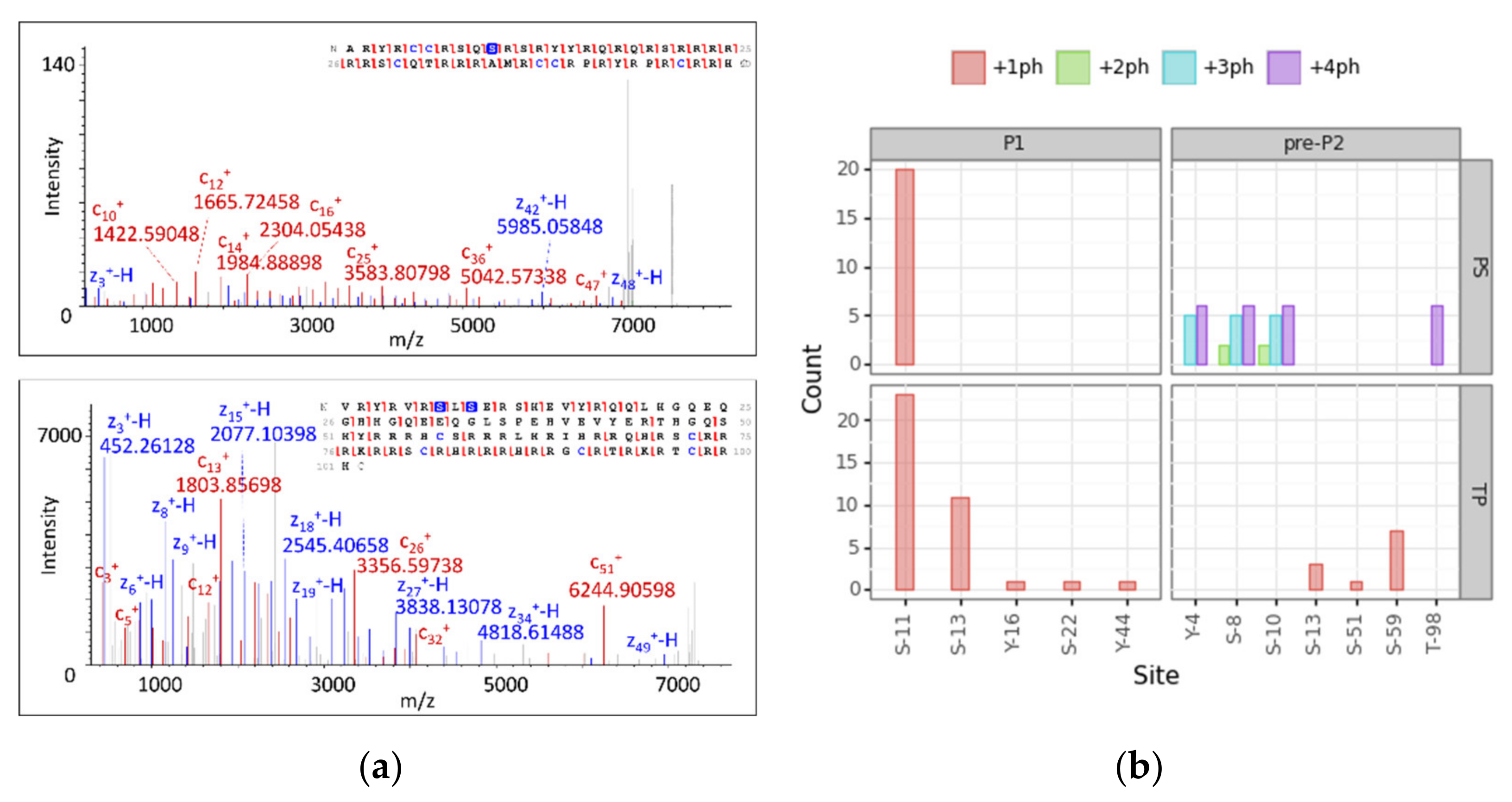
3.4. Modification Site Assignment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oliva, R.; Dixon, G.H. Vertebrate protamine genes and the histone-to-protamine replacement reaction. Prog. Nucleic Acid Res. Mol. Biol. 1991, 40, 25–94. [Google Scholar] [CrossRef] [PubMed]
- Balhorn, R. The protamine family of sperm nuclear proteins. Genome Biol. 2007, 8, 227. [Google Scholar] [CrossRef]
- Castillo, J.; Simon, L.; de Mateo, S.; Lewis, S.; Oliva, R. Protamine/DNA ratios and DNA damage in native and density gradient centrifuged sperm from infertile patients. J. Androl. 2011, 32, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Oliva, R. Protamines and male infertility. Hum. Reprod. Update 2006, 12, 417–435. [Google Scholar] [CrossRef] [Green Version]
- Mezquita, C. Chromatin composition, structure and function in spermatogenesis. Revis. Biol. Celular 1985, 5, 1–124. [Google Scholar]
- Kimmins, S.; Sassone-Corsi, P. Chromatin remodelling and epigenetic features of germ cells. Nature 2005, 434, 583–589. [Google Scholar] [CrossRef]
- Oliva, R.; Krawetz, S.A.; GHD Consortium. Gordon Henry Dixon. 25 March 1930–24 July 2016. Biogr. Mems Fell. R. Soc. 2020. [Google Scholar] [CrossRef]
- Hud, N.V.; Downing, K.H.; Balhorn, R. A constant radius of curvature model for the organization of DNA in toroidal condensates. Proc. Natl. Acad. Sci. USA 1995, 92, 3581–3585. [Google Scholar] [CrossRef] [Green Version]
- Ward, W.S. Function of sperm chromatin structural elements in fertilization and development. Mol. Hum. Reprod. 2010, 16, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Balhorn, R.; Corzett, M.; Mazrimas, J.A. Formation of intraprotamine disulfides in vitro. Arch. Biochem. Biophys. 1992, 296, 384–393. [Google Scholar] [CrossRef]
- Björndahl, L.; Kvist, U. Human sperm chromatin stabilization: A proposed model including zinc bridges. Mol. Hum. Reprod. 2010, 16, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Oliva, R.; Castillo, J. Proteomics and the genetics of sperm chromatin condensation. Asian J. Androl. 2011, 13, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Zini, A.; Agarwal, A. Sperm Chromatin: Biological and Clinical Applications in Male Infertility and Assisted Reproduction; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2011; ISBN 9781441968579. [Google Scholar]
- Lüke, L.; Tourmente, M.; Roldan, E.R.S. Sexual Selection of Protamine 1 in Mammals. Mol. Biol. Evol. 2016, 33, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Kasinsky, H.E.; Eirín-López, J.M.; Ausió, J. Protamines: Structural complexity, evolution and chromatin patterning. Protein Pept. Lett. 2011, 18, 755–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, J.; Estanyol, J.M.; Ballescá, J.L.; Oliva, R. Human sperm chromatin epigenetic potential: Genomics, proteomics, and male infertility. Asian J. Androl. 2015, 17, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Jodar, M.; Oliva, R. The contribution of human sperm proteins to the development and epigenome of the preimplantation embryo. Hum. Reprod. Update 2018, 24, 535–555. [Google Scholar] [CrossRef] [Green Version]
- Gatewood, J.M.; Cook, G.R.; Balhorn, R.; Bradbury, E.M.; Schmid, C.W. Sequence-specific packaging of DNA in human sperm chromatin. Science 1987, 236, 962–964. [Google Scholar] [CrossRef] [PubMed]
- Arpanahi, A.; Brinkworth, M.; Iles, D.; Krawetz, S.A.; Paradowska, A.; Platts, A.E.; Saida, M.; Steger, K.; Tedder, P.; Miller, D. Endonuclease-sensitive regions of human spermatozoal chromatin are highly enriched in promoter and CTCF binding sequences. Genome Res. 2009, 19, 1338–1349. [Google Scholar] [CrossRef] [Green Version]
- Hammoud, S.S.; Nix, D.A.; Zhang, H.; Purwar, J.; Carrell, D.T.; Cairns, B.R. Distinctive chromatin in human sperm packages genes for embryo development. Nature 2009, 460, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Castillo, J.; Amaral, A.; Azpiazu, R.; Vavouri, T.; Estanyol, J.M.; Ballescà, J.L.; Oliva, R. Genomic and proteomic dissection and characterization of the human sperm chromatin. Mol. Hum. Reprod. 2014, 20, 1041–1053. [Google Scholar] [CrossRef] [Green Version]
- Jodar, M.; Oliva, R. Protamine alterations in human spermatozoa. Adv. Exp. Med. Biol. 2014, 791, 83–102. [Google Scholar] [CrossRef] [PubMed]
- Ni, K.; Spiess, A.-N.; Schuppe, H.-C.; Steger, K. The impact of sperm protamine deficiency and sperm DNA damage on human male fertility: A systematic review and meta-analysis. Andrology 2016, 4, 789–799. [Google Scholar] [CrossRef]
- Barrachina, F.; Soler-Ventura, A.; Oliva, R.; Jodar, M. Sperm Nucleoproteins (Histones and Protamines). In A Clinician’s Guide to Sperm DNA and Chromatin Damage; Springer: Cham, Switzerland, 2018; pp. 31–51. [Google Scholar]
- Nanassy, L.; Liu, L.; Griffin, J.; Carrell, D.T. The clinical utility of the protamine 1/protamine 2 ratio in sperm. Protein Pept. Lett. 2011, 18, 772–777. [Google Scholar] [CrossRef]
- De Yebra, L.; Oliva, R. Rapid analysis of mammalian sperm nuclear proteins. Anal. Biochem. 1993, 209, 201–203. [Google Scholar] [CrossRef]
- Soler-Ventura, A.; Castillo, J.; de la Iglesia, A.; Jodar, M.; Barrachina, F.; Ballesca, J.L.; Oliva, R. Mammalian Sperm Protamine Extraction and Analysis: A Step-By-Step Detailed Protocol and Brief Review of Protamine Alterations. Protein Pept. Lett. 2018, 25, 424–433. [Google Scholar] [CrossRef]
- Bogle, O.A.; Kumar, K.; Attardo-Parrinello, C.; Lewis, S.E.M.; Estanyol, J.M.; Ballescà, J.L.; Oliva, R. Identification of protein changes in human spermatozoa throughout the cryopreservation process. Andrology 2017, 5, 10–22. [Google Scholar] [CrossRef] [PubMed]
- De Mateo, S.; Ramos, L.; van der Vlag, J.; de Boer, P.; Oliva, R. Improvement in chromatin maturity of human spermatozoa selected through density gradient centrifugation. Int. J. Androl. 2011, 34, 256–267. [Google Scholar] [CrossRef]
- Wang, T.; Gao, H.; Li, W.; Liu, C. Essential Role of Histone Replacement and Modifications in Male Fertility. Front. Genet. 2019, 10, 962. [Google Scholar] [CrossRef]
- Soler-Ventura, A.; Gay, M.; Jodar, M.; Vilanova, M.; Castillo, J.; Arauz-Garofalo, G.; Villarreal, L.; Ballescà, J.L.; Vilaseca, M.; Oliva, R. Characterization of Human Sperm Protamine Proteoforms through a Combination of Top-Down and Bottom-Up Mass Spectrometry Approaches. J. Proteome Res. 2020, 19, 221–237. [Google Scholar] [CrossRef]
- LeDuc, R.D.; Taylor, G.K.; Kim, Y.-B.; Januszyk, T.E.; Bynum, L.H.; Sola, J.V.; Garavelli, J.S.; Kelleher, N.L. ProSight PTM: An integrated environment for protein identification and characterization by top-down mass spectrometry. Nucleic Acids Res. 2004, 32, W340–W345. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.M.; Pesavento, J.J.; Mizzen, C.A.; Kelleher, N.L.; Pevzner, P.A. Interpreting top-down mass spectra using spectral alignment. Anal. Chem. 2008, 80, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Kou, Q.; Xun, L.; Liu, X. TopPIC: A software tool for top-down mass spectrometry-based proteoform identification and characterization. Bioinformatics 2016, 32, 3495–3497. [Google Scholar] [CrossRef] [PubMed]
- Kou, Q.; Wu, S.; Tolic, N.; Paša-Tolic, L.; Liu, Y.; Liu, X. A mass graph-based approach for the identification of modified proteoforms using top-down tandem mass spectra. Bioinformatics 2017, 33, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Cooper, T.G.; Noonan, E.; von Eckardstein, S.; Auger, J.; Baker, H.W.G.; Behre, H.M.; Haugen, T.B.; Kruger, T.; Wang, C.; Mbizvo, M.T.; et al. World Health Organization reference values for human semen characteristics. Hum. Reprod. Update 2010, 16, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Mengual, L.; Ballescá, J.L.; Ascaso, C.; Oliva, R. Marked differences in protamine content and P1/P2 ratios in sperm cells from percoll fractions between patients and controls. J. Androl. 2003, 24, 438–447. [Google Scholar] [CrossRef]
- Aebersold, R.; Agar, J.N.; Jonathan Amster, I.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- 3.9.1 Documentation. Available online: https://docs.python.org/3/ (accessed on 30 December 2020).
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 3–8 July 2010; pp. 56–61. [Google Scholar]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Kibirige, H.; Lamp, G.; Katins, J.; Gdowding; Austin, O.; Matthias, K.; Funnell, T.; Finkernagel, F.; Arnfred, J.; Blanchard, D.; et al. has2k1/Plotnine; v0.7.1; Zenodo: Genève, Switzerland, 2020. [Google Scholar] [CrossRef]
- Waskom, M.; Gelbart, M.; Botvinnik, O.; Ostblom, J.; Hobson, P.; Lukauskas, S.; Gemperline, D.C.; Augspurger, T.; Halchenko, Y.; Warmenhoven, J.; et al. Mwaskom/Seaborn; v0.11.1; Zenodo: Genève, Switzerland, 2020. [Google Scholar] [CrossRef]
- Schubert, E.; Sander, J.; Ester, M.; Kriegel, H.P.; Xu, X. DBSCAN Revisited, Revisited. ACM Trans. Database Syst. 2017, 42, 1–21. [Google Scholar] [CrossRef]
- Ester, M.; Kriegel, H.-P.; Sander, J.; Xu, X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise; Association for the Advancement of Artificial Intelligence: Palo Alto, CA, USA, 1996. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Van de Ven, B. Bokeh. Available online: https://bokeh.org/ (accessed on 29 January 2021).
- Smith, L.M.; Thomas, P.M.; Shortreed, M.R.; Schaffer, L.V.; Fellers, R.T.; LeDuc, R.D.; Tucholski, T.; Ge, Y.; Agar, J.N.; Anderson, L.C.; et al. A five-level classification system for proteoform identifications. Nat. Methods 2019, 16, 939–940. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, F.; Rousseaux-Prevost, R.; Sautiere, P.; Rousseaux, J. P2 protamines from human sperm are zinc -finger proteins with one CYS2/HIS2 motif. Biochem. Biophys. Res. Commun. 1992, 182, 540–547. [Google Scholar] [CrossRef]
- Roskamp, K.W.; Azim, S.; Kassier, G.; Norton-Baker, B.; Sprague-Piercy, M.A.; Miller, R.J.D.; Martin, R.W. Human γS-Crystallin-Copper Binding Helps Buffer against Aggregation Caused by Oxidative Damage. Biochemistry 2020, 59, 2371–2385. [Google Scholar] [CrossRef]
- Liu, H.; Ponniah, G.; Zhang, H.-M.; Nowak, C.; Neill, A.; Gonzalez-Lopez, N.; Patel, R.; Cheng, G.; Kita, A.Z.; Andrien, B. In vitro and in vivo modifications of recombinant and human IgG antibodies. MAbs 2014, 6, 1145–1154. [Google Scholar] [CrossRef] [Green Version]
- Fellers, R.T.; Greer, J.B.; Early, B.P.; Yu, X.; LeDuc, R.D.; Kelleher, N.L.; Thomas, P.M. ProSight Lite: Graphical software to analyze top-down mass spectrometry data. Proteomics 2015, 15, 1235–1238. [Google Scholar] [CrossRef] [PubMed]
- De Yebra, L.; Ballescà, J.L.; Vanrell, J.A.; Bassas, L.; Oliva, R. Complete selective absence of protamine P2 in humans. J. Biol. Chem. 1993, 268, 10553–10557. [Google Scholar] [CrossRef]
- Torregrosa, N.; Domínguez-Fandos, D.; Camejo, M.I.; Shirley, C.R.; Meistrich, M.L.; Ballescà, J.L.; Oliva, R. Protamine 2 precursors, protamine 1/protamine 2 ratio, DNA integrity and other sperm parameters in infertile patients. Hum. Reprod. 2006, 21, 2084–2089. [Google Scholar] [CrossRef] [PubMed]
- De Mateo, S.; Gázquez, C.; Guimerà, M.; Balasch, J.; Meistrich, M.L.; Ballescà, J.L.; Oliva, R. Protamine 2 precursors (Pre-P2), protamine 1 to protamine 2 ratio (P1/P2), and assisted reproduction outcome. Fertil. Steril. 2009, 91, 715–722. [Google Scholar] [CrossRef]
- LeDuc, R.D.; Fellers, R.T.; Early, B.P.; Greer, J.B.; Thomas, P.M.; Kelleher, N.L. The C-score: A Bayesian framework to sharply improve proteoform scoring in high-throughput top down proteomics. J. Proteome Res. 2014, 13, 3231–3240. [Google Scholar] [CrossRef]
- Gusse, M.; Sautière, P.; Bélaiche, D.; Martinage, A.; Roux, C.; Dadoune, J.P.; Chevaillier, P. Purification and characterization of nuclear basic proteins of human sperm. Biochim. Biophys. Acta 1986, 884, 124–134. [Google Scholar] [CrossRef]
- Pruslin, F.H.; Imesch, E.; Winston, R.; Rodman, T.C. Phosphorylation state of protamines 1 and 2 in human spermatids and spermatozoa. Gamete Res. 1987, 18, 179–190. [Google Scholar] [CrossRef]
- Arkhis, A.; Martinage, A.; Sautiere, P.; Chevaillier, P. Molecular structure of human protamine P4 (HP4), a minor basic protein of human sperm nuclei. Eur. J. Biochem. 1991, 200, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Papoutsopoulou, S.; Nikolakaki, E.; Chalepakis, G.; Kruft, V.; Chevaillier, P.; Giannakouros, T. SR protein-specific kinase 1 is highly expressed in testis and phosphorylates protamine 1. Nucleic Acids Res. 1999, 27, 2972–2980. [Google Scholar] [CrossRef] [Green Version]
- Chirat, F.; Arkhis, A.; Martinage, A.; Jaquinod, M.; Chevaillier, P.; Sautière, P. Phosphorylation of human sperm protamines HP1 and HP2: Identification of phosphorylation sites. Biochim. Biophys. Acta 1993, 1203, 109–114. [Google Scholar] [CrossRef]
- Pirhonen, A.; Linnala-Kankkunen, A.; Mäenpää, P.H. Identification of phosphoseryl residues in protamines from mature mammalian spermatozoa. Biol. Reprod. 1994, 50, 981–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Proteoform | mT (Da) | mC (Da)(mean) | mC (Da)(std) | ΔmCT (Da) | PF Count | PrSM Count | ||
|---|---|---|---|---|---|---|---|---|---|
| PS | TP | PS | TP | ||||||
| P|000 | P1 | 7029.6 | 7029.45 | 1.39 | −0.14 | 2 | 5 | 247 | 206 |
| P|001 | P1 + 1(61) | - | 7090.92 | 1.92 | 61.32 | 5 | 4 | 156 | 97 |
| P|002 | P1 + 1ph | - | 7108.86 | 1.58 | 79.27 | 3 | 5 | 71 | 74 |
| P|003 | P1 + 2(61) | - | 7151.59 | 1.56 | 122 | 6 | 3 | 71 | 46 |
| P|004 | P1 + 1ph + 1(61) | - | 7170.39 | 1.67 | 140.79 | 2 | 3 | 51 | 32 |
| P|005 | P1 + 3(61) | - | 7212.91 | 1.85 | 183.32 | 4 | 3 | 64 | 41 |
| P|006 | P1 + 1ph + 2(61) | - | 7231.82 | 1.66 | 202.23 | 6 | 2 | 29 | 20 |
| P|007 | P1 + 1ph + 3(61) | - | 7293.52 | 2.73 | 263.92 | 4 | 5 | 42 | 29 |
| P|008 | P1 + 2ph + 2(61) | - | 7310.92 | 1.43 | 281.32 | 3 | 2 | 7 | 5 |
| P|009 | HP3 | 7539.07 | 7538.54 | 1.26 | −0.53 | 2 | 2 | 72 | 53 |
| P|010 | HP3 + 1(61) | - | 7599.63 | 1.20 | 60.56 | 1 | 2 | 17 | 13 |
| P|011 | HP2 | 7933.28 | 7932.73 | 1.08 | −0.55 | 1 | 4 | 142 | 119 |
| P|012 | HP2 + 1(61) | - | 7994.03 | 1.35 | 60.75 | 1 | 1 | 64 | 51 |
| P|013 | HP4 + 1(61) | - | 8122.66 | 0.49 | 60.34 | 2 | 16 | ||
| P|014 | HPS2 | 9002.75 | 9002.09 | 1.10 | −0.66 | 2 | 3 | 69 | 54 |
| P|015 | HPS2 + 1(61) | - | 9063.4 | 1.99 | 60.66 | 2 | 3 | 31 | 10 |
| P|016 | HPS1 − 1Q | - | 9172.08 | 1.09 | −128.83 | 3 | 1 | 23 | 19 |
| P|017 | HPS1 +1Pyroglu | - | 9283.32 | 1.27 | −17.59 | 9 | 3 | 26 | 21 |
| P|018 | HPS1 | 9300.91 | 9301.17 | 1.07 | 0.25 | 1 | 3 | 79 | 70 |
| P|019 | HPS1 + 1(61) | - | 9363.59 | 1.51 | 62.67 | 3 | 2 | 23 | 18 |
| P|020 | HPI2 | 10,654.46 | 10,653.97 | 1.01 | −0.49 | 1 | 1 | 6 | 6 |
| P|021 | pre-P2 | 13,196.82 | 13,195.97 | 1.67 | −0.85 | 1 | 4 | 58 | 51 |
| P|022 | pre-P2 + 1ph | - | 13,275.94 | 1.80 | 79.12 | 3 | 4 | 27 | 22 |
| P|023 | pre-P2 + 2ph | - | 13,355.92 | 1.54 | 159.1 | 1 | 5 | 28 | 29 |
| P|024 | pre-P2 + 3ph | - | 13,435.64 | 1.59 | 238.82 | 3 | 3 | 14 | 17 |
| P|025 | pre-P2 + 4ph | - | 13,515.5 | 1.19 | 318.68 | 2 | 2 | 8 | 6 |
| P|026 | Unknown | - | 13,873.22 | 0.78 | - | 2 | 28 | ||
| Unclustered | Unknown | - | - | - | - | 57 | 89 | 166 | 94 |
| ProSight Lite Fragmentation Pattern | Form | Mod. | me (Da) | mt (Da) | Δm (Da) | P-Score | %Res. Cleav. | Match Frag. |
|---|---|---|---|---|---|---|---|---|
(a) Originally assigned by PS BioRep A—TechRep 02—Scan 349 | HPS2 | - | 9063.83 | 9002.75 | 61.084 | 1.10 × 10−91 | 74 | 62 |
(b) Manual reassignation BioRep A—TechRep R02—Scan 349 | HPS2 | +1(61) | 9063.83 | 9063.76 | 0.076 | 7.60·× 10−155 | 82 | 95 |
(c) Originally assigned by PS BioRep A—TechRep R02—Scan 539 | HPS2 | +2ph | 9282.89 | 9162.68 | 120.213 | 7.90·× 10−38 | 52 | 35 |
(d) Manual reassignation BioRep A—TechRep R02—Scan 539 | HPS1 | +1pyroGlu | 9282.89 | 9283.89 | −0.992 | 2.80·× 10−103 | 74 | 75 |
(e) Originally assigned by PS BioRep B—TechRep R04—Scan 545 | HPS1 | +1ph +1AcNterm | 9283.87 | 9422.89 | −139.022 | 9.70·× 10−45 | 59 | 40 |
(f) Manual reassignation BioRep B—TechRep R04—Scan 545 | HPS1 | +1pyroGlu | 9283.87 | 9283.89 | −0.019 | 1.18·× 10−118 | 79 | 84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arauz-Garofalo, G.; Jodar, M.; Vilanova, M.; de la Iglesia Rodriguez, A.; Castillo, J.; Soler-Ventura, A.; Oliva, R.; Vilaseca, M.; Gay, M. Protamine Characterization by Top-Down Proteomics: Boosting Proteoform Identification with DBSCAN. Proteomes 2021, 9, 21. https://doi.org/10.3390/proteomes9020021
Arauz-Garofalo G, Jodar M, Vilanova M, de la Iglesia Rodriguez A, Castillo J, Soler-Ventura A, Oliva R, Vilaseca M, Gay M. Protamine Characterization by Top-Down Proteomics: Boosting Proteoform Identification with DBSCAN. Proteomes. 2021; 9(2):21. https://doi.org/10.3390/proteomes9020021
Chicago/Turabian StyleArauz-Garofalo, Gianluca, Meritxell Jodar, Mar Vilanova, Alberto de la Iglesia Rodriguez, Judit Castillo, Ada Soler-Ventura, Rafael Oliva, Marta Vilaseca, and Marina Gay. 2021. "Protamine Characterization by Top-Down Proteomics: Boosting Proteoform Identification with DBSCAN" Proteomes 9, no. 2: 21. https://doi.org/10.3390/proteomes9020021
APA StyleArauz-Garofalo, G., Jodar, M., Vilanova, M., de la Iglesia Rodriguez, A., Castillo, J., Soler-Ventura, A., Oliva, R., Vilaseca, M., & Gay, M. (2021). Protamine Characterization by Top-Down Proteomics: Boosting Proteoform Identification with DBSCAN. Proteomes, 9(2), 21. https://doi.org/10.3390/proteomes9020021