
A Novel Method for Creating a Synthetic L-DOPA Proteome and In Vitro Evidence of Incorporation



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Tyrosinase Method Optimisation
2.3. Protein Extraction and Processing
2.4. Control Peptide Lysate Preparation
2.4.1. Tyrosinase Conversion Proof of Concept
2.4.2. Microscale Whole Proteome Tyrosinase Conversion
2.5. MALDI Analysis of Synthetic UCP-5 Conversion
2.6. Q-Exactive Plus LC–MS/MS
2.7. Data Analysis
3. Results
3.1. Tyrosinase Conversion of Synthetic Peptide UCP5
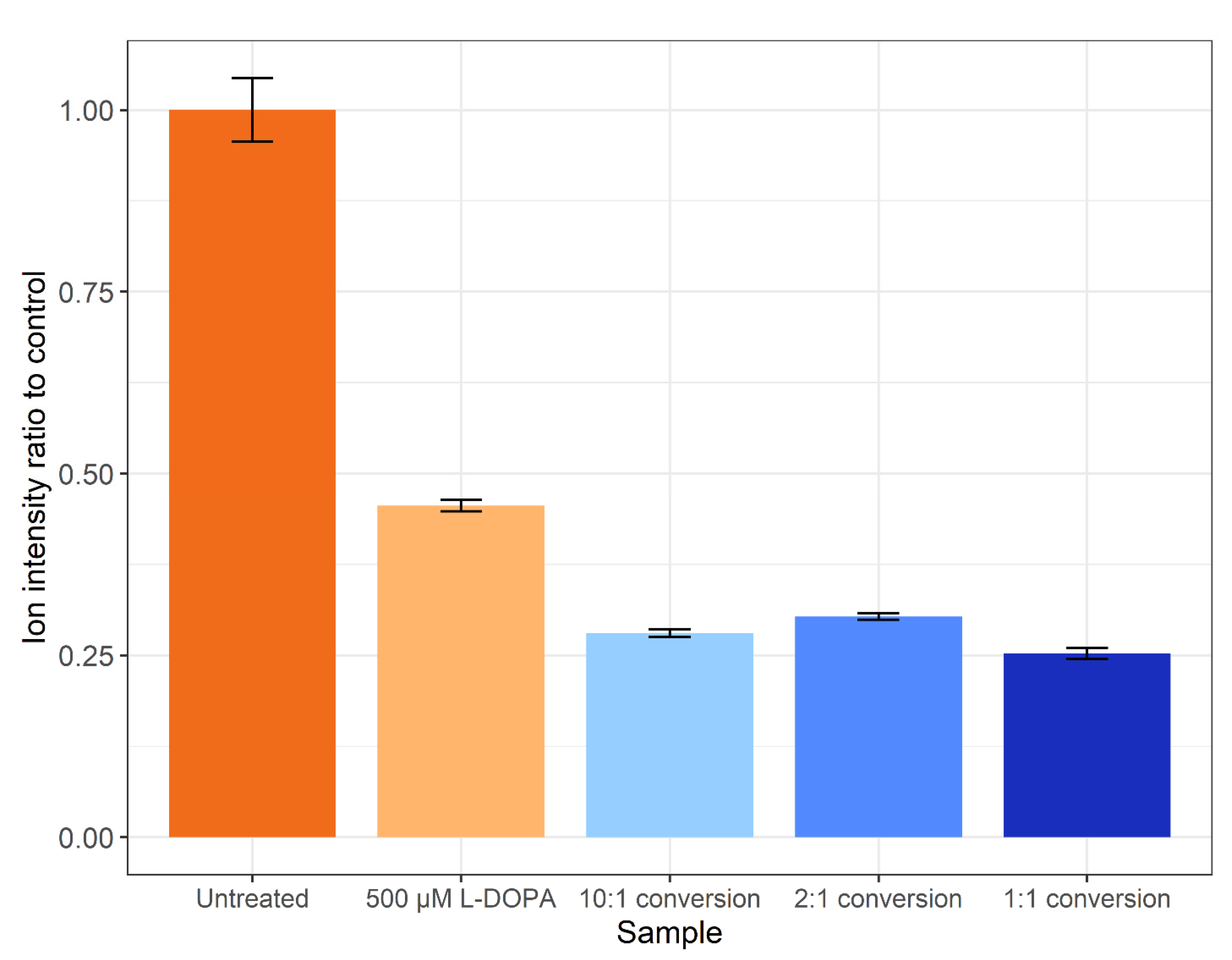
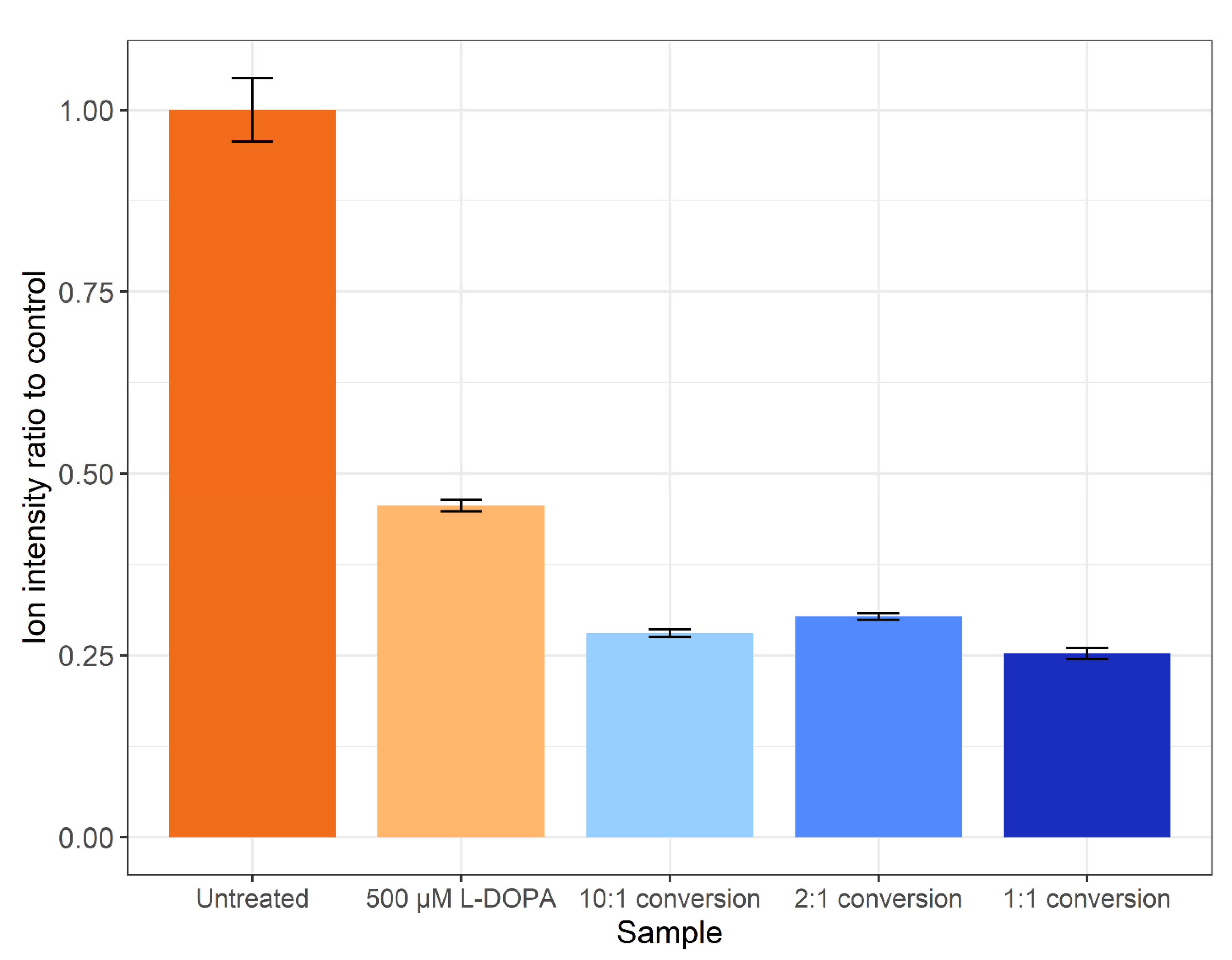
3.2. Proteome Analysis
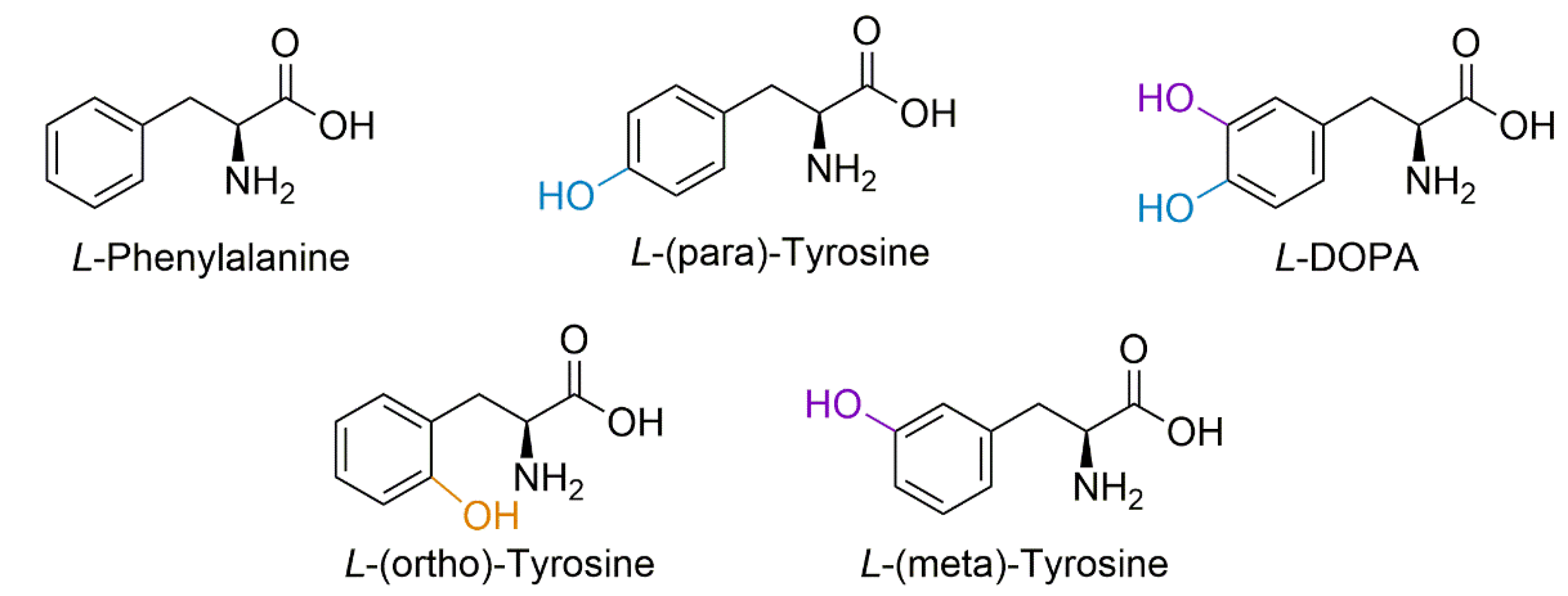
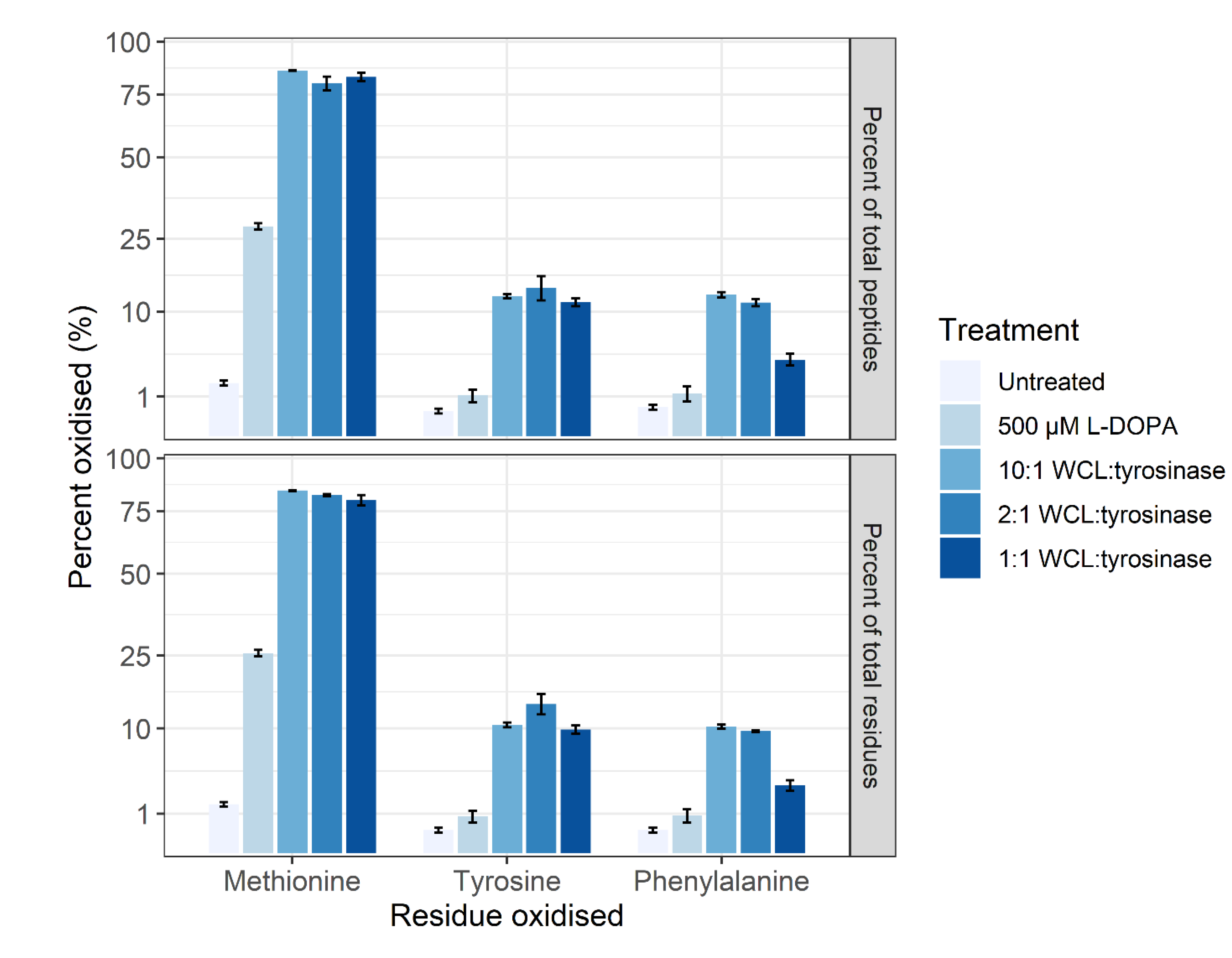
3.3. Proteoform Analysis
3.4. Biological Insights from Pathway Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rascol, O.; Payoux, P.; Ory, F.; Ferreira, J.J.; Brefel-Courbon, C.; Montastruc, J.-L. Limitations of current Parkinson’s disease therapy. Ann. Neurol. 2003, 53, S3–S12. [Google Scholar] [CrossRef]
- Parkkinen, L.; O’Sullivan, S.S.; Kuoppamäki, M.; Collins, C.; Kallis, C.; Holton, J.L.; Williams, D.R.; Revesz, T.; Lees, A.J. Does levodopa accelerate the pathologic process in Parkinson disease brain? Neurology 2011, 77, 1420–1426. [Google Scholar] [CrossRef]
- Alexander, T.; Sortwell, C.E.; Sladek, C.D.; Roth, R.H.; Steece-Collier, K. Comparison of neurotoxicity following repeated administration of L-dopa, D-dopa, and dopamine to embryonic mesencephalic dopamine neurons in cultures derived from Fisher 344 and Sprague-Dawley donors. Cell Transplant. 1997, 6, 309–315. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I.; Ogawa, N. Dopamine- or L-DOPA-induced neurotoxicity: The role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox. Res. 2003, 5, 165–176. [Google Scholar] [CrossRef]
- Chan, S.W.; Dunlop, R.A.; Rowe, A.; Double, K.L.; Rodgers, K.J. L-DOPA is incorporated into brain proteins of patients treated for Parkinson’s disease, inducing toxicity in human neuroblastoma cells in vitro. Exp. Neurol. 2012, 238, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Colamartino, M.; Santoro, M.; Duranti, G.; Sabatini, S.; Ceci, R.; Testa, A.; Padua, L.; Cozzi, R. Evaluation of Levodopa and Carbidopa Antioxidant Activity in Normal Human Lymphocytes In Vitro: Implication for Oxidative Stress in Parkinson’s Disease. Neurotox. Res. 2014, 27, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, S.; Samardzic, K.; Raymond, B.B.; Djordjevic, S.P.; Rodgers, K.J. L-DOPA causes mitochondrial dysfunction in vitro: A novel mechanism of L-DOPA toxicity uncovered. Int. J. Biochem. Cell Biol. 2019, 117, 105624. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.-H.; Park, H.-H.; Choi, N.-Y.; Lee, K.-Y.; Kim, S.; Lee, Y.J.; Kim, H.-T. Protective effects of statins on l-DOPA neurotoxicity due to the activation of phosphatidylinositol 3-kinase and free radical scavenging in PC12 cell culture. Brain Res. 2011, 1370, 53–63. [Google Scholar] [CrossRef]
- Kostrzewa, R.M.; Kostrzewa, J.P.; Brus, R. Neuroprotective and neurotoxic roles of levodopa (L-DOPA) in neurodegenerative disorders relating to Parkinson’s disease. Amino Acids 2002, 23, 57–63. [Google Scholar] [CrossRef]
- Lipski, J.; Nistico, R.; Berretta, N.; Guatteo, E.; Bernardi, G.; Mercuri, N.B. L-DOPA: A scapegoat for accelerated neurodegeneration in Parkinson’s disease? Prog. Neurobiol. 2011, 94, 389–407. [Google Scholar] [CrossRef] [PubMed]
- Mytilineou, C.; Han, S.-K.; Cohen, G. Toxic and Protective Effects of l-DOPA on Mesencephalic Cell Cultures. J. Neurochem. 1993, 61, 1470–1478. [Google Scholar] [CrossRef]
- Park, K.H.; Choi, N.-Y.; Koh, S.-H.; Park, H.-H.; Kim, Y.S.; Kim, M.-J.; Lee, S.-J.; Yu, H.-J.; Lee, K.-Y.; Lee, Y.J.; et al. L-DOPA neurotoxicity is prevented by neuroprotective effects of erythropoietin. NeuroToxicology 2011, 32, 879–887. [Google Scholar] [CrossRef]
- Pedrosa, R.; Soares-Da-Silva, P. Oxidative and non-oxidative mechanisms of neuronal cell death and apoptosis by L-3,4-dihydroxyphenylalanine (L -DOPA) and dopamine. Br. J. Pharmacol. 2002, 137, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Perveen, A.; Khan, H.Y.; Hadi, S.M.; Damanhouri, G.A.; Alharrasi, A.; Tabrez, S. Pro-oxidant DNA Breakage Induced by the Interaction of L-DOPA with Cu(II): A Putative Mechanism of Neurotoxicity. In Genedis 2014: Neurodegeneration; Vlamos, P., Alexiou, A., Eds.; Springer Int Publishing Ag: Cham, Switzerland, 2015; pp. 37–51. [Google Scholar]
- Song, J.; Kim, B.C.; Nguyen, D.-T.T.; Samidurai, M.; Choi, S.-M. Levodopa (L-DOPA) attenuates endoplasmic reticulum stress response and cell death signaling through DRD2 in SH-SY5Y neuronal cells under α-synuclein-induced toxicity. Neuroscience 2017, 358, 336–348. [Google Scholar] [CrossRef]
- Basma, A.N.; Morris, E.J.; Nicklas, W.J.; Geller, H. l-DOPA Cytotoxicity to PC12 Cells in Culture Is via Its Autoxidation. J. Neurochem. 2002, 64, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.J.; Hume, P.M.; Morris, J.G.L.; Dean, R.T. Evidence for L-dopa incorporation into cell proteins in patients treated with levodopa. J. Neurochem. 2006, 98, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.J.; Shiozawa, N. Misincorporation of amino acid analogues into proteins by biosynthesis. Int. J. Biochem. Cell Biol. 2008, 40, 1452–1466. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.-H.; Kim, S.H.; Kim, H.-T. Role of glycogen synthase kinase-3 in l -DOPA-induced neurotoxicity. Expert Opin. Drug Metab. Toxicol. 2009, 5, 1359–1368. [Google Scholar] [CrossRef]
- Jami, M.-S.; Pal, R.; Hoedt, E.; Neubert, T.A.; Larsen, J.P.; Møller, S.G. Proteome analysis reveals roles of L-DOPA in response to oxidative stress in neurons. BMC Neurosci. 2014, 15, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, K.; Headlam, M.J.; Mouradov, D.; Watt, S.J.; Beck, J.L.; Rodgers, K.J.; Dean, R.T.; Huber, T.; Otting, G.; Dixon, N.E. Translational incorporation of L-3,4-dihydroxyphenylalanine into proteins. FEBS J. 2005, 272, 3162–3171. [Google Scholar] [CrossRef]
- Rodgers, K.J.; Dean, R.T. Metabolism of protein-bound DOPA in mammals. Int. J. Biochem. Cell Biol. 2000, 32, 945–955. [Google Scholar] [CrossRef]
- Dunlop, R.A.; Rodgers, K.J.; Dean, R.T. Recent developments in the intracellular degradation of oxidized proteins1,2. Free. Radic. Biol. Med. 2002, 33, 894–906. [Google Scholar] [CrossRef]
- Rodgers, K.J.; Hume, P.M.; Dunlop, R.A.; Dean, R.T. Biosynthesis and turnover of DOPA-containing proteins by human cells. Free. Radic. Biol. Med. 2004, 37, 1756–1764. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, R.A.; Dean, R.T.; Rodgers, K.J. The impact of specific oxidized amino acids on protein turnover in J774 cells. Biochem. J. 2008, 410, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matayatsuk, C.; Poljak, A.; Bustamante, S.; Smythe, G.A.; Kalpravidh, R.W.; Sirankapracha, P.; Fucharoen, S.; Wilairat, P. Quantitative determination of ortho- and meta-tyrosine as biomarkers of protein oxidative damage in beta-thalassemia. Redox Rep. 2007, 12, 219–228. [Google Scholar] [CrossRef]
- Pattison, D.I.; Dean, R.T.; Davies, M.J. Oxidation of DNA, proteins and lipids by DOPA, protein-bound DOPA, and related catechol(amine)s. Toxicology 2002, 177, 23–37. [Google Scholar] [CrossRef]
- Steele, J.; Italiano, C.; Phillips, C.; Violi, J.; Pu, L.; Rodgers, K.; Padula, M. Misincorporation Proteomics Technologies: A Review. Proteomes 2021, 9, 2. [Google Scholar] [CrossRef]
- Searle, B.C.; Pino, L.K.; Egertson, J.D.; Ting, Y.S.; Lawrence, R.T.; MacLean, B.X.; Villén, J.; MacCoss, M.J. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zolg, D.P.; Wilhelm, M.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Delanghe, B.; Bailey, D.J.; Gessulat, S.; Ehrlich, H.-C.; Weininger, M.; et al. Building ProteomeTools based on a complete synthetic human proteome. Nat. Methods 2017, 14, 259–262. [Google Scholar] [CrossRef] [Green Version]
- Walker, E.J.; Bettinger, J.Q.; Welle, K.A.; Hryhorenko, J.R.; Ghaemmaghami, S. Global analysis of methionine oxidation provides a census of folding stabilities for the human proteome. Proc. Natl. Acad. Sci. USA 2019, 116, 6081–6090. [Google Scholar] [CrossRef] [Green Version]
- Bettinger, J.Q.; Welle, K.A.; Hryhorenko, J.R.; Ghaemmaghami, S. Quantitative Analysis of in Vivo Methionine Oxidation of the Human Proteome. J. Proteome Res. 2020, 19, 624–633. [Google Scholar] [CrossRef]
- Carballo-Carbajal, I.; Laguna, A.; Romero-Giménez, J.; Cuadros, T.; Bové, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Peñuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Cieńska, M.; Labus, K.; Lewańczuk, M.; Koźlecki, T.; Liesiene, J.; Bryjak, J. Effective L-Tyrosine Hydroxylation by Native and Immobilized Tyrosinase. PLoS ONE 2016, 11, e0164213. [Google Scholar] [CrossRef]
- Ito, S.; Kato, T.; Shinpo, K.; Fujita, K. Oxidation of tyrosine residues in proteins by tyrosinase. Formation of protein-bonded 3,4-dihydroxyphenylalanine and 5-S-cysteinyl-3,4-dihydroxyphenylalanine. Biochem. J. 1984, 222, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Vincent, S.G.; Cunningham, P.R.; Stephens, N.L.; Halayko, A.J.; Fisher, J.T. Quantitative densitometry of proteins stained with Coomassie Blue using a Hewlett Packard scanjet scanner and Scanplot software. Electrophoresis 1997, 18, 67–71. [Google Scholar] [CrossRef]
- Roediger, B.; Lee, Q.; Tikoo, S.; Cobbin, J.C.; Henderson, J.M.; Jormakka, M.; O’Rourke, M.B.; Padula, M.P.; Pinello, N.; Henry, M.; et al. An Atypical Parvovirus Drives Chronic Tubulointerstitial Nephropathy and Kidney Fibrosis. Cell 2018, 175, 530–543. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.W.; Ho, J.W.; Liu, H.-F.; So, D.H.; Tse, Z.H.; Chan, K.-H.; Ramsden, D.B.; Ho, S.-L. Mitochondrial neuronal uncoupling proteins: A target for potential disease-modification in Parkinson’s disease. Transl. Neurodegener. 2012, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Tran, N.H.; Qiao, R.; Xin, L.; Chen, X.; Liu, C.; Zhang, X.; Shan, B.; Ghodsi, A.; Li, M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat. Methods 2019, 16, 63–66. [Google Scholar] [CrossRef]
- Bloom, J.I.; Triantafyllidis, A.; Burton, P.; Infusini, G.; Webb, A.I. Mass Dynamics 1.0: A streamlined, web-based environment for analyzing, sharing and integrating Label-Free Data. bioRxiv 2021. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Hwang, D.S.; Gim, Y.; Yoo, H.J.; Cha, H.J. Practical recombinant hybrid mussel bioadhesive fp-151. Biomaterials 2007, 28, 3560–3568. [Google Scholar] [CrossRef]
- Do, H.; Kang, E.; Yang, B.; Cha, H.J.; Choi, Y.S. A tyrosinase, mTyr-CNK, that is functionally available as a monophenol monooxygenase. Sci. Rep. 2017, 7, 17267. [Google Scholar] [CrossRef] [Green Version]
- Burzio, L.A.; Waite, J. The Other Topa: Formation of 3,4,5-Trihydroxyphenylalanine in Peptides. Anal. Biochem. 2002, 306, 108–114. [Google Scholar] [CrossRef]
- Rodgers, K.J.; Wang, H.; Fu, S.; Dean, R.T. Biosynthetic incorporation of oxidized amino acids into proteins and their cellular proteolysis. Free Radic. Biol. Med. 2002, 32, 766–775. [Google Scholar] [CrossRef]
- Ghesquière, B.; Jonckheere, V.; Colaert, N.; Van Durme, J.; Timmerman, E.; Goethals, M.; Schymkowitz, J.; Rousseau, F.; Vandekerckhove, J.; Gevaert, K. Redox Proteomics of Protein-bound Methionine Oxidation. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, R.A.; Brunk, U.T.; Rodgers, K.J. Proteins containing oxidized amino acids induce apoptosis in human monocytes. Biochem. J. 2011, 435, 207–216. [Google Scholar] [CrossRef]
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative DNA Damage in the Parkinsonian Brain: An Apparent Selective Increase in 8-Hydroxyguanine Levels in Substantia Nigra. J. Neurochem. 2002, 69, 1196–1203. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxidative Med. Cell. Longev. 2019, 2019, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Rees, H.D.; Weintraub, S.T.; Levey, A.I.; Chin, L.-S.; Li, L. Oxidative Modifications and Aggregation of Cu,Zn-Superoxide Dismutase Associated with Alzheimer and Parkinson Diseases. J. Biol. Chem. 2005, 280, 11648–11655. [Google Scholar] [CrossRef] [Green Version]
- Corona, J.C.; Duchen, M.R. Impaired mitochondrial homeostasis and neurodegeneration: Towards new therapeutic targets? J. Bioenerg. Biomembr. 2015, 47, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Perentis, R.J.; Caldwell, G.; Caldwell, K.A. Gene-by-environment interactions that disrupt mitochondrial homeostasis cause neurodegeneration in C. elegans Parkinson’s models. Cell Death Dis. 2018, 9, 555. [Google Scholar] [CrossRef] [Green Version]
- Kang, I.; Chu, C.T.; Kaufman, B.A. The mitochondrial transcription factor TFAM in neurodegeneration: Emerging evidence and mechanisms. FEBS Lett. 2018, 592, 793–811. [Google Scholar] [CrossRef] [Green Version]
- Markaki, M.; Tavernarakis, N. Mitochondrial turnover and homeostasis in ageing and neurodegeneration. FEBS Lett. 2020, 594, 2370–2379. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Mena, M.; Casarejos, M.; Paíno, C.; De Yébenes, J. Toxic effects of L-DOPA on mesencephalic cell cultures: Protection with antioxidants. Brain Res. 1995, 682, 133–143. [Google Scholar] [CrossRef]
- Toth, C.; Breithaupt, K.; Ge, S.; Duan, Y.; Terris, J.M.; Thiessen, A.; Wiebe, S.; Zochodne, D.W.; Suchowersky, O. Levodopa, methylmalonic acid, and neuropathy in idiopathic Parkinson disease. Ann. Neurol. 2010, 68, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Marek, K. Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar]
- Searle, B.C.; Swearingen, K.E.; Barnes, C.A.; Schmidt, T.; Gessulat, S.; Küster, B.; Wilhelm, M. Generating high quality libraries for DIA MS with empirically corrected peptide predictions. Nat. Commun. 2020, 11, 1548. [Google Scholar] [CrossRef] [Green Version]
- Ping, L.; Kundinger, S.R.; Duong, D.M.; Yin, L.; Gearing, M.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Global quantitative analysis of the human brain proteome and phosphoproteome in Alzheimer’s disease. Sci. Data 2020, 7, 315. [Google Scholar] [CrossRef]
- Su, Z.; Burchfield, J.G.; Yang, P.; Humphrey, S.J.; Yang, G.; Francis, D.; Yasmin, S.; Shin, S.-Y.; Norris, D.M.; Kearney, A.L.; et al. Global redox proteome and phosphoproteome analysis reveals redox switch in Akt. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification | Control | Treatment | Conversion |
|---|---|---|---|
| peptides | 34 | 101 | 532 |
| proteins | 37 | 75 | 317 |
| Localisation | Sequence | Protein Accession | PTM Site Ascore |
|---|---|---|---|
| Control | AAGGDGDDSLY(+15.99)PIAVLIDELR | P30154|2AAB_HUMAN | Y11:L-DOPA:1000.00 |
| DOPA | DLYANTVLSGGTTMY(+15.99)PGIADR | P60709|ACTB_HUMAN:P63261|ACTG_HUMAN | Y15:L-DOPA:27.62 |
| DOPA | GINPDEAVAY(+15.99) GAAVQAGVLSGDQ(+0.98)DTGDLVLLDVC(+71.04)PLTLGIETVGGVMTK | P11021|GRP78_HUMAN | Y10:L-DOPA:1000.00;Q23:Deamidation (NQ):0.00;C34:Propionamide:1000.00 |
| DOPA | DLYAN(+.98)TVLSGGTTMY(+15.99) PGIADR | P60709|ACTB_HUMAN:P63261|ACTG_HUMAN | N5:Deamidation (NQ):1000.00;Y15:L-DOPA:10.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steele, J.R.; Strange, N.; Rodgers, K.J.; Padula, M.P. A Novel Method for Creating a Synthetic L-DOPA Proteome and In Vitro Evidence of Incorporation. Proteomes 2021, 9, 24. https://doi.org/10.3390/proteomes9020024
Steele JR, Strange N, Rodgers KJ, Padula MP. A Novel Method for Creating a Synthetic L-DOPA Proteome and In Vitro Evidence of Incorporation. Proteomes. 2021; 9(2):24. https://doi.org/10.3390/proteomes9020024
Chicago/Turabian StyleSteele, Joel Ricky, Natalie Strange, Kenneth J. Rodgers, and Matthew P. Padula. 2021. "A Novel Method for Creating a Synthetic L-DOPA Proteome and In Vitro Evidence of Incorporation" Proteomes 9, no. 2: 24. https://doi.org/10.3390/proteomes9020024
APA StyleSteele, J. R., Strange, N., Rodgers, K. J., & Padula, M. P. (2021). A Novel Method for Creating a Synthetic L-DOPA Proteome and In Vitro Evidence of Incorporation. Proteomes, 9(2), 24. https://doi.org/10.3390/proteomes9020024