
Proteomes Are of Proteoforms: Embracing the Complexity



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract

1. Introduction
2. What Is a Proteome?
3. Proteomics
4. Discovery Proteomics
4.1. Bottom-up
4.2. Top-Down
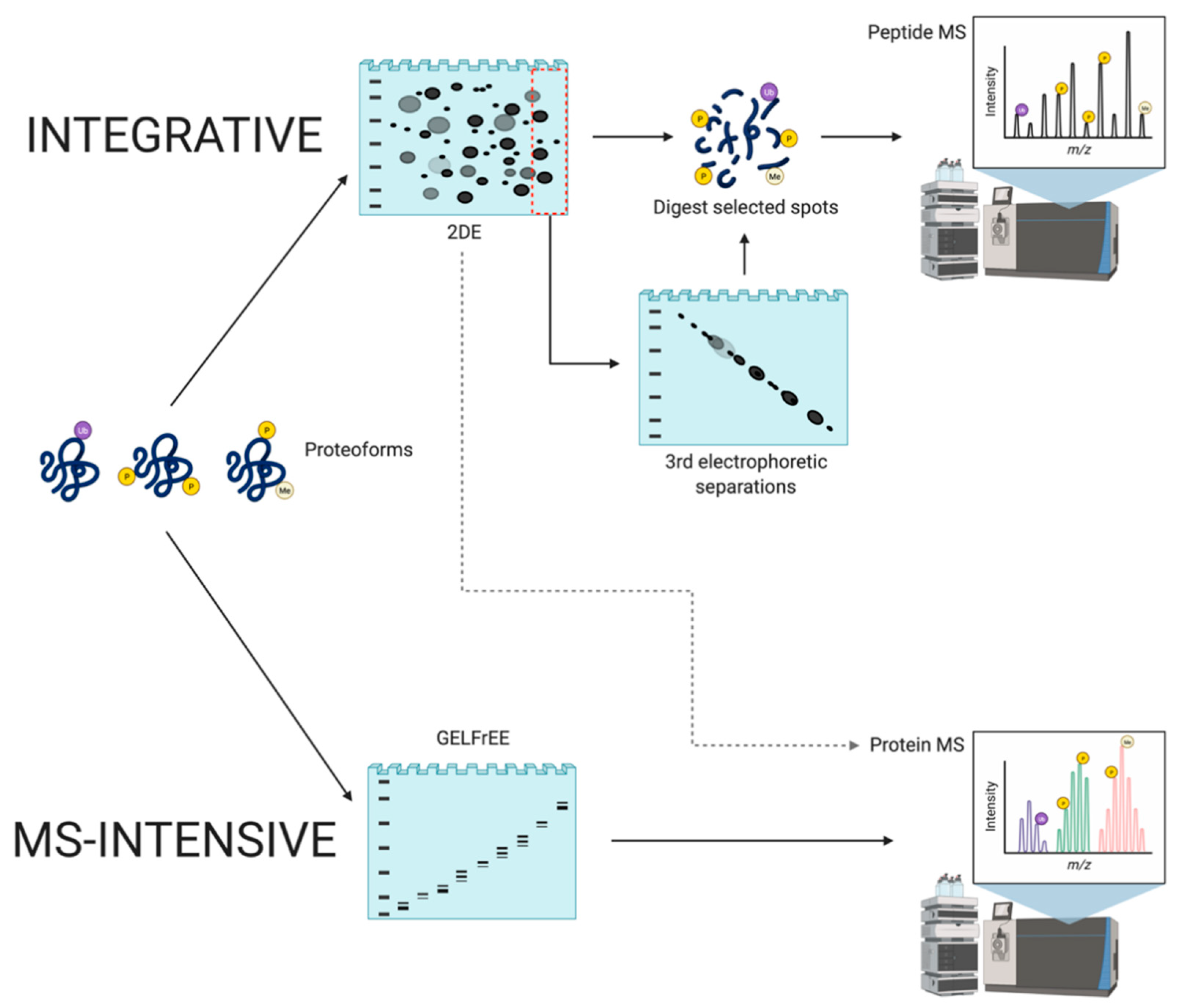
4.2.1. Integrative
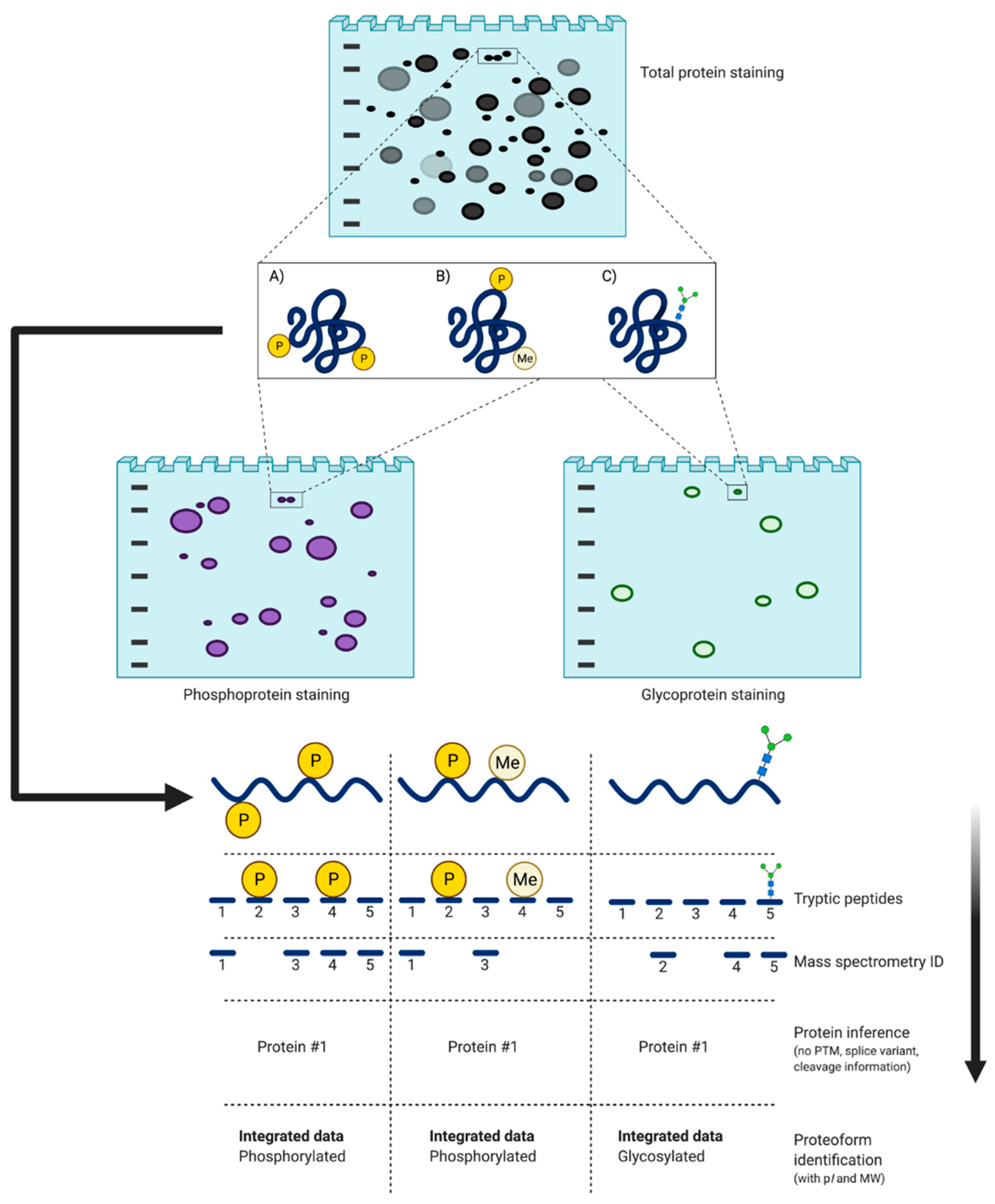
4.2.2. 2DE: Addressing the Dogma
4.2.3. MS-Intensive
4.2.4. So, What Does Top-Down Really Mean?
4.3. Additional Analytical Variations on Peptide MS Analyses
5. Targeted Proteomics
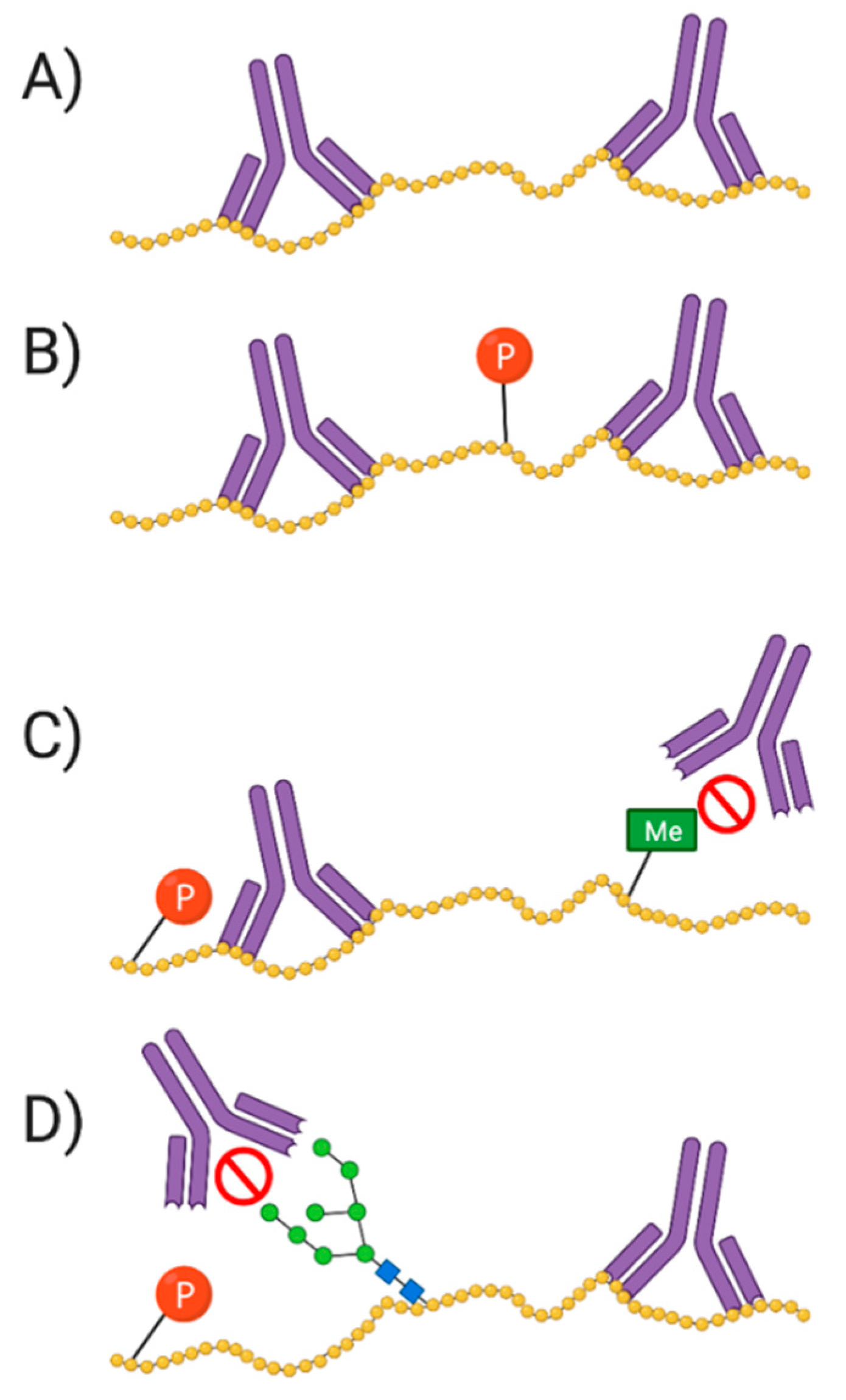
5.1. Antibodies
5.2. Immunoassays
5.3. Mass Spectromtery
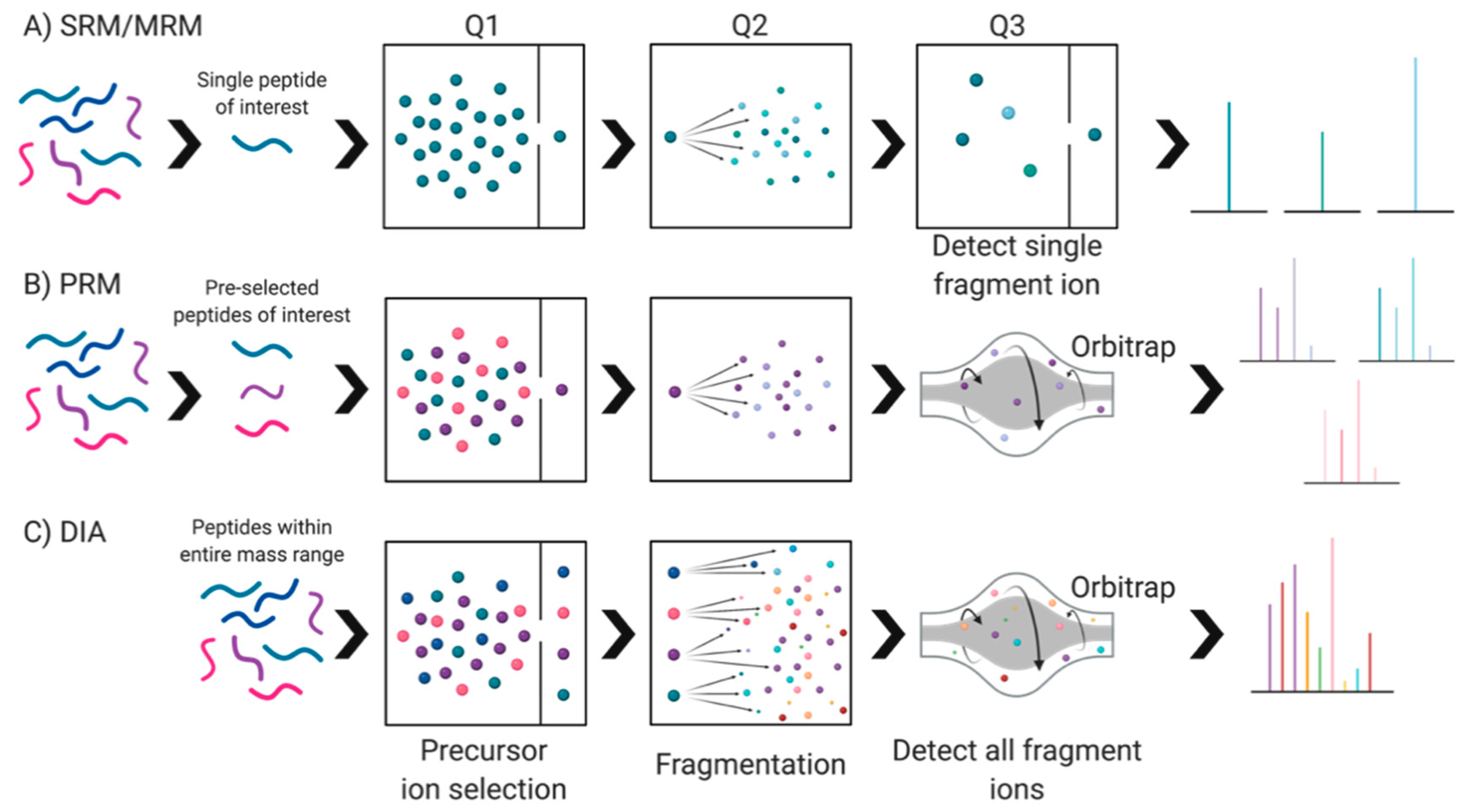
5.3.1. Label-Based
5.3.2. Label-Free
6. What Next?
For a successful technology, reality must take precedence over public relations, for Nature cannot be fooled.Richard P. Feynman
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Fey, S.J.; Larsen, P.M. 2D or not 2D. Curr. Opin. Chem. Biol. 2001, 5, 26–33. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Sanchez, J.-C.; Gooley, A.A.; Appel, R.D.; Humphery-Smith, I.; Hochstrasser, D.F.; Williams, K.L. Progress with proteome projects: Why all proteins expressed by a genome should be identified and how to do it. Biotechnol. Genet. Eng. Rev. 1996, 13, 19–50. [Google Scholar] [CrossRef]
- Duncan, M.W.; Yergey, A.L.; Gale, P.J.; Kate, Y. Quantifying proteins by mass spectrometry. LC-GC N. Am. 2014, 32, 726–735. [Google Scholar]
- Jungblut, P.R.; Holzhütter, H.; Apweiler, R.; Schlüter, H. The speciation of the proteome. Chem. Cent. J. 2008, 2, 16. [Google Scholar] [CrossRef]
- Jungblut, P.R.; Thiede, B.; Schlüter, H. Towards deciphering proteomes via the proteoform, protein speciation, moonlighting and protein code concepts. J. Proteom. 2016, 134, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Timp, W.; Timp, G. Beyond mass spectrometry, the next step in proteomics. Sci. Adv. 2020, 6, eaax8978. [Google Scholar] [CrossRef] [PubMed]
- Vanderperre, B.; Lucier, J.-F.; Bissonnette, C.; Motard, J.; Tremblay, G.; Vanderperre, S.; Wisztorski, M.; Salzet, M.; Boisvert, F.-M.; Roucou, X. Direct detection of alternative open reading frames translation products in human significantly expands the proteome. PLoS ONE 2013, 8, e70698. [Google Scholar] [CrossRef] [PubMed]
- Delcourt, V.; Brunelle, M.; Roy, A.V.; Jacques, J.-F.; Salzet, M.; Fournier, I.; Roucou, X. The protein coded by a short open reading frame, not by the annotated coding sequence, is the main gene product of the dual-coding gene MIEF1. Mol. Cell. Proteom. 2018, 17, 2402–2411. [Google Scholar] [CrossRef]
- Brunet, M.A.; Brunelle, M.; Lucier, J.-F.; Delcourt, V.; Levesque, M.; Grenier, F.; Samandi, S.; Leblanc, S.; Aguilar, J.-D.; Dufour, P.; et al. OpenProt: A more comprehensive guide to explore eukaryotic coding potential and proteomes. Nucleic Acids Res. 2019, 47, D403–D410. [Google Scholar] [CrossRef]
- Randles, L.G.; Dawes, G.J.S.; Wensley, B.G.; Steward, A.; Nickson, A.A.; Clarke, J. Understanding pathogenic single-nucleotide polymorphisms in multidomain proteins—studies of isolated domains are not enough. FEBS J. 2013, 280, 1018–1027. [Google Scholar] [CrossRef][Green Version]
- Robert, F.; Pelletier, J. Exploring the impact of single-nucleotide polymorphisms on translation. Front. Genet. 2018, 9, 507. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Habib, G.; Yang, C.Y.; Gu, Z.W.; Lee, B.R.; Weng, S.A.; Silberman, S.R.; Cai, S.J.; Deslypere, J.P.; Rosseneu, M. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science 1987, 238, 363–366. [Google Scholar] [CrossRef]
- Hospattankar, A.V.; Higuchi, K.; Law, S.W.; Meglin, N.; Brewer, H.B. Identification of a novel in-frame translational stop codon in human intestine ApoB mRNA. Biochem. Biophys. Res. Commun. 1987, 148, 279–285. [Google Scholar] [CrossRef]
- Powell, L.M.; Wallis, S.C.; Pease, R.J.; Edwards, Y.H.; Knott, T.J.; Scott, J. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell 1987, 50, 831–840. [Google Scholar] [CrossRef]
- Kim, D.D.Y.; Kim, T.T.Y.; Walsh, T.; Kobayashi, Y.; Matise, T.C.; Buyske, S.; Gabriel, A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004, 14, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, A.; de Magalhaes, J.P.; Kraytsberg, Y.; Richfield, E.K.; Levanon, E.Y.; Khrapko, K. Age-related gene-specific changes of A-to-I mRNA editing in the human brain. Mech. Ageing Dev. 2010, 131, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Knoop, V. When you can’t trust the DNA: RNA editing changes transcript sequences. Cell. Mol. Life Sci. 2011, 68, 567–586. [Google Scholar] [CrossRef]
- Schlüter, H.; Apweiler, R.; Holzhütter, H.-G.; Jungblut, P.R. Finding one’s way in proteomics: A protein species nomenclature. Chem. Cent. J. 2009, 3, 11. [Google Scholar] [CrossRef]
- Gorr, T.A.; Vogel, J. Western blotting revisited: Critical perusal of underappreciated technical issues. Proteom. Clin. Appl. 2015, 9, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Issaq, H.J.; Veenstra, T.D. The role of electrophoresis in disease biomarker discovery. Electrophoresis 2007, 28, 1980–1988. [Google Scholar] [CrossRef]
- Anjo, S.I.; Santa, C.; Manadas, B. SWATH-MS as a tool for biomarker discovery: From basic research to clinical applications. Proteomics 2017, 17, 1600278. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Y.; Lin, S.; Deng, W.; Peng, D.; Cui, Q.; Xue, Y. PTMD: A database of human disease-associated post-translational modifications. Genom. Proteom. Bioinform. 2018, 16, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef]
- UNIMOD: Protein Identifications for Mass Spectrometry. Available online: http://www.unimod.org/modifications_list.php? (accessed on 1 July 2021).
- Chapman, J.D.; Goodlett, D.R.; Masselon, C.D. Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass Spectrom. Rev. 2013, 33, 452–470. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Kelleher, N.L. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef]
- Oliveira, B.M.; Coorssen, J.R.; Martins-de-Souza, D. 2DE: The phoenix of proteomics. J. Proteom. 2014, 104, 140–150. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Yergey, A. Proteomics is analytical chemistry: Fitness-for-purpose in the application of top-down and bottom-up analyses. Proteomes 2015, 3, 440–453. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Li, B.; Zhan, X.; Schlüter, H.; Jungblut, P.R.; Coorssen, J.R. Innovating the concept and practice of two-dimensional gel electrophoresis in the analysis of proteomes at the proteoform level. Proteomes 2019, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, C.J. Moonlighting proteins. Trends Biochem. Sci. 1999, 24, 8–11. [Google Scholar] [CrossRef]
- Jeffery, C.J. Why study moonlighting proteins? Front. Genet. 2015, 6, 211. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, C.J. Protein species and moonlighting proteins: Very small changes in a protein’s covalent structure can change its biochemical function. J. Proteom. 2016, 134, 19–24. [Google Scholar] [CrossRef]
- Jeffery, C.J. Protein moonlighting: What is it, and why is it important? Philos. Transactions. Biol. Sci. 2018, 373, 20160523. [Google Scholar] [CrossRef]
- Jaffe, E.K.; Lawrence, S.H. The morpheein model of allostery: Evaluating proteins as potential morpheeins. Methods Mol. Biol. 2012, 796, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.K. Wrangling shape-shifting morpheeins to tackle disease and approach drug discovery. Front. Mol. Biosci. 2020, 7, 582966. [Google Scholar] [CrossRef]
- Chen, J.; Brunner, A.-D.; Cogan, J.Z.; Nuñez, J.K.; Fields, A.P.; Adamson, B.; Itzhak, D.N.; Li, J.Y.; Mann, M.; Leonetti, M.D.; et al. Pervasive functional translation of noncanonical human open reading frames. Science 2020, 367, 1140–1146. [Google Scholar] [CrossRef]
- Frith, M.C.; Forrest, A.R.; Nourbakhsh, E.; Pang, K.C.; Kai, C.; Kawai, J.; Carninci, P.; Hayashizaki, Y.; Bailey, T.L.; Grimmond, S.M. The abundance of short proteins in the mammalian proteome. PLoS Genet. 2006, 2, e52. [Google Scholar] [CrossRef]
- Eguen, T.; Straub, D.; Graeff, M.; Wenkel, S. MicroProteins: Small size—Big impact. Trends Plant Sci. 2015, 20, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Khitun, A.; Na, Z.; Dumitrescu, D.G.; Kubica, M.; Olatunji, E.; Slavoff, S.A. Comparative proteomic profiling of unannotated microproteins and alternative proteins in human cell lines. J. Proteome Res. 2020, 19, 3418–3426. [Google Scholar] [CrossRef]
- Schlesinger, D.; Elsässer, S.J. Revisiting sORFs: Overcoming challenges to identify and characterize functional microproteins. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Chandramouly, G.; Zhao, J.; McDevitt, S.; Rusanov, T.; Hoang, T.; Borisonnik, N.; Treddinick, T.; Lopezcolorado, F.W.; Kent, T.; Siddique, L.A.; et al. Polθ reverse transcribes RNA and promotes RNA-templated DNA repair. Sci. Adv. 2021, 7, eabf1771. [Google Scholar] [CrossRef] [PubMed]
- Gygi, S.P.; Rochon, Y.; Franza, B.R.; Aebersold, R. Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 1999, 19, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Chan, J.; Fang, H.; Robinson, A.E.; Anand, S.; Gabriel, S.; Graubert, A.; Hadley, K.; Meier, S.R.; Nedzel, J.L.; et al. A quantitative proteome map of the human body. Cell 2020, 183, 269–283.e219. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Beyer, A.; Aebersold, R. On the dependency of cellular protein levels on mRNA abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Wanjek, C. Systems Biology as Defined by NIH. NIH Catal. 2011, 19, 10–12. [Google Scholar]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis—a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef]
- Elliott, M.H.; Smith, D.S.; Parker, C.E.; Borchers, C. Current trends in quantitative proteomics. J. Mass Spectrom. 2009, 44, 1637–1660. [Google Scholar] [CrossRef]
- Naryzhny, S. Towards the full realization of 2DE power. Proteomes 2016, 4, 33. [Google Scholar] [CrossRef]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The size of the human proteome: The width and depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef] [PubMed]
- Marcus, K.; Rabilloud, T. How do the different proteomic strategies cope with the complexity of biological regulations in a multi-omic world? Critical appraisal and suggestions for improvements. Proteomes 2020, 8, 23. [Google Scholar] [CrossRef]
- Marcus, K.; Lelong, C.; Rabilloud, T. What room for two-dimensional gel-based proteomics in a shotgun proteomics world? Proteomes 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S. Inventory of proteoforms as a current challenge of proteomics: Some technical aspects. J. Proteom. 2019, 191, 22–28. [Google Scholar] [CrossRef]
- Lander, E.; Linton, L.; Birren, B.; Nusbaum, C.; Zody, M.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; Fitzhugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.; Adams, M.D.; Myers, E.; Li, P.; Mural, R.; Sutton, G.; Smith, H.; Yandell, M.; Evans, C.; Holt, R.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Abascal, F.; Juan, D.; Jungreis, I.; Martinez, L.; Rigau, M.; Jose, M.R.; Vazquez, J.; Tress, M.L. Loose ends: Almost one in five human genes still have unresolved coding status. Nucleic Acids Res. 2018, 46, 7070–7084. [Google Scholar] [CrossRef]
- Sherman, R.M.; Forman, J.; Antonescu, V.; Puiu, D.; Daya, M.; Rafaels, N.; Boorgula, M.P.; Chavan, S.; Vergara, C.; Ortega, V.E.; et al. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet. 2019, 51, 30–35. [Google Scholar] [CrossRef]
- Sherman, R.M.; Salzberg, S.L. Pan-genomics in the human genome era. Nat. Rev. Genet. 2020, 21, 243–254. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. bioRxiv 2021. [Google Scholar] [CrossRef]
- Clamp, M.; Fry, B.; Kamal, M.; Xie, X.; Cuff, J.; Lin, M.F.; Kellis, M.; Lindblad-Toh, K.; Lander, E.S. Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl. Acad. Sci. USA 2007, 104, 19428–19433. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu. Rev. Biochem. 2011, 80, 273–299. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.D.; Aebersold, R.H. Proteomics: The first decade and beyond. Nat. Genet. Suppl. 2003, 33, 311–323. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Aebersold, R. Interpretation of shotgun proteomic data: The protein inference problem. Mol. Cell. Proteom. 2005, 4, 1419–1440. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Blank, P.S.; Albertorio, F.; Bezrukov, L.; Kolosova, I.; Backlund, P.S.; Zimmerberg, J. Quantitative femto- to attomole immunodetection of regulated secretory vesicle proteins critical to exocytosis. Anal. Biochem. 2002, 307, 54–62. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Blank, P.S.; Albertorio, F.; Bezrukov, L.; Kolosova, I.; Chen, X.; Backlund, J.P.S.; Zimmerberg, J. Regulated secretion: SNARE density, vesicle fusion and calcium dependence. J. Cell Sci. 2003, 116, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Domon, B.; Aebersold, R. Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 2010, 28, 710–721. [Google Scholar] [CrossRef]
- Borràs, E.; Sabidó, E. What is targeted proteomics? A concise revision of targeted acquisition and targeted data analysis in mass spectrometry. Proteomics 2017, 17, 1700180. [Google Scholar] [CrossRef] [PubMed]
- Moradian, A.; Kalli, A.; Sweredoski, M.J.; Hess, S. The top-down, middle-down, and bottom-up mass spectrometry approaches for characterization of histone variants and their post-translational modifications. Proteomics 2014, 14, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Resing, K.A.; Ahn, N.G. Proteomics strategies for protein identification. FEBS Lett. 2005, 579, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, S.; Nice, E.C.; Deutsch, E.W.; Lane, L.; Omenn, G.S.; Pennington, S.R.; Paik, Y.-K.; Overall, C.M.; Corrales, F.J.; Cristea, I.M.; et al. A high-stringency blueprint of the human proteome. Nat. Commun. 2020, 11, 5301. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S.; Lane, L.; Overall, C.M.; Cristea, I.M.; Corrales, F.J.; Lindskog, C.; Paik, Y.-K.; Van Eyk, J.E.; Liu, S.; Pennington, S.R.; et al. Research on the human proteome reaches a major milestone: >90% of predicted human proteins now credibly detected, according to the HUPO Human Proteome Project. J. Proteome Res. 2020, 19, 4735–4746. [Google Scholar] [CrossRef]
- Couso, J.P. Finding smORFs: Getting closer. Genome Biol. 2015, 16, 189. [Google Scholar] [CrossRef]
- Gilmore, J.M.; Washburn, M.P. Advances in shotgun proteomics and the analysis of membrane proteomes. J. Proteom. 2010, 73, 2078–2091. [Google Scholar] [CrossRef] [PubMed]
- Matallana-Surget, S.; Leroy, B.; Wattiez, R. Shotgun proteomics: Concept, key points and data mining. Expert Rev. Proteom. 2010, 7, 5–7. [Google Scholar] [CrossRef]
- Carvalho, P.C.; Han, X.; Xu, T.; Cociorva, D.; Carvalho Mda, G.; Barbosa, V.C.; Yates, J.R., 3rd. XDIA: Improving on the label-free data-independent analysis. Bioinformatics 2010, 26, 847–848. [Google Scholar] [CrossRef]
- Bell, A.J.; Deutsch, E.W.; Au, C.E.; Kearney, R.E.; Beavis, R.; Sechi, S.; Nilsson, T.; Bergeron, J.J.M. A HUPO test sample study reveals common problems in mass spectrometry–based proteomics. Nat. Methods 2009, 6, 423–430. [Google Scholar] [CrossRef] [PubMed]
- The, M.; Tasnim, A.; Käll, L. How to talk about protein-level false discovery rates in shotgun proteomics. Proteomics 2016, 16, 2461–2469. [Google Scholar] [CrossRef]
- Chait, B.T. Mass spectrometry: Bottom-up or top-down? Science 2006, 314, 65–66. [Google Scholar] [CrossRef]
- Streng, A.S.; de Boer, D.; Bouwman, F.G.; Mariman, E.C.M.; Scholten, A.; van Dieijen-Visser, M.P.; Wodzig, W.K.W.H. Development of a targeted selected ion monitoring assay for the elucidation of protease induced structural changes in cardiac troponin T. J. Proteom. 2016, 136, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, K.G.; Levitsky, L.I.; Pyatnitskiy, M.A.; Ilina, I.Y.; Bubis, J.A.; Solovyeva, E.M.; Zgoda, V.G.; Gorshkov, M.V.; Moshkovskii, S.A. Cysteine alkylation methods in shotgun proteomics and their possible effects on methionine residues. J. Proteom. 2021, 231, 104022. [Google Scholar] [CrossRef]
- Wright, E.P.; Partridge, M.A.; Padula, M.P.; Gauci, V.J.; Malladi, C.S.; Coorssen, J.R. Top-down proteomics: Enhancing 2D gel electrophoresis from tissue processing to high-sensitivity protein detection. Proteomics 2014, 14, 872–889. [Google Scholar] [CrossRef]
- Yates, J.R.; Kelleher, N.L. Top down proteomics. Anal. Chem. 2013, 85, 6151. [Google Scholar] [CrossRef]
- Lobas, A.A.; Karpov, D.S.; Kopylov, A.T.; Solovyeva, E.M.; Ivanov, M.V.; Ilina, I.Y.; Lazarev, V.N.; Kuznetsova, K.G.; Ilgisonis, E.V.; Zgoda, V.G.; et al. Exome-based proteogenomics of HEK-293 human cell line: Coding genomic variants identified at the level of shotgun proteome. Proteomics 2016, 16, 1980–1991. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef]
- Jungblut, P.R. The proteomics quantification dilemma. J. Proteom. 2014, 107, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Bogdanow, B.; Zauber, H.; Selbach, M. Systematic errors in peptide and protein identification and quantification by modified peptides. Mol. Cell. Proteom. 2016, 15, 2791–2801. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Mann, M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol. Cell. Proteom. 2013, 12, 3444–3452. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, J.A.; Bohlander, P.; Dai, M.; Filius, M.; Howard, C.J.; van Kooten, X.F.; Ohayon, S.; Pomorski, A.; Schmid, S.; Aksimentiev, A.; et al. The emerging landscape of single-molecule protein sequencing technologies. Nat. Methods 2021, 18, 604–617. [Google Scholar] [CrossRef]
- Shi, T.; Song, E.; Nie, S.; Rodland, K.D.; Liu, T.; Qian, W.J.; Smith, R.D. Advances in targeted proteomics and applications to biomedical research. Proteomics 2016, 16, 2160–2182. [Google Scholar] [CrossRef]
- Guo, T.; Kouvonen, P.; Koh, C.C.; Gillet, L.C.; Wolski, W.E.; Röst, H.L.; Rosenberger, G.; Collins, B.C.; Blum, L.C.; Gillessen, S.; et al. Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat. Med. 2015, 21, 407–413. [Google Scholar] [CrossRef]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [CrossRef]
- Coorssen, J.; Yergey, A. Approaches to top-down proteomics: In honour of Prof. Patrick H. O’Farrell. Proteomes 2017, 5, 18. [Google Scholar] [CrossRef]
- Gygi, S.P.; Corthals, G.L.; Zhang, Y.; Rochon, Y.; Aebersold, R. Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc. Natl. Acad. Sci. USA 2000, 97, 9390–9395. [Google Scholar] [CrossRef]
- Timms, J.F.; Cramer, R. Difference gel electrophoresis. Proteomics 2008, 8, 4886–4897. [Google Scholar] [CrossRef]
- Butt, R.H.; Coorssen, J.R. Postfractionation for enhanced proteomic analyses: Routine electrophoretic methods increase the resolution of standard 2D-PAGE. J. Proteome Res. 2005, 4, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.H.; Coorssen, J.R. Pre-extraction sample handling by automated frozen disruption significantly improves subsequent proteomic analyses. J. Proteome Res. 2006, 5, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Thiede, B.; Koehler, C.J.; Strozynski, M.; Treumann, A.; Stein, R.; Zimny-Arndt, U.; Schmid, M.; Jungblut, P.R. High resolution quantitative proteomics of HeLa cells protein species using stable isotope labeling with amino acids in cell culture(SILAC), two-dimensional gel electrophoresis(2DE) and nano-liquid chromatograpohy coupled to an LTQ-OrbitrapMass spectrometer. Mol. Cell. Proteom. 2012, 12, 529–538. [Google Scholar] [CrossRef]
- Butt, R.H.; Coorssen, J.R. Coomassie blue as a near-infrared fluorescent stain: A systematic comparison with Sypro Ruby for in-gel protein detection. Mol. Cell. Proteom. 2013, 12, 3834–3850. [Google Scholar] [CrossRef]
- Gauci, V.J.; Padula, M.P.; Coorssen, J.R. Coomassie blue staining for high sensitivity gel-based proteomics. J. Proteom. 2013, 90, 96–106. [Google Scholar] [CrossRef]
- Rogowska-Wrzesinska, A.; Le Bihan, M.-C.; Thaysen-Andersen, M.; Roepstorff, P. 2D gels still have a niche in proteomics. J. Proteom. 2013, 88, 4–13. [Google Scholar] [CrossRef]
- Wright, E.P.; Prasad, K.A.G.; Padula, M.P.; Coorssen, J.R. Deep imaging: How much of the proteome does current top-down technology already resolve? PLoS ONE 2014, 9, e86058. [Google Scholar] [CrossRef]
- Noaman, N.; Abbineni, P.S.; Withers, M.; Coorssen, J.R. Coomassie staining provides routine (sub)femtomole in-gel detection of intact proteoforms: Expanding opportunities for genuine Top-down Proteomics. Electrophoresis 2017, 38, 3086–3099. [Google Scholar] [CrossRef]
- Zhan, X.; Yang, H.; Peng, F.; Li, J.; Mu, Y.; Long, Y.; Cheng, T.; Huang, Y.; Li, Z.; Lu, M.; et al. How many proteins can be identified in a 2DE gel spot within an analysis of a complex human cancer tissue proteome? Electrophoresis 2018, 39, 965–980. [Google Scholar] [CrossRef]
- Naryzhny, S.; Klopov, N.; Ronzhina, N.; Zorina, E.; Zgoda, V.; Kleyst, O.; Belyakova, N.; Legina, O. A database for inventory of proteoform profiles: “2DE-pattern”. Electrophoresis 2020, 41, 1118–1124. [Google Scholar] [CrossRef]
- Bjellqvist, B.; Ek, K.; Giorgio Righetti, P.; Gianazza, E.; Görg, A.; Westermeier, R.; Postel, W. Isoelectric focusing in immobilized pH gradients: Principle, methodology and some applications. J. Biochem. Biophys. Methods 1982, 6, 317–339. [Google Scholar] [CrossRef]
- Chevalier, F. Highlights on the capacities of “Gel-based” proteomics. (Review) (Report). Proteome Sci. 2010, 8, 23. [Google Scholar] [CrossRef]
- Pergande, M.R.; Cologna, S.M. Isoelectric point separations of peptides and proteins. Proteomes 2017, 5, 4. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Naryzhny, S.N.; Zgoda, V.G.; Maynskova, M.A.; Novikova, S.E.; Ronzhina, N.L.; Vakhrushev, I.V.; Khryapova, E.V.; Lisitsa, A.V.; Tikhonova, O.V.; Ponomarenko, E.A.; et al. Combination of virtual and experimental 2DE together with ESI LC-MS/MS gives a clearer view about proteomes of human cells and plasma. Electrophoresis 2016, 37, 302–309. [Google Scholar] [CrossRef]
- D’Silva, A.M.; Hyett, J.A.; Coorssen, J.R. Proteomic analysis of first trimester maternal serum to identify candidate biomarkers potentially predictive of spontaneous preterm birth. J. Proteom. 2018, 178, 31–42. [Google Scholar] [CrossRef]
- Kurgan, N.; Noaman, N.; Pergande, M.R.; Cologna, S.M.; Coorssen, J.R.; Klentrou, P. Changes to the human serum proteome in response to high intensity interval exercise: A sequential top-down proteomic analysis. Front. Physiol. 2019, 10, 362. [Google Scholar] [CrossRef]
- Zhu, K.; Zhao, J.; Lubman, D.M.; Miller, F.R.; Barder, T.J. Protein pI shifts due to posttranslational modifications in the separation and characterization of proteins. Anal. Chem. 2005, 77, 2745–2755. [Google Scholar] [CrossRef]
- Hsieh, E.J.; Bereman, M.S.; Durand, S.; Valaskovic, G.A.; MacCoss, M.J. Effects of column and gradient lengths on peak capacity and peptide identification in nanoflow LC-MS/MS of complex proteomic samples. J. Am. Soc. Mass Spectrom. 2013, 24, 148–153. [Google Scholar] [CrossRef]
- Shishkova, E.; Alexander, S.H.; Coon, J.J. Now, more than ever, proteomics needs better chromatography. Cell Syst. 2016, 3, 321–324. [Google Scholar] [CrossRef]
- Harris, L.R.; Churchward, M.A.; Butt, R.H.; Coorssen, J.R. Assessing detection methods for gel-based proteomic analyses. J. Proteome Res. 2007, 6, 1418–1425. [Google Scholar] [CrossRef]
- Gauci, V.J.; Wright, E.P.; Coorssen, J.R. Quantitative proteomics: Assessing the spectrum of in-gel protein detection methods. J. Chem. Biol. 2011, 4, 3–29. [Google Scholar] [CrossRef]
- Neuhoff, V.; Stamm, R.; Eibl, H. Clear background and highly sensitive protein staining with Coomassie Blue dyes in polyacrylamide gels: A systematic analysis. Electrophoresis 1985, 6, 427–448. [Google Scholar] [CrossRef]
- Colignon, B.; Raes, M.; Dieu, M.; Delaive, E.; Mauro, S. Evaluation of three-dimensional gel electrophoresis to improve quantitative profiling of complex proteomes. Proteomics 2013, 13, 2077–2082. [Google Scholar] [CrossRef]
- Marouga, R.; David, S.; Hawkins, E. The development of the DIGE system: 2D fluorescence difference gel analysis technology. Anal. Bioanal. Chem. 2005, 382, 669–678. [Google Scholar] [CrossRef]
- Unlü, M.; Morgan, M.E.; Minden, J.S. Difference gel electrophoresis: A single gel method for detecting changes in protein extracts. Electrophoresis 1997, 18, 2071–2077. [Google Scholar] [CrossRef]
- Minden, J.S.; Dowd, S.R.; Meyer, H.E.; Stühler, K. Difference gel electrophoresis. Electrophoresis 2009, 30, S156–S161. [Google Scholar] [CrossRef]
- McNamara, L.E.; Kantawong, F.A.; Dalby, M.J.; Riehle, M.O.; Burchmore, R. Preventing and troubleshooting artefacts in saturation labelled fluorescence 2-D difference gel electrophoresis (saturation DiGE). Proteomics 2011, 11, 4610–4621. [Google Scholar] [CrossRef]
- Hacker, S.M.; Backus, K.M.; Lazear, M.R.; Forli, S.; Correia, B.E.; Cravatt, B.F. Global profiling of lysine reactivity and ligandability in the human proteome. Nat. Chem. 2017, 9, 1181–1190. [Google Scholar] [CrossRef]
- Tonge, R.; Shaw, J.; Middleton, B.; Rowlinson, R.; Rayner, S.; Young, J.; Pognan, F.; Hawkins, E.; Currie, I.; Davison, M. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics 2001, 1, 377–396. [Google Scholar] [CrossRef]
- Shaw, J.; Rowlinson, R.; Nickson, J.; Stone, T.; Sweet, A.; Williams, K.; Tonge, R. Evaluation of saturation labelling two-dimensional difference gel electrophoresis fluorescent dyes. Proteomics 2003, 3, 1181–1195. [Google Scholar] [CrossRef]
- McNamara, L.E.; Dalby, M.J.; Riehle, M.O.; Burchmore, R. Fluorescence two-dimensional difference gel electrophoresis for biomaterial applications. J. R. Soc. Interface 2010, 7, S107–S118. [Google Scholar] [CrossRef][Green Version]
- Herbert, B.; Galvani, M.; Hamdan, M.; Olivieri, E.; MacCarthy, J.; Pedersen, S.; Righetti, P.G. Reduction and alkylation of proteins in preparation of two-dimensional map analysis: Why, when, and how? Electrophoresis 2001, 22, 2046–2057. [Google Scholar] [CrossRef]
- Smejkal, G.B.; Li, C.; Robinson, M.H.; Lazarev, A.V.; Lawrence, N.P.; Chernokalskaya, E. Simultaneous reduction and alkylation of protein disulfides in a centrifugal ultrafiltration device prior to two-dimensional gel electrophoresis. J. Proteome Res. 2006, 5, 983–987. [Google Scholar] [CrossRef]
- Kurien, B.T.; Scofield, R.H. Common artifacts and mistakes made in electrophoresis. Methods Mol. Biol. 2012, 869, 633–640. [Google Scholar] [CrossRef]
- Sitek, B.; Lüttges, J.; Marcus, K.; Klöppel, G.; Schmiegel, W.; Meyer, H.E.; Hahn, S.A.; Stühler, K. Application of fluorescence difference gel electrophoresis saturation labelling for the analysis of microdissected precursor lesions of pancreatic ductal adenocarcinoma. Proteomics 2005, 5, 2665–2679. [Google Scholar] [CrossRef] [PubMed]
- Gade, D.; Thiermann, J.; Markowsky, D.; Rabus, R. Evaluation of two-dimensional difference gel electrophoresis for protein profiling. J. Mol. Microbiol. Biotechnol. 2003, 5, 240–251. [Google Scholar] [CrossRef]
- Lanouette, S.; Mongeon, V.; Figeys, D.; Couture, J.F. The functional diversity of protein lysine methylation. Mol. Syst. Biol. 2014, 10, 724-n/a. [Google Scholar] [CrossRef] [PubMed]
- Stastna, M.; Van Eyk, J.E. Posttranslational modifications of lysine and evolving role in heart pathologies-Recent developments. Proteomics 2015, 15, 1164–1180. [Google Scholar] [CrossRef] [PubMed]
- Couvertier, S.M.; Zhou, Y.; Weerapana, E. Chemical-proteomic strategies to investigate cysteine posttranslational modifications. Biochim. Biophys. Acta 2014, 1844, 2315–2330. [Google Scholar] [CrossRef] [PubMed]
- Shannon, D.A.; Weerapana, E. Orphan PTMs: Rare, yet functionally important modifications of cysteine. Biopolymers 2014, 101, 156–164. [Google Scholar] [CrossRef]
- Churchward, M.A.; Butt, R.H.; Lang, J.C.; Hsu, K.K.; Coorssen, J.R. Enhanced detergent extraction for analysis of membrane proteomes by two-dimensional gel electrophoresis. Proteome Sci. 2005, 3, 5. [Google Scholar] [CrossRef]
- Takemori, N.; Takemori, A.; Wongkongkathep, P.; Nshanian, M.; Loo, R.R.O.; Lermyte, F.; Loo, J.A. Top-down/bottom-up mass spectrometry workflow using dissolvable polyacrylamide gels. Anal. Chem. 2017, 89, 8244–8250. [Google Scholar] [CrossRef]
- Binz, P.-A.; Müller, M.; Hoogland, C.; Zimmermann, C.; Pasquarello, C.; Corthals, G.; Sanchez, J.-C.; Hochstrasser, D.F.; Appel, R.D. The molecular scanner: Concept and developments. Curr. Opin. Biotechnol. 2004, 15, 17–23. [Google Scholar] [CrossRef]
- Papasotiriou, D.G.; Markoutsa, S.; Gorka, J.; Schleiff, E.; Karas, M.; Meyer, B. MALDI analysis of proteins after extraction from dissolvable ethylene glycol diacrylate cross-linked polyacrylamide gels. Electrophoresis 2013, 34, 2484–2494. [Google Scholar] [CrossRef]
- Takemori, A.; Ishizaki, J.; Nakashima, K.; Shibata, T.; Kato, H.; Kodera, Y.; Suzuki, T.; Hasegawa, H.; Takemori, N. BAC-DROP: Rapid digestion of proteome fractionated via dissolvable polyacrylamide gel electrophoresis and its applicaiton to bottom-up proteomics workflow. J. Proteome Res. 2021, 20, 1535–1543. [Google Scholar] [CrossRef]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in top-down proteomics and the analysis of proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. [Google Scholar] [CrossRef]
- Huguet, R.; Mullen, C.; Srzentić, K.; Greer, J.B.; Fellers, R.T.; Zabrouskov, V.; Syka, J.E.P.; Kelleher, N.L.; Fornelli, L. Proton transfer charge reduction enables high-throughput top-down analysis of large proteoforms. Anal. Chem. 2019, 91, 15732–15739. [Google Scholar] [CrossRef]
- Kafader, J.O.; Durbin, K.R.; Melani, R.D.; Des Soye, B.J.; Schachner, L.F.; Senko, M.W.; Compton, P.D.; Kelleher, N.L. Individual ion mass spectrometry enhances the sensitivity and sequence coverage of top-down mass spectrometry. J. Proteome Res. 2020, 19, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Jin, Y.; Wu, Z.; Tucholski, T.; Brown, K.A.; Zhang, L.; Zhang, Y.; Ge, Y. Bridge hybrid monolithic column coupled to high-resolution mass spectrometry for top-down proteomics. Anal. Chem. 2019, 91, 1743–1747. [Google Scholar] [CrossRef] [PubMed]
- Melby, J.A.; Roberts, D.S.; Larson, E.J.; Brown, K.A.; Bayne, E.F.; Jin, S.; Ge, Y. Novel strategies to address the challenges in top-down proteomics. J. Am. Soc. Mass Spectrom. 2021, 32, 1278–1294. [Google Scholar] [CrossRef] [PubMed]
- Melchior, K.; Tholey, A.; Heisel, S.; Keller, A.; Lenhof, H.-P.; Meese, E.; Huber, C.G. Protein- versus peptide fractionation in the first dimension of two-dimensional high-performance liquid chromatography-matrix-assisted laser desorption/ionization tandem mass spectrometry for qualitative proteome analysis of tissue samples. J. Chromatogr. A 2010, 1217, 6159–6168. [Google Scholar] [CrossRef]
- Tran, J.C.; Zamdborg, L.; Ahlf, D.R.; Lee, J.E.; Catherman, A.D.; Durbin, K.R.; Tipton, J.D.; Vellaichamy, A.; Kellie, J.F.; Li, M.; et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 2011, 480, 254. [Google Scholar] [CrossRef]
- Camerini, S.; Mauri, P. The role of protein and peptide separation before mass spectrometry analysis in clinical proteomics. J. Chromatogr. A 2015, 1381, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.C.; Doucette, A.A. Gel-Eluted Liquid Fraction Entrapment Electrophoresis: An electrophoretic method for broad molecular weight range proteome separation. Anal. Chem. 2008, 80, 1568–1573. [Google Scholar] [CrossRef]
- Chrambach, A.; Jovin, T.M. Selected buffer systems for moving boundary electrophoresis on gels at various pH values, presented in a simplified manner. Electrophoresis 1983, 4, 190–204. [Google Scholar] [CrossRef]
- Model 491 Prep Cell and Mini Prep Cell. Available online: https://www.bio-rad.com/en-ca/product/model-491-prep-cell-mini-prep-cell?ID=230a0852-ae4f-4861-b463-194663fdc7ac (accessed on 1 July 2021).
- Mehrotra, J.; Mittal, A.; Dhindsa, M.S.; Sinha, S. Fractionation of mycobacterial integral membrane proteins by continuous elution SDS–PAGE reveals the immunodominance of low molecular weight subunits for human T cells. Clin. Exp. Immunol. 1997, 109, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Krause, R.G.E.; Goldring, J.P.D. Crystal violet stains proteins in SDS-PAGE gels and zymograms. Anal. Biochem. 2019, 566, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Takemori, A.; Butcher, D.S.; Harman, V.M.; Brownridge, P.; Shima, K.; Higo, D.; Ishizaki, J.; Hasegawa, H.; Suzuki, J.; Yamashita, M.; et al. PEPPI-MS: Polyacrylamide-gel-based prefractionation for analysis of intact proteoforms and protein complexes by mass spectrometry. J. Proteome Res. 2020, 19, 3779–3791. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Mazumdar, S. Electrospray ionization mass spectrometry: A technique to access the information beyond the molecular weight of the analyte. Int. J. Anal. Chem. 2012, 2012, 282574. [Google Scholar] [CrossRef]
- Clarke, W. Mass Spectrometry in the Clinical Laboratory: Determining the Need and Avoiding Pitfalls. In Mass Spectrometry for the Clinical Laboratory; Nair, H., Clarke, W., Eds.; Academic Press: San Diego, CA, USA, 2017; pp. 1–15. [Google Scholar]
- Krüger, R.; Karas, M. Formation and fate of ion pairs during MALDI analysis: Anion adduct generation as an indicative tool to determine ionization processes. J. Am. Soc. Mass Spectrom. 2002, 13, 1218–1226. [Google Scholar] [CrossRef]
- Susa, A.C.; Xia, Z.; Williams, E.R. Small emitter tips for native mass spectrometry of proteins and protein complexes from nonvolatile buffers that mimic the intracellular environment. Anal. Chem. 2017, 89, 3116–3122. [Google Scholar] [CrossRef]
- Grandori, R. Origin of the conformation dependence of protein charge-state distributions in electrospray ionization mass spectrometry. J. Mass Spectrom. 2003, 38, 11–15. [Google Scholar] [CrossRef]
- Compton, P.D.; Zamdborg, L.; Thomas, P.M.; Kelleher, N.L. On the scalability and requirements of whole protein mass spectrometry. Anal. Chem. 2011, 83, 6868–6874. [Google Scholar] [CrossRef]
- Comisarow, M.B.; Marshall, A.G. Theory of Fourier transform ion cyclotron resonance mass spectroscopy. I. Fundamental equations and low-pressure line shape. J. Chem. Phys. 1976, 64, 110–119. [Google Scholar] [CrossRef]
- Nikolaev, E.N.; Kostyukevich, Y.I.; Vladimirov, G.N. Fourier transform ion cyclotron resonance (FT ICR) mass spectrometry: Theory and simulations. Mass Spectrom. Rev. 2016, 35, 219–258. [Google Scholar] [CrossRef]
- Smith, D.F.; Podgorski, D.C.; Rodgers, R.P.; Blakney, G.T.; Hendrickson, C.L. 21 tesla FT-ICR mass spectrometer for ultrahigh-resolution analysis of complex organic mixtures. Anal. Chem. 2018, 90, 2041–2047. [Google Scholar] [CrossRef]
- Marshall, A.G.; Comisarow, M.B.; Parisod, G. Relaxation and spectral line shape in Fourier transform ion resonance spectroscopy. J. Chem. Phys. 1979, 71, 4434–4444. [Google Scholar] [CrossRef]
- Shaw, J.B.; Lin, T.-Y.; Leach Iii, F.E.; Tolmachev, A.V.; Tolić, N.; Robinson, E.W.; Koppenaal, D.W.; Paša-Tolić, L. 21 Tesla Fourier transform ion cyclotron resonance mass spectrometer greatly expands mass spectrometry toolbox. J. Am. Soc. Mass Spectrom. 2016, 27, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, E.; Lioznov, A. How to increase further the resolving power of the ultrahigh magnetic field FT ICR instruments? The new concept of the FT ICR cell-the open dynamically harmonized cell as a part of the vacuum system wall. Anal. Chem. 2021, 93, 1249–1253. [Google Scholar] [CrossRef]
- Marshall, A.G.; Hendrickson, C.L.; Jackson, G.S. Fourier transform ion cyclotron resonance mass spectrometry: A primer. Mass Spectrom. Rev. 1998, 17, 1–35. [Google Scholar] [CrossRef]
- Perry, R.H.; Cooks, R.G.; Noll, R.J. Orbitrap mass spectrometry: Instrumentation, ion motion and applications. Mass Spectrom. Rev. 2008, 27, 661–699. [Google Scholar] [CrossRef]
- Makarov, A. Electrostatic axially harmonic orbital trapping: A high-performance technique of mass analysis. Anal. Chem. 2000, 72, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Makarov, A.; Denisov, E.; Kholomeev, A.; Balschun, W.; Lange, O.; Strupat, K.; Horning, S. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal. Chem. 2006, 78, 2113–2120. [Google Scholar] [CrossRef]
- Fort, K.L.; van de Waterbeemd, M.; Boll, D.; Reinhardt-Szyba, M.; Belov, M.E.; Sasaki, E.; Zschoche, R.; Hilvert, D.; Makarov, A.A.; Heck, A.J.R. Expanding the structural analysis capabilities on an Orbitrap-based mass spectrometer for large macromolecular complexes. Analyst 2018, 143, 100–105. [Google Scholar] [CrossRef]
- Shaw, J.B.; Brodbelt, J.S. Extending the isotopically resolved mass range of orbitrap mass spectrometers. Anal. Chem. 2013, 85, 8313–8318. [Google Scholar] [CrossRef]
- Harper, C.C.; Elliott, A.G.; Oltrogge, L.M.; Savage, D.F.; Williams, E.R. Multiplexed charge detection mass spectrometry for high-throughput single ion analysis of large molecules. Anal. Chem. 2019, 91, 7458–7465. [Google Scholar] [CrossRef]
- Kafader, J.O.; Beu, S.C.; Early, B.P.; Melani, R.D.; Durbin, K.R.; Zabrouskov, V.; Makarov, A.A.; Maze, J.T.; Shinholt, D.L.; Yip, P.F.; et al. STORI plots enable accurate tracking of individual ion signals. J. Am. Soc. Mass Spectrom. 2019, 30, 2200–2203. [Google Scholar] [CrossRef]
- Kafader, J.O.; Melani, R.D.; Durbin, K.R.; Ikwuagwu, B.; Early, B.P.; Fellers, R.T.; Beu, S.C.; Zabrouskov, V.; Makarov, A.A.; Maze, J.T.; et al. Multiplexed mass spectrometry of individual ions improves measurement of proteoforms and their complexes. Nat. Methods 2020, 17, 391–394. [Google Scholar] [CrossRef]
- Li, Z.; Adams, R.M.; Chourey, K.; Hurst, G.B.; Hettich, R.L.; Pan, C. Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. J. Proteome Res. 2012, 11, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Pan, H.; Chen, X. Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom. Rev. 2009, 28, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Cristobal, A.; Marino, F.; Post, H.; van den Toorn, H.W.P.; Mohammed, S.; Heck, A.J.R. Toward an optimized workflow for middle-down proteomics. Anal. Chem. 2017, 89, 3318–3325. [Google Scholar] [CrossRef]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantification in Saccharomyces cerecisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef]
- Chong, P.K.; Gan, C.S.; Pham, T.K.; Wright, P.C. Isobaric tags for relative and absolute quantitation (iTRAQ) reproducibility: Implication of multiple injections. J. Proteome Res. 2006, 5, 1232–1240. [Google Scholar] [CrossRef]
- Mahoney, D.W.; Therneau, T.M.; Heppelmann, C.J.; Higgins, L.; Benson, L.M.; Zenka, R.M.; Jagtap, P.; Nelsestuen, G.L.; Bergen, H.R.; Oberg, A.L. Relative quantification: Characterization of bias, variability and fold changes in mass spectrometry data from iTRAQ-labeled peptides. J. Proteome Res. 2011, 10, 4325–4333. [Google Scholar] [CrossRef]
- Evans, C.; Noirel, J.; Ow, S.Y.; Salim, M.; Pereira-Medrano, A.G.; Couto, N.; Pandhal, J.; Smith, D.; Pham, T.K.; Karunakaran, E.; et al. An insight into iTRAQ: Where do we stand now? Anal. Bioanal. Chem. 2012, 404, 1011–1027. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Xu, G.; Liu, M.; Xue, L.; Liu, L.; Hu, S.; Zhang, Y.; Nie, Y.; Liang, S.; et al. Mechanism study of peptide GMBP1 and its receptor GRP78 in modulating gastric cancer MDR by iTRAQ-based proteomic analysis. BMC Cancer 2015, 15, 358. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Zhao, X.; Dang, X. Mass spectrometry-based iTRAQ analysis of serum markers in patients with pancreatic cancer. Oncol. Lett. 2020, 19, 4106–4114. [Google Scholar] [CrossRef] [PubMed]
- Beati, H.; Langlands, A.; ten Have, S.; Müller, H.A.J. SILAC-based quantitative proteomic analysis of Drosophila gastrula stage embryos mutant for fibroblast growth factor signalling. Fly 2020, 14, 10–28. [Google Scholar] [CrossRef] [PubMed]
- Schober, F.A.; Atanassov, I.; Moore, D.; Calvo-Garrido, J.; Moedas, M.F.; Wedell, A.; Freyer, C.; Wredenberg, A. Stable isotope labeling of amino acids in flies (SILAF) reveals differential phosphorylation of mitochondrial proteins upon loss of OXPHOS subunits. Mol. Cell. Proteom. 2021, 20, 100065. [Google Scholar] [CrossRef]
- Issaq, H.J.; Conrads, T.P.; Janini, G.M.; Veenstra, T.D. Methods for fractionation, separation and profiling of proteins and peptides. Electrophoresis 2002, 23, 3048–3061. [Google Scholar] [CrossRef]
- Fang, X.; Zhang, W.-W. Affinity separation and enrichment methods in proteomic analysis. J. Proteom. 2008, 71, 284–303. [Google Scholar] [CrossRef]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.-C.; Yates, J.R. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef]
- Pinkse, M.W.H.; Uitto, P.M.; Hilhorst, M.J.; Ooms, B.; Heck, A.J.R. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal. Chem. 2004, 76, 3935–3943. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef]
- Nielsen, M.L.; Vermeulen, M.; Bonaldi, T.; Cox, J.; Moroder, L.; Mann, M. Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat. Methods 2008, 5, 459–460. [Google Scholar] [CrossRef]
- Harlan, R.; Zhang, H. Targeted proteomics: A bridge between discovery and validation. Expert Rev. Proteom. 2014, 11, 657–661. [Google Scholar] [CrossRef]
- Parker, C.E.; Borchers, C.H. Mass spectrometry based biomarker discovery, verification, and validation—Quality assurance and control of protein biomarker assays. Mol. Oncol. 2014, 8, 840–858. [Google Scholar] [CrossRef]
- Paulovich, A.G.; Whiteaker, J.R.; Hoofnagle, A.N.; Wang, P. The interface between biomarker discovery and clinical validation: The tar pit of the protein biomarker pipeline. Proteom. Clin. Appl. 2008, 2, 1386–1402. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Cheloha, R.W.; Harmand, T.J.; Wijne, C.; Schwartz, T.U.; Ploegh, H.L. Exploring cellular biochemistry with nanobodies. J. Biol. Chem. 2020, 295, 15307–15327. [Google Scholar] [CrossRef]
- Li, D.; Morisseau, C.; McReynolds, C.B.; Duflot, T.; Bellien, J.; Nagra, R.M.; Taha, A.Y.; Hammock, B.D. Development of improved double-nanobody sandwich ELISAs for human soluble epoxide hydrolase detection in peripheral blood mononuclear cells of diabetic patients and the prefrontal cortex of Multiple Sclerosis patients. Anal. Chem. 2020, 92, 7334–7342. [Google Scholar] [CrossRef]
- Lipman, N.S.; Jackson, L.R.; Trudel, L.J.; Weis-Garcia, F. Monoclonal versus polyclonal antibodies: Distinguishing characteristics, applications, and information resources. ILAR J. 2005, 46, 258–268. [Google Scholar] [CrossRef]
- Ascoli, C.A.; Aggeler, B. Overlooked benefits of using polyclonal antibodies. BioTechniques 2018, 65, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Koide, S. Next-generation antibodies for post-translational modifications. Curr. Opin. Struct. Biol. 2018, 51, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Haab, B.; Paulovich, A.G.; Anderson, N.L.; Clark, A.M.; Downing, G.J.; Hermjakob, H.; LaBaer, J.; Uhlen, M. A reagent resource to identify proteins and peptides of interest for the cancer community. Mol. Cell. Proteom. 2006, 5, 1996. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Targeted proteomics. Nat. Methods 2013, 10, 19–22. [Google Scholar] [CrossRef]
- Gilda, J.E.; Ghosh, R.; Cheah, J.X.; West, T.M.; Bodine, S.C.; Gomes, A.V. Western blotting inaccuracies with unverified antibodies: Need for a Western blotting minimal reporting standard (WDBMRS). PLoS ONE 2015, 10, e0135392. [Google Scholar] [CrossRef]
- Bjerner, J.; Nustad, K.; Norum, L.F.; Olsen, K.H.; Bormer, O.P. Immunometric assay interference: Incidence and prevention. Clin. Chem. 2002, 48, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Ylikotila, J.; Hellström, J.L.; Eriksson, S.; Vehniäinen, M.; Välimaa, L.; Takalo, H.; Bereznikova, A.; Pettersson, K. Utilization of recombinant Fab fragments in a cTnI immunoassay conducted in spot wells. Clin. Biochem. 2006, 39, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Mann, M. Can proteomics retire the western blot? J. Proteome Res. 2008, 7, 3065. [Google Scholar] [CrossRef] [PubMed]
- Krastins, B.; Prakash, A.; Sarracino, D.A.; Nedelkov, D.; Niederkofler, E.E.; Kiernan, U.A.; Nelson, R.; Vogelsang, M.S.; Vadali, G.; Garces, A.; et al. Rapid development of sensitive, high-throughput, quantitative and highly selective mass spectrometric targeted immunoassays for clinically important proteins in human plasma and serum. Clin. Biochem. 2013, 46, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef]
- Butler, T.A.J.; Paul, J.W.; Chan, E.-C.; Smith, R.; Tolosa, J.M. Misleading Westerns: Common quantification mistakes in Western blot densitometry and proposed corrective measures. BioMed Res. Int. 2019, 2019, 5214821. [Google Scholar] [CrossRef]
- Hause, R.J.; Kim, H.D.; Leung, K.K.; Jones, R.B. Targeted protein-omic methods are bridging the gap between proteomic and hypothesis-driven protein analysis approaches. Expert Rev Proteom. 2011, 8, 565–575. [Google Scholar] [CrossRef] [PubMed]
- D’Silva, A.M.; Hyett, J.A.; Coorssen, J.R. First trimester protein biomarkers for risk of spontaneous preterm birth: Identifying a critical need for more rigorous approaches to biomarker identification and validation. Fetal Diagn. Ther. 2020, 47, 497–506. [Google Scholar] [CrossRef]
- Mollica, J.P.; Oakhill, J.S.; Lamb, G.D.; Murphy, R.M. Are genuine changes in protein expression being overlooked? Reassessing Western blotting. Anal. Biochem. 2009, 386, 270–275. [Google Scholar] [CrossRef]
- Taylor, S.C.; Posch, A. The design of a quantitative Western blot experiment. BioMed Res. Int. 2014, 2014, 361590. [Google Scholar] [CrossRef]
- Kurien, B.T.; Scofield, R.H. Western Blotting: An Introduction. In Western Blotting: Methods and Protocols; Kurien, B.T., Scofield, R.H., Walker, J.M., Eds.; Methods in Molecular Biology Humana Press: New York, NY, USA, 2015; pp. 17–30. [Google Scholar]
- Macphee, D.J. Methodological considerations for improving Western blot analysis. J. Pharmacol. Toxicol. Methods 2010, 61, 171–177. [Google Scholar] [CrossRef]
- Peferoen, M.; Huybrechts, R.; De Loof, A. Vacuum-blotting: A new simple and efficient transfer of proteins from sodium dodecyl sulfate—polyacrylamide gels to nitrocellulose. FEBS Lett. 1982, 145, 369–372. [Google Scholar] [CrossRef]
- Bass, J.J.; Wilkinson, D.J.; Rankin, D.; Phillips, B.E.; Szewczyk, N.J.; Smith, K.; Atherton, P.J. An overview of technical considerations for Western blotting applications to physiological research. Scand. J. Med. Sci. Sports 2017, 27, 4–25. [Google Scholar] [CrossRef]
- Wang, G.; de Jong, R.N.; van den Bremer, E.T.J.; Parren, P.W.H.I.; Heck, A.J.R. Enhancing accuracy in molecular weight determination of highly heterogeneously glycosylated proteins by native tandem mass spectrometry. Anal. Chem. 2017, 89, 4793–4797. [Google Scholar] [CrossRef]
- Alegria-Schaffer, A.; Lodge, A.; Vattem, K. Performing and Optimizing Western Blots with an Emphasis on Chemiluminescent Detection. Available online: https://www.sciencedirect.com/science/article/abs/pii/S0076687909630330 (accessed on 1 July 2021).
- Dorri, Y. Immunoblotting of Antigens: Whole, Strip, and New-Line Nitrocellulose Membrane Immunoblotting Using the Chemiluminescence Technique. In Detection of Blotted Proteins: Methods and Protocols, 1st ed.; Kurien, B.T., Scofield, R.H., Walker, J.M., Eds.; Methods in Molecular Biology Humana Press: New York, NY, USA, 2015; pp. 79–94. [Google Scholar]
- Desai, S.; Dworecki, B.R.; Nlend, M.C. Direct Immunodetection of Antigens Within the Precast Polyacrylamide Gel. In Detection of Blotted Proteins: Methods and Protocols, 1st ed.; Kurien, B.T., Scofield, R.H., Walker, J.M., Eds.; Methods in Molecular Biology Humana Press: New York, NY, USA, 2015; pp. 101–114. [Google Scholar]
- Gingrich, J.C.; Davis, D.R.; Nguyen, Q. Multiplex detection and quantitation of proteins on Western blots using fluorescent probes. BioTechniques 2000, 29, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Hao, Y.; Xue, J.; Liu, X.; Xu, X.; Liu, L. A colorimetric enzyme-linked immunosorbent assay with CuO nanoparticles as signal labels based on the growth of gold nanoparticles in situ. Nanomaterials 2018, 9, 4. [Google Scholar] [CrossRef]
- Wu, C.; Shan, Y.; Wu, X.; Wang, S.; Liu, F. Quantitative protein detection using single molecule imaging enzyme-linked immunosorbent assay (iELISA). Anal. Biochem. 2019, 587, 113466. [Google Scholar] [CrossRef] [PubMed]
- Rissin, D.M.; Walt, D.R. Digital concentration readout of single enzyme molecules using femotliter arrays and Poisson statistics. Nano Lett. 2006, 6, 520–523. [Google Scholar] [CrossRef]
- Rissin, D.M.; Walt, D.R. Digital readout of target binding with attomole detection limits via enzyme amplification in femotliter arrays. J. Am. Chem. Soc. 2006, 128, 6286–6287. [Google Scholar] [CrossRef] [PubMed]
- Rissin, D.M.; Gorris, H.H.; Walt, D.R. Distinct and long-lived activity states of single enzyme molecules. J. Am. Chem. Soc. 2008, 130, 5349–5353. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Milutinovic, M.D.; Walt, D.R. Single molecule array (Simoa) assay with optimal antibody pairs for cytokine detection in human serum samples. Analyst 2015, 140, 6277–6282. [Google Scholar] [CrossRef]
- Rissin, D.M.; Kan, C.W.; Campbell, T.G.; Howes, S.C.; Fournier, D.R.; Song, L.; Piech, T.; Patel, P.P.; Chang, L.; Rivnak, A.J.; et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 2010, 28, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Coons, A.H.; Creech, H.J.; Jones, R.N. Immunological properties of an antibody containing a fluorescent group. Proc. Soc. Exp. Biol. Med. 1941, 47, 200–202. [Google Scholar] [CrossRef]
- Gatter, K.C.; Abdulaziz, Z.; Beverley, P.; Corvalan, J.R.F.; Ford, C.; Lane, E.B.; Mota, M.; Nash, J.R.G.; Pulford, K.; Stein, H.; et al. Use of monoclonal antibodies for the histopathological diagnosis of human malignancy. J. Clin. Pathol. 1982, 35, 1253–1267. [Google Scholar] [CrossRef]
- Lohse, J.; Petersen, K.H.; Woller, N.C.; Pedersen, H.C.; Skladtchikova, G.; Jørgensen, R.M. Improved catalyzed reporter deposition, iCARD. Bioconjugate Chem. 2014, 25, 1036–1042. [Google Scholar] [CrossRef]
- Jensen, K.; Krusenstjerna-Hafstrøm, R.; Lohse, J.; Petersen, K.H.; Derand, H. A novel quantitative immunohistochemistry method for precise protein measurements directly in formalin-fixed, paraffin-embedded specimens: Analytical performance measuring HER2. Mod. Pathol. 2017, 30, 180–193. [Google Scholar] [CrossRef]
- Anand, S.; Samuel, M.; Ang, C.-S.; Keerthikumar, S.; Mathivanan, S. Label-based and label-free strategies for protein quantification. In Proteome Bioinformatics; Keerthikumar, S., Mathivanan, S., Eds.; Springer: New York, NY, USA, 2017; pp. 31–43. [Google Scholar]
- Bantscheff, M.; Schirle, M.; Sweetman, G.; Rick, J.; Kuster, B. Quantitative mass spectrometry in proteomics: A critical review. Anal. Bioanal. Chem. 2007, 389, 1017–1031. [Google Scholar] [CrossRef]
- Gouw, J.W.; Krijgsveld, J.; Heck, A.J.R. Quantitative proteomics by metabolic labeling of model organsisms. Mol. Cell. Proteom. 2010, 9, 11–24. [Google Scholar] [CrossRef]
- Chahrour, O.; Cobice, D.; Malone, J. Stable isotope labelling methods in mass spectrometry-based quantitative proteomics. J. Pharm. Biomed. Anal. 2015, 113, 2–20. [Google Scholar] [CrossRef]
- Ankney, J.A.; Muneer, A.; Chen, X. Relative and absolute quantitation in mass spectrometry-based proteomics. Annu. Rev. Anal. Chem. 2018, 11, 49–77. [Google Scholar] [CrossRef]
- Xie, F.; Liu, T.; Qian, W.-J.; Petyuk, V.A.; Smith, R.D. Liquid chromatography-mass spectrometry-based quantitative proteomics. J. Biol. Chem. 2011, 286, 25443–25449. [Google Scholar] [CrossRef]
- Quijada, J.V.; Schmitt, N.D.; Salisbury, J.P.; Auclair, J.R.; Agar, J.N. Heavy sugar and heavy water create tunable intact protein mass increases for quantitative mass spectrometry in any feed and organism. Anal. Chem. 2016, 88, 11139–11146. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Rauniyar, N.; Gao, B.; McClatchy, D.B.; Yates, J.R. Comparison of protein expression ratios observed by sixplex and duplex TMT labeling method. J. Proteome Res. 2013, 12, 1031–1039. [Google Scholar] [CrossRef]
- Zecha, J.; Satpathy, S.; Kanashova, T.; Avanessian, S.C.; Kane, M.H.; Clauser, K.R.; Mertins, P.; Carr, S.A.; Kuster, B. TMT labeling for masses: A robust and cost-efficient, in-solution labeling approach. Mol. Cell. Proteom. 2019, 18, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Megger, D.A.; Pott, L.L.; Ahrens, M.; Padden, J.; Bracht, T.; Kuhlmann, K.; Eisenacher, M.; Meyer, H.E.; Sitek, B. Comparison of label-free and label-based strategies for proteome analysis of hepatoma cell lines. Biochim. Biophys. Acta 2014, 1844, 967–976. [Google Scholar] [CrossRef]
- Rauniyar, N. Parallel reaction monitoring: A targeted experiment performed using high resolution and high mass accuracy mass spectrometry. Int. J. Mol. Sci. 2015, 16, 28566–28581. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Shen, S.; Li, J.; Hu, Q.; Nie, L.; Tu, C.; Wang, X.; Orsburn, B.; Wang, J.; Qu, J. An IonStar experimental strategy for MS1 ion current-based quantification using ultrahigh-field orbitrap: Reproducible, in-depth, and accurate protein measurement in large cohorts. J. Proteome Res. 2017, 16, 2445–2456. [Google Scholar] [CrossRef] [PubMed]
- Lange, V.; Picotti, P.; Domon, B.; Aebersold, R. Selected reaction monitoring for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2008, 4, 222. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Zhang, Y.; Fu, J.; Wang, Y.; Li, Y.; Yang, Q.; Yao, L.; Xue, W.; Zhu, F. Computational advances in the label-free quantification of cancer proteomics data. Curr. Pharm. Des. 2018, 24, 3842–3858. [Google Scholar] [CrossRef] [PubMed]
- Sticker, A.; Goeminne, L.; Martens, L.; Clement, L. Robust summarization and inference in proteome-wide label-free quantification. Mol. Cell. Proteom. 2020, 19, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Rezai, T.; Krastins, B.; Sarracino, D.; Athanas, M.; Russo, P.; Ross, M.M.; Zhang, H.; Tian, Y.; Kulasingam, V.; et al. Platform for establishing interlaboratory reproducibility of selected reaction monitoring-based mass spectrometry peptide assays. J. Proteome Res. 2010, 9, 6678–6688. [Google Scholar] [CrossRef] [PubMed]
- Kockmann, T.; Trachsel, C.; Panse, C.; Wahlander, A.; Selevsek, N.; Grossmann, J.; Wolski, W.E.; Schlapbach, R. Targeted proteomics coming of age—SRM, PRM and DIA performance evaluated from a core facility perspective. Proteomics 2016, 16, 2183–2192. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Noble, W.S.; Wolf-Yadlin, A. Technical advances in proteomics: New developments in data-independent acquisition. F1000 Res. 2016, 5, 419. [Google Scholar] [CrossRef] [PubMed]
- Sen, M.K.; Almuslehi, M.S.M.; Shortland, P.J.; Mahns, D.A.; Coorssen, J.R. Proteomics of Multiple Sclerosis: Inherent issues in defining the pathoetiology and identifying (early) biomarkers. Int. J. Mol. Sci. 2021, 22, 7377. [Google Scholar] [CrossRef]
- Rabilloud, T. When 2D is not enough, go for an extra dimension. Proteomics 2013, 13, 2065–2068. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, J.; Wang, F.; Mao, J.; Zhang, Z.; Liu, Z.; Huang, G.; Cheng, K.; Zou, H. High-sensitivity N-glycoproteomic analysis of mouse brain tissue by protein extraction with a mild detergent of N-dodecyl β-D-maltoside. Anal. Chem. 2015, 87, 2054–2057. [Google Scholar] [CrossRef]
- Epstein, J.A.; Blank, P.S.; Searle, B.C.; Catlin, A.D.; Cologna, S.M.; Olson, M.T.; Backlund, P.S.; Coorssen, J.R.; Yergey, A.L. ProteinProcessor: A probabilistic analysis using mass accuracy and the MS spectrum. Proteomics 2016, 16, 2480–2490. [Google Scholar] [CrossRef]
- Cesnik, A.J.; Shortreed, M.R.; Schaffer, L.V.; Knoener, R.A.; Frey, B.L.; Scalf, M.; Solntsev, S.K.; Dai, Y.; Gasch, A.P.; Smith, L.M. Proteoform Suite: Software for constructing, quantifying, and visualizing proteoform families. J. Proteome Res. 2018, 17, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, D.P.; Rawlins, C.M.; DeHart, C.J.; Fornelli, L.; Schachner, L.F.; Lin, Z.; Lippens, J.L.; Aluri, K.C.; Sarin, R.; Chen, B.; et al. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat. Methods 2019, 16, 587–594. [Google Scholar] [CrossRef]
- Hardman, G.; Perkins, S.; Brownridge, P.J.; Clarke, C.J.; Byrne, D.P.; Campbell, A.E.; Kalyuzhnyy, A.; Myall, A.; Eyers, P.A.; Jones, A.R.; et al. Strong anion exchange-mediated phosphoproteomics reveals extensive human non-canonical phosphorylation. EMBO J. 2019, 38, e100847. [Google Scholar] [CrossRef]
- Meyer, J.G. Fast proteome identification and quantification from data-dependent acquistion-tandem mass spectrometry (DDA MS/MS) using free software tools. Methods Protoc. 2019, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Ahmad, S.; Majumder, S.; Jenkins, C.; Orsburn, B. Bolt: A new age peptide search engine for comprehensive MS/MS sequencing through vast protein databases in minutes. J. Am. Soc. Mass Spectrom. 2019, 30, 2408–2418. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hou, J.; Tanner, J.J.; Cheng, J. Bioinformatics methods for mass spectrometry-based proteomics data analysis. Int. J. Mol. Sci. 2020, 21, 2873. [Google Scholar] [CrossRef]
- Hubler, S.L.; Kumar, P.; Mehta, S.; Easterly, C.; Johnson, J.E.; Jagtap, P.D.; Griffin, T.J. Challenges in peptide-spectrum matching: A robust and reproducible statistical framework for removing low-accuracy, high-scoring hits. J. Proteome Res. 2020, 19, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Teclemariam, E.T.; Pergande, M.R.; Cologna, S.M. Considerations for mass spectrometry-based multi-omic analysis of clinical samples. Expert Rev. Proteom. 2020, 17, 99–107. [Google Scholar] [CrossRef]
- Yu, F.; Haynes, S.E.; Teo, G.C.; Avtonomov, D.M.; Polasky, D.A.; Nesvizhskii, A.I. Fast quantitative analysis of timsTOF PASEF data with MSFragger and IonQuant. Mol. Cell. Proteom. 2020, 19, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Cappelletti, V.; Hauser, T.; Piazza, I.; Pepelnjak, M.; Malinovska, L.; Fuhrer, T.; Li, Y.; Dörig, C.; Boersema, P.; Gillet, L.; et al. Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ. Cell 2021, 184, 545–559.e522. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Perez-Riverol, Y.; Carver, J.; Kawano, S.; Mendoza, L.; Van Den Bossche, T.; Gabriels, R.; Binz, P.-A.; Pullman, B.; Sun, Z.; et al. Universal Spectrum Identifier for mass spectra. Nat. Methods 2021, 18, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Geiszler, D.J.; Kong, A.T.; Avtonomov, D.M.; Yu, F.; Leprevost, F.D.V.; Nesvizhskii, A.I. PTM-Shepherd: Analysis and summarization of post-translational and chemical modifications from open search results. Mol. Cell. Proteom. 2021, 20, 100018. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Park, M.A.; Mann, M. Trapped ion mobility spectrometry (TIMS) and parallel accumulation—Serial fragmentation (PASEF) in proteomics. Mol. Cell. Proteom. 2021, 100138. [Google Scholar] [CrossRef]
- Onisiforou, A.; Spyrou, G.M. Identification of viral-mediated pathogenic mechanisms in neurodegenerative diseases using network-based approaches. Brief. Bioinform. 2021. [Google Scholar] [CrossRef] [PubMed]
- Riffle, M.; Hoopmann, M.R.; Jaschob, D.; Zhong, G.; Moritz, R.L.; MacCoss, M.J.; Davis, T.N.; Isoherranen, N.; Zelter, A. Discovery and visualization of uncharacterized drug-protein adducts using mass spectrometry. bioRxiv 2021. [Google Scholar] [CrossRef]
- Theodorakis, E.; Antonakis, A.N.; Baltsavia, I.; Georgios, A.P.; Samiotaki, M.; Grigoris, D.A.; Theodosiou, T.; Acuto, O.; Efstathiou, G.; Iliopoulos, I. ProteoSign v2: A faster and evolved user-friendly online tool for statistical analyses of differential proteomics. Nucleic Acids Res. 2021, 49, W573–W577. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbonara, K.; Andonovski, M.; Coorssen, J.R. Proteomes Are of Proteoforms: Embracing the Complexity. Proteomes 2021, 9, 38. https://doi.org/10.3390/proteomes9030038
Carbonara K, Andonovski M, Coorssen JR. Proteomes Are of Proteoforms: Embracing the Complexity. Proteomes. 2021; 9(3):38. https://doi.org/10.3390/proteomes9030038
Chicago/Turabian StyleCarbonara, Katrina, Martin Andonovski, and Jens R. Coorssen. 2021. "Proteomes Are of Proteoforms: Embracing the Complexity" Proteomes 9, no. 3: 38. https://doi.org/10.3390/proteomes9030038
APA StyleCarbonara, K., Andonovski, M., & Coorssen, J. R. (2021). Proteomes Are of Proteoforms: Embracing the Complexity. Proteomes, 9(3), 38. https://doi.org/10.3390/proteomes9030038