

Categorical Variable Mapping Considerations in Classification Problems: Protein Application

Abstract
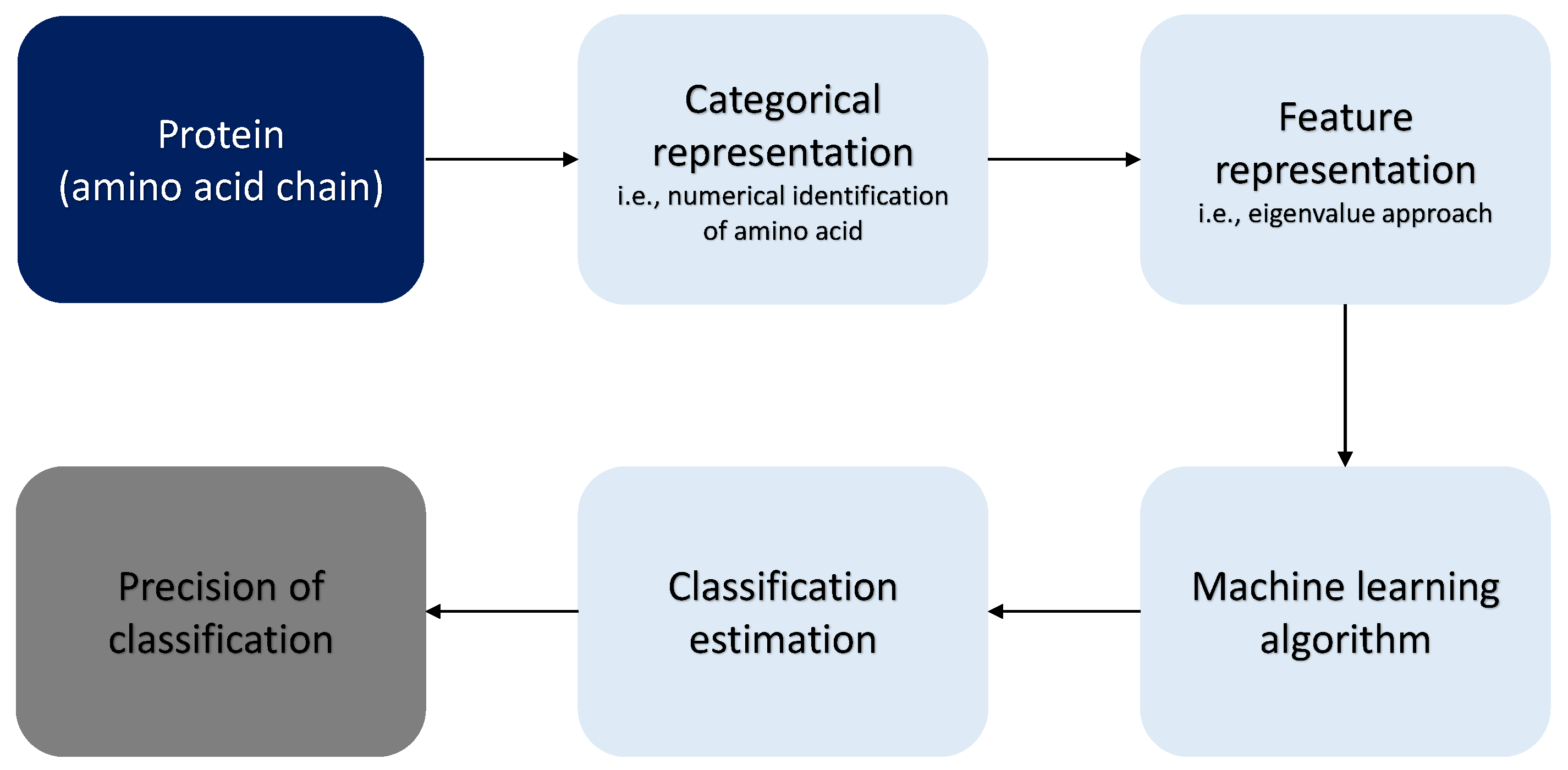
:1. Introduction
2. Literature Review
3. Materials and Methods
3.1. Comparable Mappings
3.2. Optimization
- 1.
- Chose the number of simulations , the required accuracy , the maximum number of iterations , and the maximum number of neurons .
- 2.
- Define a penalty function P. For example,where a is the number of neurons, and is a constant.
- 3.
- Obtain a randomly generated number of neurons , with .
- 4.
- Store a classification vector (target vector) with and the mapping into a matrix X.
- 5.
- 6.
- Train the network with the training dataset .
- 7.
- Estimate the classification estimations .
- 8.
- Estimate as follows:
- 9.
- Estimate the accuracy (Equation (27)) and calculate the additional metrics of precision (Pr), recall (Rec), and F1-score (F1) using Equations (28)–(30), respectively.In this notation, TP, FP, and FN are the true-positive, false-positive, and false-negative values, respectively.
- 10.
- The estimated adjusted accuracy is expressed in Equation (31):This term penalizes an overly complex model with too many neurons.
- 11.
- Compare the results of iterations and choose the model.
- 12.
- Iterate until or .
- 13.
- Repeat k times generating ={ }.
- 14.
- Select .
- 15.
- Calculate the classification estimations (Equation (33)) with the testing dataset for the mode .
- 16.
- Repeat step 7 with to obtain the testing dataset accuracy.
3.3. Data
3.4. Numerical Simulations
4. Results
4.1. Assumption 1
4.2. Assumption 2
4.3. Assumption 3
4.4. Assumption 4
4.5. Optimization
5. Conclusions and Recommendations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M1–M2 | M1–M3 | M1–M4 | M1–M5 |
|---|---|---|---|
| 0.97479 | 0.97479 | 0.31285 | 0.67508 |
| 0.97479 | 0.97479 | 0.031047 | 0.67508 |
| 1.00000 | 0.67508 | 0.67508 | 0.97479 |
| 1.00000 | 0.67508 | 0.11084 | 0.11084 |
| 1.00000 | 0.31285 | 1.89 × 10 | 1.89 × 10 |
| 0.97479 | 0.31285 | 0.97479 | 0.67508 |
| 1.00000 | 0.67508 | 0.31285 | 0.97479 |
| 0.97479 | 0.97479 | 0.11084 | 0.31285 |
| 0.97479 | 0.97479 | 0.67508 | 0.67508 |
| 1.00000 | 0.97479 | 0.97479 | 1.00000 |
| 0.97479 | 0.97479 | 0.97479 | 0.97479 |
| 1.00000 | 1.00000 | 1.00000 | 1.00000 |
| 1.00000 | 0.31285 | 0.67508 | 0.11084 |
| 1.00000 | 0.97479 | 1.00000 | 0.67508 |
| 1.00000 | 1.00000 | 1.00000 | 1.00000 |
| 1.00000 | 0.97479 | 0.67508 | 0.97479 |
| 1.00000 | 0.67508 | 0.97479 | 0.97479 |
| 0.97479 | 0.67508 | 0.67508 | 0.67508 |
| 1.00000 | 0.67508 | 0.031047 | 0.67508 |
| 1.00000 | 0.97479 | 0.11084 | 0.31285 |
| 1.00000 | 0.97479 | 0.11084 | 0.03105 |
| 0.67508 | 0.67508 | 0.31285 | 0.67508 |
| 0.97479 | 0.67508 | 0.11084 | 0.67508 |
| M1–M6 | M1–M7 | M1–M8 | M1–M9 | M1–M10 |
|---|---|---|---|---|
| 0.67508 | 0.67508 | 0.67508 | 0.67508 | 0.31285 |
| 0.97479 | 0.67508 | 0.67508 | 0.67508 | 0.31285 |
| 1.00000 | 0.97479 | 0.67508 | 0.31285 | 0.31285 |
| 0.31285 | 0.11084 | 0.67508 | 0.31285 | 0.31285 |
| 1.00000 | 0.67508 | 1.00000 | 1.00000 | 0.67508 |
| 0.67508 | 0.031047 | 0.97479 | 0.67508 | 0.31285 |
| 0.67508 | 0.97479 | 1.00000 | 0.97479 | 0.11084 |
| 0.31285 | 0.67508 | 0.67508 | 0.67508 | 0.67508 |
| 0.31285 | 0.11084 | 0.31285 | 0.31285 | 0.67508 |
| 1.00000 | 0.97479 | 0.67508 | 1.00000 | 0.97479 |
| 0.97479 | 0.97479 | 0.97479 | 0.97479 | 0.97479 |
| 1.00000 | 1.00000 | 1.00000 | 1.00000 | 1.00000 |
| 0.97479 | 0.97479 | 0.67508 | 0.31285 | 0.97479 |
| 0.67508 | 0.67508 | 0.97479 | 1.00000 | 0.67508 |
| 1.00000 | 1.00000 | 1.00000 | 1.00000 | 1.00000 |
| 0.31285 | 1.00000 | 0.67508 | 0.97479 | 0.97479 |
| 0.31285 | 0.97479 | 0.67508 | 0.97479 | 0.97479 |
| 0.67508 | 0.97479 | 1.00000 | 0.97479 | 0.67508 |
| 0.11084 | 0.0068986 | 0.11084 | 0.031047 | 0.31285 |
| 1.00000 | 0.67508 | 0.31285 | 0.31285 | 0.97479 |
| 1.00000 | 1.00000 | 0.31285 | 0.67508 | 0.67508 |
| M1–M11 | M1–M12 | M1–M13 | M1–M14 |
|---|---|---|---|
| 1.00000 | 0.67508 | 0.11084 | 0.67508 |
| 1.00000 | 0.67508 | 0.31285 | 0.97479 |
| 0.67508 | 0.67508 | 0.31285 | 0.67508 |
| 1.00000 | 0.0012162 | 0.67508 | 0.0068986 |
| 0.97479 | 0.0068986 | 0.97479 | 1.89 × 10 |
| 0.67508 | 0.31285 | 0.97479 | 0.31285 |
| 0.97479 | 0.67508 | 0.97479 | 0.31285 |
| 0.67508 | 0.0068986 | 0.11084 | 0.031047 |
| 0.97479 | 0.00017012 | 0.67508 | 0.0068986 |
| 0.97479 | 0.31285 | 0.67508 | 0.97479 |
| 0.97479 | 0.97479 | 0.97479 | 1.00000 |
| 1.00000 | 1.00000 | 1.00000 | 1.00000 |
| 0.97479 | 0.67508 | 0.67508 | 0.31285 |
| 0.97479 | 1.00000 | 1.00000 | 0.97479 |
| 1.00000 | 1.00000 | 1.00000 | 1.00000 |
| 0.97479 | 0.67508 | 0.97479 | 0.97479 |
| 0.97479 | 0.97479 | 1.00000 | 0.97479 |
| 0.67508 | 0.31285 | 0.67508 | 1.00000 |
| 1.00000 | 0.67508 | 0.67508 | 0.97479 |
| 0.31285 | 0.11084 | 0.97479 | 0.67508 |
| 0.97479 | 0.31285 | 0.97479 | 0.031047 |
| 0.67508 | 0.67508 | 0.67508 | 0.31285 |
| 0.31285 | 0.67508 | 0.67508 | 0.67508 |
| M1–M15 | M1–M16 |
|---|---|
| 0.31285 | 0.67508 |
| 0.031047 | 0.67508 |
| 0.11084 | 0.31285 |
| 0.0012162 | 0.00017012 |
| 1.89 × 10 | 1.89 × 10 |
| 1.89 × 10 | 1.89 × 10 |
| 0.31285 | 0.31285 |
| 0.67508 | 0.31285 |
| 0.97479 | 0.11084 |
| 0.97479 | 0.97479 |
| 0.97479 | 0.31285 |
| 1.00000 | 1.00000 |
| 0.31285 | 0.0012162 |
| 0.67508 | 0.67508 |
| 1.00000 | 1.00000 |
| 0.67508 | 0.67508 |
| 0.67508 | 0.97479 |
| 0.97479 | 1.00000 |
| 0.031047 | 0.67508 |
| 0.97479 | 0.11084 |
| 0.00017012 | 0.00017012 |
| 0.11084 | 0.67508 |
| 0.31285 | 0.97479 |















References
- Carleo, G.; Cirac, I.; Cranmer, K.; Daudet, L.; Schuld, M.; Tishby, N.; Vogt-Maranto, L.; Zdeborová, L. Machine learning and the physical sciences. Rev. Mod. Phys. 2019, 91, 45002–45041. [Google Scholar] [CrossRef] [Green Version]
- Radovic, A.; Williams, M.; Rousseau, D.; Kagan, M.; Bonacorsi, D.; Himmel, A.; Aurisano, A.; Terao, K.; Wongjirad, T. Machine learning at the energy and intensity frontiers of particle physics. Nature 2018, 560, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Karniadakis, G.E.; Kevrekidis, I.G.; Lu, L.; Perdikaris, P.; Wang, S.; Yang, L. Physics-informed machine learning. Nat. Rev. Phys. 2021, 3, 422–440. [Google Scholar] [CrossRef]
- Jimenez, J.; Doerr, S.; Martinez-Rosell, G.; Rose, A.S.; DeFabritis, G. Deepsite: Protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 2017, 19, 3036–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pages, G.; Charmettant, B.; Grudinin, S. Protein model quality assessment using 3D oriented convolutional neural networks. Bioinformatics 2019, 35, 3313–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Terashi, G.; Christoffer, C.W.; Zhu, M.; Kihara, D. Protein docking model evaluation by 3D deep convolutional neural network. Bioinformatics 2020, 36, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein-ligand scoring with convolutional neural networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef] [Green Version]
- Keith, J.A.; Vassilev-Galindo, V.; Cheng, B.; Chmiela, S.; Gastegger, M.; Muller, K.; Tkatchenko, A. Combining machine learning and computational chemistry for predictive insights into chemical systems. Chem. Rev. 2021, 121, 9816–9872. [Google Scholar] [CrossRef]
- Artrith, N.; Butler, K.T.; Coudert, F.; Han, S.; Isayev, O.; Jain, A.; Walsh, A. Best practices in machine learning for chemistry. Nat. Chem. 2021, 13, 505–508. [Google Scholar] [CrossRef]
- Amershi, S.; Begel, A.; Bird, C.; DeLine, R.; Gall, H.; Kamar, E.; Nagappan, N.; Nushi, B.; Zimmermann, T. Software engineering for machine learning: A case study. In Proceedings of the 2019 IEEE/ACM 41st International Conference on Software Engineering: Software Engineering in Practice (ICSE-SEIP), Montreal, QC, Canada, 25–31 May 2019; pp. 291–300. [Google Scholar]
- Park, C.; Took, C.C.; Seong, J. Machine learning in biomedical engineering. Biomed. Eng. Lett. 2018, 8, 1–3. [Google Scholar] [CrossRef]
- Zhang, D.; Tsai, J. Machine learning and software engineering. Softw. Qual. J. 2003, 11, 87–119. [Google Scholar] [CrossRef]
- Rodriguez-Galiano, V.; Sanchez-Castillo, M.; Chica-Olmo, M.; Chica-Rivas, M. Machine learning predictive models for mineral prospectivity: An evaluation of neural networks, random forest, regression trees and support vector machines. Ore Geol. Rev. 2015, 71, 804–818. [Google Scholar] [CrossRef]
- Blanco-Justicia, A.; Domingo-Ferrer, J. Machine learning explainability through comprehensible decision trees. In Proceedings of the International Cross-Domain Conference for Machine Learning and Knowledge Extraction, Canterbury, UK, 26–29 August 2019; pp. 15–26. [Google Scholar]
- Allen, J.P.; Snitkin, E.; Pincus, N.B.; Hauser, A.R. Forest and trees: Exploring bacterial virulence with genome-wide association studies and machine learning. Trends Microbiol. 2021, 29, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Wang, H.; Bell, D.; Bi, Y.; Greer, K. OTM Confederated International Conferences “On the Move to Meaningful Internet Systems”; Springer: Berlin/Heidelberg, Germany, 2003; pp. 986–996. [Google Scholar]
- Lee, T.; Wood, W.T.; Phrampus, B.J. A machine learning (kNN) approach to predicting global seafloor total organic carbon. Wiley Online Libr. 2019, 33, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Li, X.; Zong, M.; Zhu, X.; Cheng, D. Learning k for knn classification. ACM Trans. Intell. Syst. Technol. 2017, 8, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Noble, W.S. What is a support vector machine? Nat. Biol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support vector machine. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Pisner, D.A.; Schnyer, D.M. Support vector machine. In Chapter 6—Machine Learning; Academic Press: Cambridge, MA, USA, 2020; pp. 101–121. [Google Scholar]
- Qi, D.; Majda, A.J. Using machine learning to predict extreme events in complex systems. Proc. Natl. Acad. Sci. USA 2020, 117, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Qi, D.; Majda, A.J. Introduction to Focus Issue: When machine learning meets complex systems: Networks, chaos, and nonlinear dynamics. Chaos Interdiscip. J. Nonlinear Sci. 2020, 30, 063151. [Google Scholar]
- Wood, D. A transparent open-box learning network provides insight to complex systems and a performance benchmark for more-opaque machine learning algorithms. Adv. Geo-Energy Res. 2018, 2, 148–162. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Hu, F.; Liu, Y.; Witherell, P.; Wang, C.; Rosen, D.W.; Simpson, T.; Lu, Y.; Tang, Q. Research and application of machine learning for additive manufacturing. Addit. Manuf. 2022, 52, 102691. [Google Scholar] [CrossRef]
- Rudin, C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat. Mach. Intell. 2019, 1, 206–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGovern, A.; Lagerquist, R.; Gagne, D.J.; Jergensen, G.E.; Elmore, K.L.; Homeyer, C.R.; Smith, T. Making the black box more transparent: Understanding the physical implications of machine learning. Nat. Mach. Intell. 2019, 100, 2175–2199. [Google Scholar] [CrossRef]
- Zhou, Z. Learnware: On the future of machine learning. Front. Comput. Sci. 2016, 10, 589–590. [Google Scholar] [CrossRef]
- Cerda, P.; Varoquaux, G.; Kégl, B. Similarity encoding for learning with dirty categorical variables. Mach. Learn. 2018, 8, 1477–1494. [Google Scholar] [CrossRef] [Green Version]
- Cerda, P.; Varoquaux, G.; Kégl, B. Encoding high-cardinality string categorical variables. IEEE Trans. Knowl. Data Eng. 2020, 34, 1164–1176. [Google Scholar] [CrossRef]
- Sonego, P.; Pacurar, M.; Dhir, S.; Kertesz-Farkas, A.; Kocsor, A.; Gáspári, Z.; Leunissen, J.A.M.; Pongor, S. A protein classification benchmark collection for machine learning. Nucleic Acids Res. 2007, 35, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Garibaldi, J.M.; Kirst, J.D. Supervised machine learning algorithms for protein structure classification. Comput. Biol. Chem. 2009, 33, 216–223. [Google Scholar] [CrossRef]
- Muller, B.; Joachim, R.; Strickland, M.T. Neural Networks an Introduction; Springer Science & Business Media: Abingdon, UK, 1995. [Google Scholar]
- Anderson, J.A. An Introduction to Neural Networks; MIT Press: Cambridge, MA, USA, 1995. [Google Scholar]
- Miller, W.T.; Werbos, P.J.; Sutton, R.S. Neural Networks for Control; MIT Press: Cambridge, MA, USA, 1995. [Google Scholar]
- Le, T.; Hoai, A.; Le, H.M.; Pham, D.T. Feature selection in machine learning: An exact penalty approach using a difference of convex function algorithm. Mach. Learn. 2015, 101, 163–186. [Google Scholar]
- Jiang, M.; Meng, Z.; Shen, R. Partial Exactness for the Penalty Function of Biconvex Programming. Entropy 2021, 23, 132. [Google Scholar] [CrossRef]
- Roelofs, R.; Shankar, V.; Recht, B.; Fridovich-Keil, S.; Hardt, M.; Miller, J.; Schmidt, L. A meta-analysis of overfitting in machine learning. Adv. Neural Inf. Process. Syst. 2019, 32, 1–11. [Google Scholar]
- Peng, Y.; Nagata, M.H. An empirical overview of nonlinearity and overfitting in machine learning using COVID-19 data. Chaos Solitons Fractals 2020, 139, 110055. [Google Scholar] [CrossRef]
- De Prisco, R.; Guarino, A.; Lettieri, N.; Malandrino, D.; Zaccagnino, R. Providing music service in ambient intelligence: Experiments with gym users. Expert Syst. Appl. 2021, 177, 114951. [Google Scholar] [CrossRef]
- Kamerzell, T.J.; Middaugh, C.R. Prediction machines: Applied machine learning for therapeutic protein design and development. J. Pharm. Sci. 2021, 110, 665–681. [Google Scholar] [CrossRef]
- Xu, Y.; Verma, D.; Sheridan, R.P.; Liaw, A.; Ma, J.; Marshall, N.M.; McIntosh, J.; Sherer, E.C.; Svetnik, V.; Johnston, J.M. Deep Dive into Machine Learning Models for Protein Engineering. J. Chem. Inf. Model. 2020, 60, 2773–2790. [Google Scholar] [CrossRef]
- Salau, A.O.; Jain, S. Adaptive diagnostic machine learning technique for classification of cell decisions for AKT protein. Inform. Med. Unlocked 2021, 23, 100511. [Google Scholar] [CrossRef]
- Salau, A.O.; Jain, S. Computational modeling and experimental analysis for the diagnosis of cell survival/death for Akt protein. J. Genet. Eng. Biotechnol. 2020, 18, 1–10. [Google Scholar] [CrossRef]
- Jain, S.; Salau, A.O. An image feature selection approach for dimensionality reduction based on kNN and SVM for AkT proteins. Cogent Eng. 2019, 6, 1599537. [Google Scholar] [CrossRef]
- Hancock, J.T.; Khoshgoftaar, T.M. Survey on categorical data for neural networks. J. Big Data 2020, 7, 1–41. [Google Scholar] [CrossRef] [Green Version]
- Ofer, D.; Brandes, N.; Linial, M. The language of proteins: NLP, machine learning & protein sequences. Comput. Struct. Biotechnol. J. 2021, 19, 1750–1758. [Google Scholar]
- McDowall, J.; Hunter, S. InterPro protein classification. Bioinform. Comp. Proteom. 2011, 694, 37–47. [Google Scholar]
- Nanni, L.; Lumini, A.; Brahnam, S. An empirical study of different approaches for protein classification. Sci. World J. 2014, 2014, 236717. [Google Scholar] [CrossRef]
- Diplaris, S.; Tsoumakas, G.; Mitkas, P.A.; Vlahavas, I. Protein classification with multiple algorithms. Panhellenic Conf. Inform. 2005, 7, 448–456. [Google Scholar]
- Cai, C.Z.; Han, L.Y.; Ji, Z.L.; Chen, X.; Chen, Y.Z. SVM-Prot: Web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 2003, 31, 3692–3697. [Google Scholar] [CrossRef] [Green Version]
- Bock, J.R.; Gough, D.A. Predicting protein–protein interactions from primary structure. Bioinformatics 2001, 17, 455–460. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Chakrabarti, S. Classification and prediction of protein–protein interaction interface using machine learning algorithm. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Karchin, R.; Karplus, K.; Haussler, D. Classifying G-protein coupled receptors with support vector machines. Bioinformatics 2002, 18, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Gao, Y.; Wang, Y.; Pan, G. Mussel-inspired peptide mimicking: An emerging strategy for surface bioengineering of medical implants. Smart Mater. Med. 2021, 2, 26–37. [Google Scholar] [CrossRef]
- Kazemzadeh-Narbat, M.; Cheng, H.; Chabok, R.; Alvarez, M.M.; De La Fuente-Nunez, C.; Phillips, K.S.; Khademhosseini, A. Strategies for antimicrobial peptide coatings on medical devices: A review and regulatory science perspective. Crit. Rev. Biotechnol. 2021, 41, 94–120. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Bojarska, J.; Chai, T.-T.; Elnagdy, S.; Kaczmarek, K.; Matsoukas, J.; New, R.; Parang, K.; Lopez, O.P.; Parhiz, H. A global review on short peptides: Frontiers and perspectives. Molecules 2021, 26, 430. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Chiangjong, W.; Hasan, M.M.; Nantasenamat, C.; Shoombuatong, W. Review and Comparative Analysis of Machine Learning-based Predictors for Predicting and Analyzing Anti-angiogenic Peptides. Curr. Med. Chem. 2022, 29, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Jenssen, H.; Hilpert, K.; Cheung, W.A.; Pante, N.; Hancock, R.E.W.; Cherkasov, A. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 2009, 52, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Rondon-Villarreal, P.; Sierra, D.; Torres, R. Machine learning in the rational design of antimicrobial peptides. Curr. Comput. Aided Drug Des. 2014, 10, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Mousavizadegan, M.; Mohabatkar, H. An evaluation on different machine learning algorithms for classification and prediction of antifungal peptides. Med. Chem. 2016, 12, 795–800. [Google Scholar] [CrossRef]
- Sen, P.C.; Hajra, M.; Ghosh, M. Supervised classification algorithms in machine learning: A survey and review. Emerg. Technol. Model. Graph. 2020, 937, 99–111. [Google Scholar]
- Ivankov, D.N.; Finkelstein, A.V. Prediction of protein folding rates from the amino acid sequence predicted secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 8942–8944. [Google Scholar] [CrossRef] [Green Version]
- Kunt, I.D.; Crippen, G.M.; Kollman, P.A. Calculation of protein tertiary structure. J. Mol. Biol. 1976, 106, 983–994. [Google Scholar]
- Hagler, A.T.; Barry, H. On the formation of the protein tertiary structure on a computer. Proc. Natl. Acad. Sci. USA 1978, 75, 554–558. [Google Scholar] [CrossRef]
- Salau, A.O.; Jain, S. Feature Extraction: A Survey of the Types, Techniques, Applications. In Proceedings of the 2019 International Conference on Signal Processing and Communication (ICSC), Noida, India, 7–9 March 2019; Volume 75, pp. 158–164. [Google Scholar]
- Zur, R.M.; Jiang, Y.P.; Pesce, L.L. Noise injection for training artificial neural networks. A comparison with weight decay and early stopping. Med. Phys. 2009, 36, 4810–4818. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Setiono, R.; Huan, L. Effective data mining using neural networks. IEE Trans. Knowl. Data Eng. 1996, 8, 957–961. [Google Scholar]
- Torgyn, S.; Khovanova, N.A. Handling limited datasets with neural networks applications: A small data approach. Artif. Intell. Med. 2017, 75, 51–63. [Google Scholar]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2016, gkw1000, 271–281. [Google Scholar]
- Rose, P.W.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dimitropoulus, D.; Dutta, S.; Bourne, P.E. The RCSB Protein data bank: New resources for research and education. Nucleic Acids Res. 2012, 41, 475–482. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Springs, R.V.; Artymiuk, P.J.; Willet, W. Searching for 3D patterns of amino acids in 3D protein structures. J. Chem. Inf. Comput. Sci. 2003, 43, 412–421. [Google Scholar] [CrossRef]
- Abola, E.E.; Bernstein, F.C.; Frances, C.; Koetzle, T.F. The protein data bank. Neutroms in Biology; Springer: Boston, MA, USA, 1984. [Google Scholar]
- Berman, H.M.; Henrick, K.; Nakamura, H. Announcing the worldwide protein data bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Parasuraman, S. Protein data bank. J. Pharmacol. Pharmacother. 2012, 3, 351. [Google Scholar] [CrossRef]
- Sussman, J.L.; Abola, E.E.; Lin, D.; Jiang, J.; Manning, N.O. The protein data bank. Struct. Biol. Funct. Genom. 1999, 54, 251–264. [Google Scholar]
- Fauman, E.B.; Hyde, C. An optimal variant to gene distance window derived from an empirical definition of cis and trans protein QTLs. BMC Bioinform. 2022, 23, 1–11. [Google Scholar] [CrossRef]
- Guarino, A.; Malandrino, D.; Zaccagnino, R. An automatic mechanism to provide privacy awareness and control over unwittingly dissemination of online private information. Comput. Netw. 2022, 202, 108614. [Google Scholar] [CrossRef]








| N. | Algorithm | N. | Algorithm |
|---|---|---|---|
| 1 | Complex Tree (fitctree) | 13 | Fine KNN (fitcknn) |
| 2 | Medium Tree (fitctree) | 14 | Medium KNN (fitcknn) |
| 3 | Simple Tree (fitctree) | 15 | Coarse KNN (fitcknn) |
| 4 | Linear Discriminant (fitcdiscr) | 16 | Cosine KNN (fitcknn) |
| 5 | Quadratic Discriminant (fitcdiscr) | 17 | Cubic KNN (fitcknn) |
| 6 | Logistic Regression (fitglm) | 18 | Weighted KNN (fitcknn) |
| 7 | Linear SVM (fitcsvm) | 19 | Boosted Trees (fitctree) |
| 8 | Quadratic SVM (fitcsvm) | 20 | Bagged Tress (fitctree) |
| 9 | Cubic SVM (fitcsvm) | 21 | Subspace Discriminant (fitcdiscr) |
| 10 | Fine Gaussian SVM (fitcsvm) | 22 | Subspace KNN (fitcknn) |
| 11 | Medium Gaussian SVM (fitcsvm) | 23 | RUSBoosted Trees (fitctree) |
| 12 | Coarse Gaussian SVM (fitcsvm) |
| Algorithm | Eig. Acc. | M1 Acc. | p-Value (ks) | T (s) |
|---|---|---|---|---|
| Complex Tree | 82.89 | 72.04 | 0.0001 | 1.34 |
| Medium Tree | 82.83 | 72.13 | 0.0001 | 1.79 |
| Simple Tree | 82.75 | 78.11 | 0.0002 | 0.60 |
| Linear Discriminant | 17.08 | 54.44 | 0.0001 | 1.70 |
| Quadratic Discriminant | 30.37 | 17.47 | 0.6751 | 1.82 |
| Logistic regression | 82.95 | 61.87 | 0.0001 | 4.32 |
| Fine KNN | 79.88 | 76.53 | 0.0002 | 7.96 |
| Medium KNN | 81.59 | 83.18 | 0.6751 | 7.85 |
| Coarse KNN | 80.65 | 82.88 | 1.0000 | 7.69 |
| Cosine KNN | 81.83 | 82.95 | 0.6751 | 8.52 |
| Cubic KNN | 81.75 | 83.14 | 0.6751 | 7.10 |
| Weighted KNN | 83.25 | 81.83 | 0.0069 | 6.90 |
| Boosted Trees | 80.65 | 75.64 | 0.0001 | 7.64 |
| Bagged Trees | 82.83 | 81.6 | 0.0069 | 9.71 |
| Subspace Discriminant | 81.91 | 77.03 | 0.0001 | 10.98 |
| Subspace KNN | 82.59 | 78.91 | 0.0002 | 11.84 |
| RUSBoosted Trees | 81.55 | 54.74 | 0.0001 | 13.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfonso Perez, G.; Castillo, R. Categorical Variable Mapping Considerations in Classification Problems: Protein Application. Mathematics 2023, 11, 279. https://doi.org/10.3390/math11020279
Alfonso Perez G, Castillo R. Categorical Variable Mapping Considerations in Classification Problems: Protein Application. Mathematics. 2023; 11(2):279. https://doi.org/10.3390/math11020279
Chicago/Turabian StyleAlfonso Perez, Gerardo, and Raquel Castillo. 2023. "Categorical Variable Mapping Considerations in Classification Problems: Protein Application" Mathematics 11, no. 2: 279. https://doi.org/10.3390/math11020279