
Association Testing of a Group of Genetic Markers Based on Next-Generation Sequencing Data and Continuous Response Using a Linear Model Framework

Abstract
:1. Introduction
2. Methodology
2.1. Model Continuous Phenotype Using a Linear Model Framework
2.2. Uncertain Genotypes
2.3. Joint Significance Test for a Group of Common Genetic Variants
- the analytical formula for the sore function;
- the evaluation of the score function at the constrained MLE;
- the analytical formula for the observed information matrix;
- the evaluation of the observed information matrix evaluated at the constrained MLE.
2.4. Variable Collapse Test for a Group of Rare Genetic Variants
3. Results of Simulation Studies
3.1. Results of Type I Errors
3.2. Results of Type II Errors and Power Analyses
3.3. Results of Estimating Allele Frequencies
4. Discussion
5. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GC | Genotype calling |
| GLF | Genotype likelihood function |
| GLM | Generalized linear model |
| JS | Joint significance |
| LM | Linear model |
| LRT | Likelihood ratio test |
| MAD | Mean absolute deviation |
| MSE | Mean squared error |
| NGS | Next-generation sequencing |
| SKAT | Sequence kernel association test |
| VC | Variable collapse |
Appendix A. Detailed Derivation for Simplification of Log-Likelihood Under Null Hypothesis H0: β = 0
Appendix B. Analytical Formula of Score Function
Appendix C. Evaluation of Score Function at Constrained MLE
Appendix D. Analytical Formula of Observed Information Matrix
Appendix E. Evaluation of Observed Information Matrix at Constrained MLE
References
- Men, A.E.; Wilson, P.; Siemering, K.; Forrest, S. Sanger DNA sequencing. In Next Generation Genome Sequencing: Towards Personalized Medicine; John Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 1–11. [Google Scholar]
- Illumina_Inc. DNA Sequencing with Solexa® Technology. Available online: https://courses.cs.duke.edu/spring21/compsci260/resources/GenomeSequencingTechnology/Illumina.Solexa.sequencing.pdf (accessed on 15 January 2023).
- Wall, P.K.; Leebens-Mack, J.; Chanderbali, A.S.; Barakat, A.; Wolcott, E.; Liang, H.; Landherr, L.; Tomsho, L.P.; Hu, Y.; Carlson, J.E.; et al. Comparison of next generation sequencing technologies for transcriptome characterization. BMC Genom. 2009, 10, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardis, E.R. Next-generation sequencing platforms. Annu. Rev. Anal. Chem. 2013, 6, 287–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef]
- Long, K.; Cai, L.; He, L. DNA sequencing data analysis. In Computational Systems Biology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 1–13. [Google Scholar]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.H.; Asselbergs, F.W.; Williams, S.M. Bioinformatics challenges for genome-wide association studies. Bioinformatics 2010, 26, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Guo, Y.; Li, J.; Long, J.; Zhang, B.; Shyr, Y. Steps to ensure accuracy in genotype and SNP calling from Illumina sequencing data. BMC Genom. 2012, 13, S8. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Korneliussen, T.; Albrechtsen, A.; Li, Y.; Wang, J. SNP calling, genotype calling, and sample allele frequency estimation from new-generation sequencing data. PLoS ONE 2012, 7, e37558. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.M.; Knight, J. Introduction to Genetic Association Studies; CSHL Press: Cold Spring Harbor, NY, USA, 2012; Volume 2012. [Google Scholar] [CrossRef] [Green Version]
- Balding, D.J. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 2006, 7, 781–791. [Google Scholar] [CrossRef]
- Huang, E.; Aitken, K.; George, A. Association studies. In Genetics, Genomics and Breeding of Sugarcane; CRC Press: Boca Raton, FL, USA, 2010; pp. 43–68. [Google Scholar]
- Cordell, H.J.; Clayton, D.G. Genetic association studies. Lancet 2005, 366, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Abecasis, G.R.; Boehnke, M.; Lin, X. Rare-variant association analysis: Study designs and statistical tests. Am. J. Hum. Genet. 2014, 95, 5–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Via, M.; Gignoux, C.; Burchard, E.G. The 1000 Genomes Project: New opportunities for research and social challenges. Genome Med. 2010, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Boerwinkle, E.; Xiong, M. Association studies for next-generation sequencing. Genome Res. 2011, 21, 1099–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galesloot, T.E.; Van Steen, K.; Kiemeney, L.A.; Janss, L.L.; Vermeulen, S.H. A comparison of multivariate genome-wide association methods. PLoS ONE 2014, 9, e95923. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Sung, P.Y.; Lin, P.L.; Yu, Y.W.; Chung, R.H. A multi-SNP association test for complex diseases incorporating an optimal P-value threshold algorithm in nuclear families. BMC Genom. 2015, 16, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auer, P.L.; Lettre, G. Rare variant association studies: Considerations, challenges and opportunities. Genome Med. 2015, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.J.; Leal, S.M. A novel adaptive method for the analysis of next-generation sequencing data to detect complex trait associations with rare variants due to gene main effects and interactions. PLoS Genet. 2010, 6, e1001156. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.Y. Beyond rare-variant association testing: Pinpointing rare causal variants in case-control sequencing study. Sci. Rep. 2016, 6, 21824. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Akinsanmi, I.; Arafat, D.; Cradick, T.; Lee, C.M.; Banskota, S.; Marigorta, U.M.; Bao, G.; Gibson, G. A burden of rare variants associated with extremes of gene expression in human peripheral blood. Am. J. Hum. Genet. 2016, 98, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.C.; Lee, S.; Cai, T.; Li, Y.; Boehnke, M.; Lin, X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011, 89, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Wu, M.C.; Lin, X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics 2012, 13, 762–775. [Google Scholar] [CrossRef] [Green Version]
- Plagnol, V.; Cooper, J.D.; Todd, J.A.; Clayton, D.G. A method to address differential bias in genotyping in large-scale association studies. PLoS Genet. 2007, 3, e74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sham, P.C.; Purcell, S.M. Statistical power and significance testing in large-scale genetic studies. Nat. Rev. Genet. 2014, 15, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Skotte, L.; Korneliussen, T.S.; Albrechtsen, A. Association testing for next-generation sequencing data using score statistics. Genet. Epidemiol. 2012, 36, 430–437. [Google Scholar] [CrossRef]
- Yan, S.; Yuan, S.; Xu, Z.; Zhang, B.; Zhang, B.; Kang, G.; Byrnes, A.; Li, Y. Likelihood-based complex trait association testing for arbitrary depth sequencing data. Bioinformatics 2015, 31, 2955–2962. [Google Scholar] [CrossRef] [Green Version]
- Harismendy, O.; Ng, P.C.; Strausberg, R.L.; Wang, X.; Stockwell, T.B.; Beeson, K.Y.; Schork, N.J.; Murray, S.S.; Topol, E.J.; Levy, S.; et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009, 10, R32. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, W.; Liu, E.Y.; Zhou, Y.H. Single nucleotide polymorphism (SNP) detection and genotype calling from massively parallel sequencing (MPS) data. Stat. Biosci. 2013, 5, 3–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Chanda, P.; Alonso, A.; Bader, J.S.; Arking, D.E. Gene-based tests of association. PLoS Genet. 2011, 7, e1002177. [Google Scholar] [CrossRef] [Green Version]
- Weir, B.S. Genetic Data Analysis II; Sinauer Associates: Sunderland, MA, USA, 1996. [Google Scholar]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef]
- Li, Y.; Willer, C.J.; Ding, J.; Scheet, P.; Abecasis, G.R. MaCH: Using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010, 34, 816–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, H.; Xu, L.; Su, Z.; Liu, J.; Ge, W.; Shen, J.; Fang, H.; Perkins, R.; Shi, L.; Tong, W. Pitfall of genome-wide association studies: Sources of inconsistency in genotypes and their effects. J. Biomed. Sci. Eng. 2012, 5, 23768. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Li, Y. BETASEQ: A powerful novel method to control type-I error inflation in partially sequenced data for rare variant association testing. Bioinformatics 2014, 30, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of next generation sequencing data. BMC Bioinform. 2014, 15, 356. [Google Scholar] [CrossRef] [Green Version]
- Belonogova, N.M.; Svishcheva, G.R.; Axenovich, T.I. FREGAT: An R package for region-based association analysis. Bioinformatics 2016, 32, 2392–2393. [Google Scholar] [CrossRef]
- Agresti, A. Categorical Data Analysis; Wiley Series in Probability and Statistics; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- McCullagh, P.; Nelder, J. Generalized Linear Models, 2nd ed.; Chapman & Hall: London, UK, 1989. [Google Scholar]
- Baxter, M. Generalised linear models, by P. McCullagh and JA Nelder. Pp 511.£ 30. 1989. ISBN 0-412-31760-5 (Chapman and Hall). Math. Gaz. 1990, 74, 320–321. [Google Scholar] [CrossRef]
- Cox, D.R. Principles of Statistical Inference; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Young, G.A.; Smith, R.L. Essentials of Statistical Inference; Cambridge University Press: Cambridge, UK, 2005; Volume 16. [Google Scholar]
- Sul, J.H.; Han, B.; He, D.; Eskin, E. An optimal weighted aggregated association test for identification of rare variants involved in common diseases. Genetics 2011, 188, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Ionita-Laza, I.; Buxbaum, J.D.; Laird, N.M.; Lange, C. A new testing strategy to identify rare variants with either risk or protective effect on disease. PLoS Genet. 2011, 7, e1001289. [Google Scholar] [CrossRef] [Green Version]
- Schaffner, S.F.; Foo, C.; Gabriel, S.; Reich, D.; Daly, M.J.; Altshuler, D. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005, 15, 1576–1583. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Huang, K.C.; Xu, Z.; Wang, Y.; Abecasis, G.R.; Li, Y. AbCD: Arbitrary coverage design for sequencing-based genetic studies. Bioinformatics 2013, 29, 799–801. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.M.; Zheng, J.P.; Yang, D.; Liu, Z.F.; Li, Z.; Hu, Z.Z.; Li, Z.N. Recessive/dominant model: Alternative choice in case-control-based genome-wide association studies. PLoS ONE 2021, 16, e0254947. [Google Scholar] [CrossRef] [PubMed]
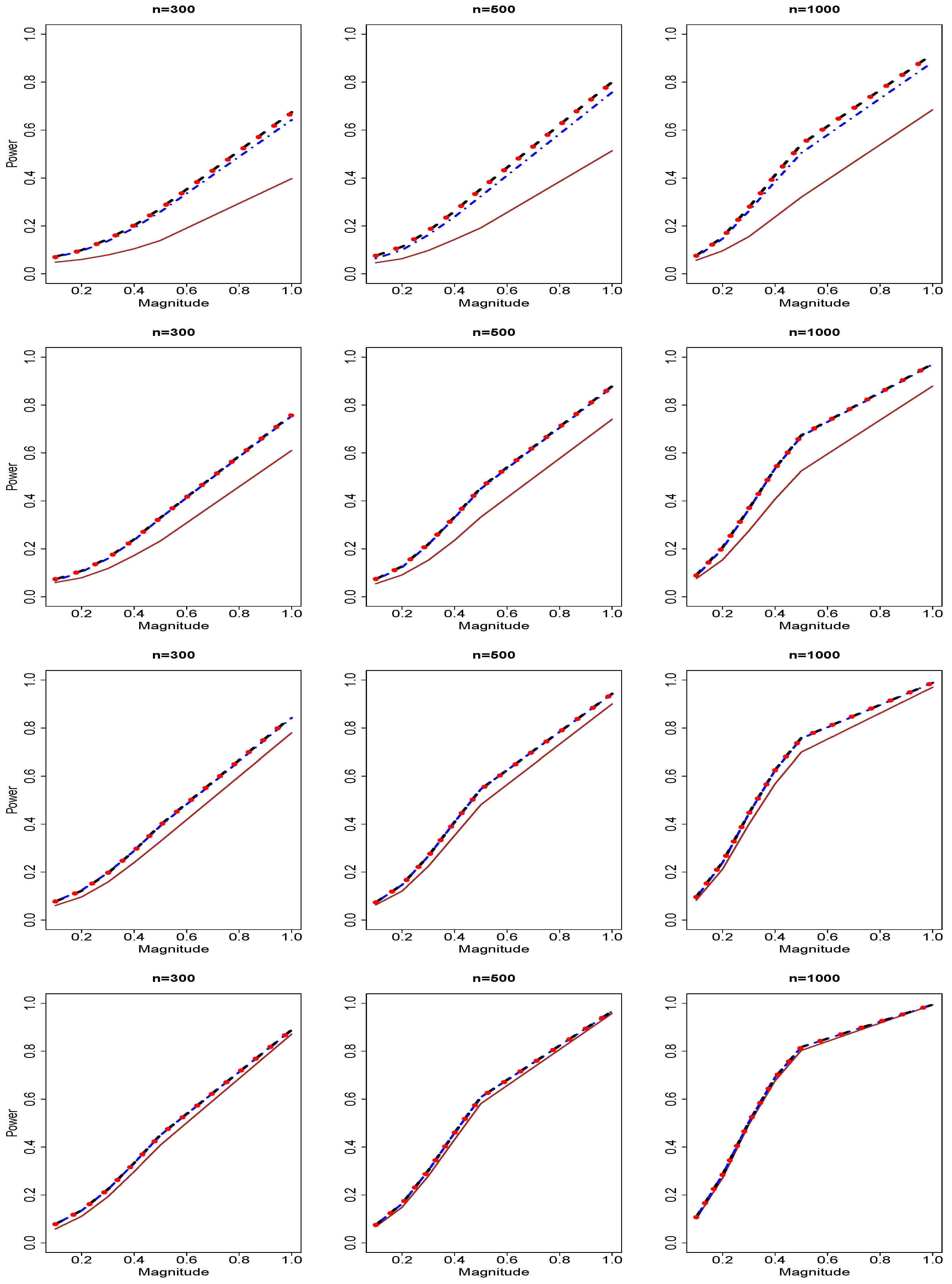
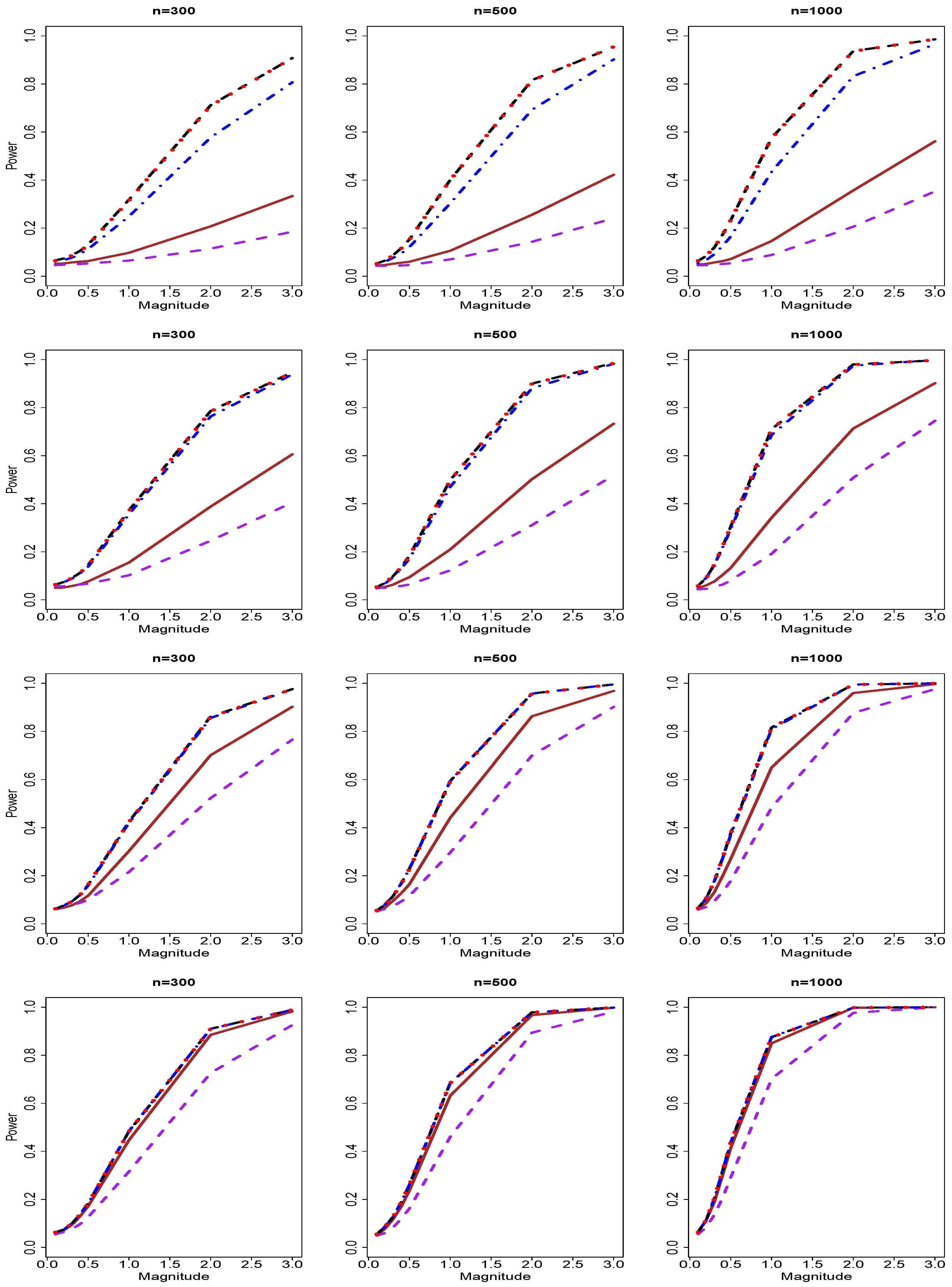

{kind=link}
{kind=link}
| Sample Size | Depth | Genotype-Based F Test | NGS JS Test 1 | NGS JS Test 2 | NGS JS Test 3 |
|---|---|---|---|---|---|
| 300 | 1 | 0.045 | 0.053 | 0.055 | 0.053 |
| 500 | 1 | 0.037 | 0.054 | 0.045 | 0.054 |
| 1000 | 1 | 0.039 | 0.043 | 0.039 | 0.043 |
| 300 | 2 | 0.047 | 0.050 | 0.047 | 0.051 |
| 500 | 2 | 0.042 | 0.049 | 0.048 | 0.049 |
| 1000 | 2 | 0.048 | 0.047 | 0.045 | 0.047 |
| 300 | 4 | 0.042 | 0.052 | 0.052 | 0.052 |
| 500 | 4 | 0.042 | 0.044 | 0.044 | 0.044 |
| 1000 | 4 | 0.041 | 0.039 | 0.039 | 0.039 |
| 300 | 10 | 0.041 | 0.050 | 0.050 | 0.050 |
| 500 | 10 | 0.042 | 0.042 | 0.042 | 0.042 |
| 1000 | 10 | 0.046 | 0.042 | 0.042 | 0.042 |
| Sample Size | Depth | Burden | SKAT | NGS VC Test 1 | NGS VC Test 2 | NGS VC Test 3 |
|---|---|---|---|---|---|---|
| 300 | 1 | 0.041 | 0.051 | 0.050 | 0.049 | 0.050 |
| 500 | 1 | 0.043 | 0.032 | 0.043 | 0.039 | 0.044 |
| 1000 | 1 | 0.044 | 0.032 | 0.041 | 0.040 | 0.042 |
| 300 | 2 | 0.036 | 0.048 | 0.048 | 0.046 | 0.048 |
| 500 | 2 | 0.044 | 0.051 | 0.039 | 0.036 | 0.040 |
| 1000 | 2 | 0.043 | 0.029 | 0.039 | 0.044 | 0.039 |
| 300 | 4 | 0.054 | 0.056 | 0.051 | 0.051 | 0.051 |
| 500 | 4 | 0.041 | 0.036 | 0.041 | 0.040 | 0.042 |
| 1000 | 4 | 0.044 | 0.049 | 0.045 | 0.043 | 0.044 |
| 300 | 10 | 0.050 | 0.043 | 0.052 | 0.053 | 0.052 |
| 500 | 10 | 0.042 | 0.043 | 0.042 | 0.042 | 0.042 |
| 1000 | 10 | 0.039 | 0.030 | 0.037 | 0.037 | 0.037 |
| Sample Size | Depth | ||||
|---|---|---|---|---|---|
| 300 | 1 | 0.12792 | 0.01389 | 0.01934 | 0.00035 |
| 300 | 2 | 0.05177 | 0.00862 | 0.00324 | 0.00014 |
| 300 | 4 | 0.01408 | 0.00506 | 0.00027 | 0.00005 |
| 300 | 10 | 0.00338 | 0.00178 | 0.00002 | 0.00001 |
| 500 | 1 | 0.13011 | 0.01047 | 0.01978 | 0.00020 |
| 500 | 2 | 0.05199 | 0.00661 | 0.00320 | 0.00008 |
| 500 | 4 | 0.01360 | 0.00394 | 0.00024 | 0.00003 |
| 500 | 10 | 0.00312 | 0.00139 | 0.00002 | 0.00000 |
| 1000 | 1 | 0.13180 | 0.00726 | 0.02011 | 0.00010 |
| 1000 | 2 | 0.05178 | 0.00468 | 0.00314 | 0.00004 |
| 1000 | 4 | 0.01326 | 0.00277 | 0.00022 | 0.00001 |
| 1000 | 10 | 0.00288 | 0.00099 | 0.00001 | 0.00000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z. Association Testing of a Group of Genetic Markers Based on Next-Generation Sequencing Data and Continuous Response Using a Linear Model Framework. Mathematics 2023, 11, 1285. https://doi.org/10.3390/math11061285
Xu Z. Association Testing of a Group of Genetic Markers Based on Next-Generation Sequencing Data and Continuous Response Using a Linear Model Framework. Mathematics. 2023; 11(6):1285. https://doi.org/10.3390/math11061285
Chicago/Turabian StyleXu, Zheng. 2023. "Association Testing of a Group of Genetic Markers Based on Next-Generation Sequencing Data and Continuous Response Using a Linear Model Framework" Mathematics 11, no. 6: 1285. https://doi.org/10.3390/math11061285