All articles published by MDPI are made immediately available worldwide under an open access license. No special
permission is required to reuse all or part of the article published by MDPI, including figures and tables. For
articles published under an open access Creative Common CC BY license, any part of the article may be reused without
permission provided that the original article is clearly cited. For more information, please refer to
https://www.mdpi.com/openaccess.
Feature papers represent the most advanced research with significant potential for high impact in the field. A Feature
Paper should be a substantial original Article that involves several techniques or approaches, provides an outlook for
future research directions and describes possible research applications.
Feature papers are submitted upon individual invitation or recommendation by the scientific editors and must receive
positive feedback from the reviewers.
Editor’s Choice articles are based on recommendations by the scientific editors of MDPI journals from around the world.
Editors select a small number of articles recently published in the journal that they believe will be particularly
interesting to readers, or important in the respective research area. The aim is to provide a snapshot of some of the
most exciting work published in the various research areas of the journal.
For rare disease drug development, the United States (US) Food and Drug Administration (FDA) has indicated that the same standards as those for drug products for common conditions will be applied. To assist the sponsors in rare disease drug development, the FDA has initiated several incentive programs to encourage the sponsors in rare disease drug development. In practice, these incentive programs may not help in achieving the study objectives due to the limited small patient population. To overcome this problem, some out-of-the-box innovative thinking and/or approaches, without jeopardizing the integrity, quality, and scientific validity of rare disease drug development, are necessarily considered. These innovative thinking and/or approaches include but are not limited to (i) sample size justification based on probability statements rather than conventional power analysis; (ii) demonstrating not-ineffectiveness and not-unsafeness rather than demonstrating effectiveness and safety with the small patient population (i.e., limited sample size) available; (iii) the use of complex innovative designs such as a two-stage seamless adaptive trial design and/or an n-of-1 trial design for flexibility and the efficient assessment of the test treatment under study; (iv) using real-world data (RWD) and real-world evidence (RWE) to support regulatory submission; and (v) conducting an individual benefit–risk assessment for a complete picture of the clinical performance of the test treatment under investigation. In this article, we provide a comprehensive summarization of this innovative thinking and these approaches for an efficient, accurate and reliable assessment of a test treatment used for treating patients with rare diseases under study. Statistical considerations including challenges and justifications are provided whenever possible. In addition, an innovative approach that combines innovative thinking and these approaches is proposed for regulatory consideration in rare disease drug development.
To approve drug products in common conditions, the United States (US) Food and Drug Administration (FDA) requires that substantial evidence regarding the safety and efficacy of the test treatment under investigation be provided for regulatory review. The FDA further indicates that substantial evidence can only be obtained through the conduct of adequate and well-controlled randomized clinical trials (RCTs). The substantial evidence must achieve clinical/statistical significance at a pre-specified level with a desired (sufficient) power (i.e., the probability of correctly concluding the test treatment is efficacious and safe when, in fact, the test treatment is), e.g., 80% or 90%. In practice, however, the traditional approach is to power the study based on a pre-selected primary efficacy endpoint and then perform safety assessments, including the tolerability of the test treatment under study, before the test treatment can be approved and released for the marketplace.
In the US, a rare disease is defined as a disorder or condition that affects less than 200,000 persons [1,2]. However, no universal definition exists worldwide. For example, in the European Union (EU), a rare disease is defined as a disorder that affects fewer than 1 in 2000 people. For the approval of a test treatment for treating patients with rare diseases, the FDA emphasizes that the same standard will be applied as that for drugs for common conditions [2]. For rare disease clinical trials, one of the major concerns is the relatively small patient population (i.e., only a limited number of subjects) available to obtain substantial evidence regarding the safety and efficacy of the test treatment under investigation. As a result, in addition to the unavailability of patient populations, most sponsors are not willing to develop rare disease drug products due to some difficulties and challenging issues that are commonly encountered. These practical issues include heterogeneity, no control arm, no universally accepted study endpoint, the unavailability of a biomarker, and an inflexible/inefficient study design for providing efficient, accurate and reliable substantial evidence regarding the safety and efficacy of the test treatment under investigation, in support of regulatory submission, review and approval.
The FDA has initiated several incentive programs to assist sponsors in rare disease drug development, which include (i) fast track designation; (ii) breakthrough therapy designation; (iii) priority review designation; and (iv) accelerated approval of rare disease drug products. The FDA’s intention is good; however, these incentive programs do not necessarily address the issue of the small population available in rare disease drug development. Thus, these incentive programs are unable to provide an efficient, accurate and reliable assessment of the test treatment under investigation to the same standard as that required for the review and approval of drug products for normal conditions. To overcome these issues, some out-of-the-box innovative thinking and approaches are necessarily applied. This innovative thinking and these approaches include but are not limited to (i) sample size justification based on probability statements, e.g., the probability monitoring procedure proposed by [3], rather than traditional power calculations; (ii) the development of a composite (therapeutic) index that may combine both clinical endpoints and biomarkers [4]; (iii) the concept of demonstrating not-ineffectiveness and/or not-unsafeness rather than demonstrating effectiveness and safety [5]; (iv) the application of complex innovative designs such as an adaptive (flexible) trial design and/or an n-of-1 trial design; (v) the use of real-world data (RWD) and real-world evidence (RWE) in supporting rare disease drug development; and (vi) performing individual benefit–risk assessment according to FDA recent guidance [6] for obtaining a complete clinical picture of the test treatment under study. These innovative thoughts and approaches would lead to a more efficient, accurate and reliable assessment of the safety and efficacy of the rare disease test treatment under study in a more efficient way, even with only a limited number of patients available.
Regarding the use of RWD and RWE in support of regulatory submission, the 21st Century Cures Act passed in December 2016 by the US Congress requires the FDA to establish a program to evaluate the potential use of RWE, which is generated from RWD, to (i) support the approval of a new indication for a drug approved under Section 505(c) and (ii) satisfy post-approval study requirements. RWE offers opportunities to develop robust evidence using high-quality data and sophisticated methods for producing causal-effect estimates regardless of whether randomization is feasible. For rare disease drug development, we propose performing a gap analysis of substantial evidence and RWE before the RWE can be used in support of regulatory submission in critical decision-making. It should be noted that the FDA does not intend to replace the required substantial evidence with the RWE derived from RWD obtained from RWS. RWE should be used in support of regulatory submission in the review and approval process of drug development.
In the next section, some undesirable characteristics regarding rare disease drug development are briefly described. To overcome these undesirable characteristics, Section 3 provides some out-of-the-box innovative thoughts and approaches to assessing the safety and efficacy of a test treatment for patients with rare diseases under investigation. In Section 4, the use and implementation of RWD/RWE in support of rare disease regulatory submissions is discussed. For regulatory consideration, Section 5 proposes an innovative approach of a two-stage seamless adaptive design combining RCT and real-world study (RWS) in rare disease drug development. Some concluding remarks are provided in the final section of this article.
2. Undesirable Characteristics of Rare Disease Drug Development
As mentioned earlier, to assist/encourage rare disease drug development, the FDA has initiated several incentive (expedited) programs. Despite the FDA’s incentive programs, some practical difficulties and challenges are inevitably encountered during the conduct of rare disease clinical trials. Commonly seen undesirable characteristics in rare disease drug development include (i) insufficient power (due to a relatively small patient population being available); (ii) heterogeneity across baseline characteristics; (iii) no control arm; (iv) no universally accepted endpoints and/or biomarkers; and (v) inflexible and inefficient study design, which are briefly described below.
2.1. Insufficient Power
In practice, for rare disease drug development, it is expected that the intended clinical trial may not achieve the desired power (i.e., the probability of correctly detecting a clinically meaningful difference or treatment effect when such a difference truly exists) to confirm the safety and efficacy of the test treatment under investigation at the 5% level of significance due to the small sample available. Thus, for rare disease clinical trials, it is not feasible to use the traditional power calculation for sample size calculation. Consequently, the sponsor must seek alternative methods to justify a much smaller sample size by achieving certain statistical assurance for the intended rare disease clinical trials.
In addition to power analysis, other methods such as precision analysis, reproducibility analysis, and probability monitoring procedures could be used for sample size calculation to achieve certain statistical assurance in the intended clinical trials. Precision analysis is used to select a sample size that controls the type I error rate within the desired precision, while reproducibility analysis is used to select a sample size that will achieve the desired probability of reproducibility. The probability monitoring procedure is used to justify a selected sample size based on the probability of crossing efficacy/safety boundaries (see, e.g., [3]).
2.2. Heterogeneity across Baseline Characteristics
In clinical trials, it is not uncommon to see heterogeneity across baseline characteristics such as demographics and/or patient characteristics at baseline. At the planning stage of the intended clinical trial, heterogeneity will impact the power analysis for sample size calculation. A much larger sample size may be required for detecting a treatment effect size (or clinically meaningful difference) in the presence of heterogeneity. Heterogeneity is an undesirable characteristic in rare disease drug development. In practice, stratified randomization is applied to prevent the biased assessment of the test treatment under study due to treatment imbalance. For rare disease clinical trials, stratified randomization is not feasible due to the small sample size. In addition, it is also suggested that power calculations should be performed by (i) maintaining the treatment effect (clinically meaningful difference) and at the same time (ii) controlling the variability associated with the response, to ensure that there is a high reproducibility probability (i.e., the result is reproducible with high probability) if the study were conducted repeatedly under similar clinical conditions/environments.
2.3. No Control Arm
In practice, rare disease clinical trials often do not include a control arm due to (i) ethical considerations and (ii) the unavailability of patients with the rare disease under study. Because of the small patient population, the FDA encourages utilizing existing historical knowledge/data to assist in rare disease (orphan) drug development. Though the requirement for substantial evidence cannot be relaxed, historical data may be used as an external control arm. One typical example is the use of RWD. The evidence generated by RWD (i.e., RWE) is a valuable source of information that reveals the real-world performance of the test treatment, including both safety and effectiveness. Due to the accelerated approval process, some rare disease clinical trials may have a shorter follow-up period compared with typical trials, which makes post-marketing evaluation (e.g., evaluation of RWD) even more important [7].
2.4. No Universally Accepted Study Endpoints or Biomarkers
In rare disease clinical trials, there often exist no universally accepted study endpoints or biomarkers to evaluate the safety and efficacy of the test treatment under study. Thus, the FDA suggests that a sponsor should define a trial endpoint by selecting a patient assessment as an outcome measure and defining when the patient would be assessed in the trial [8]. As indicated in the [8] draft guidance, endpoint selection in a clinical trial involves the knowledge and understanding of the following: (i) the range and course of clinical manifestations associated with the disease; (ii) the clinical characteristics of the specific target population, which may be a subset of the total population with a disease; (iii) the aspects of the disease that are meaningful to the patient and could be assessed to evaluate the drug’s effectiveness; and (iv) the possibility of using the accelerated approval pathway. Despite continuing efforts to develop novel surrogate endpoints, the FDA emphasized that only the usual clinical endpoints for adequate and well-controlled trials can provide substantial evidence of effectiveness supporting the marketing approval of the drug ([8,9]). Thus, it is recommended that sponsors select endpoints that consider the objectives of each trial in the context of the overall clinical development program.
A biomarker is defined as an indicator of biological and pathogenic processes, which can be used to indirectly evaluate the clinical outcome or identify the patient population. A good biomarker should have the following properties: (i) easy to measure; (ii) less affected by other treatment modalities; (iii) large effect size; and (iv) predictive of the clinical outcome of interest. Hence, such biomarkers can be used in rare disease development since they not only offer an opportunity to have a smaller sample size (due to the large effect size) but also help screen patients who may receive more benefits by taking the test drug. However, due to the limited target population and the low rate of return, the understanding of rare diseases is incomplete. As a result, it is difficult to identify the right biomarker, let alone the variation of phenotypes for certain rare diseases.
2.5. Inflexible/Inefficient Study Design
Under the restriction that only a small sample is available, the usual parallel-group design is considered not flexible and not efficient. Instead, some complex innovative designs (CIDs) like the n-of-1 trial design, adaptive trial design, master protocol, and Bayesian sequential design may be considered. Among these complex innovative designs, the n-of-1 trial design has become very popular for evaluating the difference in treatment effects within the same individual when n treatments are administered at different dosing periods. In general, as compared to a parallel-group design, the n-of-1 trial design requires fewer subjects for evaluation of the test treatment under investigation. On the other hand, adaptive trial design has the flexibility to modify the study protocol as it continues after the review of interim data. Clinical trials utilizing adaptive design methods may not only increase the probability of success in drug development but also shorten the development process.
As an example, consider dose finding for identifying the maximum tolerable dose (MTD) in phase II clinical development. A traditional “3 + 3” dose escalation design is often considered. The traditional “3 + 3” escalation design enters three patients at a new dose level and then enters another three patients when a dose-limiting toxicity (DLT) is observed. Assessment of the six patients is then performed to determine if the trial should be stopped at the level or escalated to the next dose level. Note that DLT is referred as an unacceptable or unmanageable safety profile when pre-defined by certain criteria such as Grade 3 or greater hematological toxicity according to the US National Cancer Institute’s Common Toxicity Criteria (CTC). This dose-finding design suffers the following disadvantages: (i) inefficient, (ii) often underestimates the MTD especially when the starting dose is too low, (iii) depends upon the DLT rate at MTD, and (iv) the low probability of correctly identifying the MTD.
Alternatively, it is suggested that a continued re-assessment method (CRM) and Bayesian optimal interval (BOIN) approach should be considered. In the method of CRM, the dose–response relationship is continually reassessed based on accumulative data collected from the trial. The next patient who enters the trial is then assigned to the potential MTD level. Thus, the CRM involves (i) dose toxicity modeling; (ii) dose level selection; (iii) re-assessment of model parameters; and (iv) assignment of the next patient. In addition, the CRM method in conjunction with a Bayesian approach for dose–response trials can substantially improve the CRM for dose finding. To select a more efficient dose-finding design between the “3 + 3” escalation design, the CRM design, and the BOIN design, the FDA recommends the following criteria for design selection: (i) the number of patients expected; (ii) the number of DLT expected; (iii) the toxicity rate; (iv) the probability of observing DLT prior to MTD; (v) the probability of correctly achieving the MTD; and (vi) the probability of overdosing. Based on a clinical trial simulation study, the “3 + 3” dose escalation design can be compared to the CRM design in conjunction with a Bayesian approach for a radiation therapy dose-finding trial. The results indicated that (i) CRM has an acceptable probability of correctly reaching the MTD; (ii) the “3 + 3” dose escalation design always underestimates the MTD; and (iii) CRM generally performs better than the “3 + 3” dose escalation design.
3. Innovative Thinking and Approaches for Rare Disease Clinical Trials
In rare disease clinical development, some out-of-the-box innovative thoughts are necessarily applied to overcome undesirable characteristics commonly seen in rare disease drug development. These innovative thoughts include but are not limited to (i) the use of external control; (ii) endpoint selection; (iii) sample size requirement; (iv) the concept of demonstrating not-ineffectiveness and/or not-unsafeness; (v) complex innovative design (e.g., adaptive trial design and n-of-1 trial design); (vi) the use of RWD and RWE; and (vii) individual benefit–risk assessment, which will be briefly described below.
3.1. External Control
In rare disease clinical trials, it may not be ethical or feasible to assign the limited subjects available to a placebo control given that there are no effective treatments available in the marketplace. In this case, it is suggested that the use of external control accelerates rare disease drug development by reducing/eliminating the number of subjects on placebo (e.g., [2,8]). However, one of the biggest concerns for the use of external control is likely selection bias. To minimize potential selection bias, it is suggested that appropriate statistical methods such as propensity score-matching techniques, based on matching factors such as baseline demographics and/or patient characteristics, should be considered to prevent unmeasured confounding and possible inconsistency.
Propensity score-matching is a quasi-experimental method in which the principal investigator uses statistical techniques to construct an artificial control group by matching each treated unit with a non-treated unit of similar characteristics (e.g., demographics and patient characteristics). Propensity score-matching computes the probability that a unit will enroll in a trial based on observed characteristics. It should be noted that propensity score-matching relies on the assumption that, conditional on some observable characteristics, untreated units can be compared to treated units as if the treatment has been fully randomized. In other words, propensity score-matching is used to mimic randomization to overcome issues of selection bias.
For illustration purposes, the standard framework for propensity score-matching by [10,11] is considered. For a given subject I, i = 1, …, N. In the case of a binary treatment, the treatment indicator equals one if individual subject i receives treatment; it is zero otherwise. The potential outcomes are then defined as for each subject i, where i = 1, …, N. The treatment effect for subject i can then be written as:
Since only one of the outcomes is observed for each subject i, estimating the individual treatment effect is not possible. In this case, one may focus on population average treatment effects (ATT) instead. The ATT can be defined as
Since E [Y (0)|D = 1] is not observed, we will have to find a way to estimate ATT. One commonly considered approach is to consider using the mean outcome of an untreated individual’s E [Y (0)|D = 0] to estimate ATT. However, the outcomes of individuals from the treatment and comparison groups would differ even in the absence of treatment, leading to a ‘self-selection bias’, i.e.,
Thus, the true parameter can only be identified if E [Y (0)|D = 1] – E [Y (0)|D = 0] = 0.
In randomized trials without a control arm, one must invoke some identifying assumptions to solve the problem, as stated above. For this purpose, the propensity score-matching method is commonly considered. First, a decision has to be made concerning the estimation of the propensity score. Following that, one has to choose which matching algorithm to employ and determine the region of common support. Subsequently, the matching quality has to be assessed, and treatment effects and their standard errors have to be estimated (see, e.g., [12]).
3.2. Endpoint Selection
Since there may exist no universally accepted study endpoints and/or biomarkers, some endpoints or biomarkers may achieve the study objectives and some may not. In this case, it is difficult to determine which endpoints or biomarkers are telling the truth as they more or less reflect clinical performance in terms of the safety and efficacy of the test treatment under study. Thus, we would like to propose an innovative approach by combining all of these study endpoints and/or biomarkers for the development of a so-called therapeutic index to assess the overall safety and efficacy of the test treatment under investigation. Following the idea discussed by [4,5] proposed the development of a therapeutic index using a utility function to incorporate multiple distinct endpoints in clinical trials via the following steps.
First, we identify a utility function that can incorporate all of the relevant study endpoints. Suppose there are study endpoints, denoted by Let be the clinical relevant study endpoints. An ideal (therapeutic) index (say ) that incorporates these individual clinically relevant study endpoints can then be defined as
where is a vector of weights (with ) assigned to the individual study endpoint with respect to index . is a utility function, which could be a linear or nonlinear function depending upon the relationships among . The use of a utility function is to construct index based on appropriate selections of and . In this article, we will refer to index as therapeutic index .
Note that in general individual study endpoints, can be different data types (e.g., continuous variable, binary response, or time-to-event data), and are pre-specified weights that could be different for different therapeutic indexes . The therapeutic index typically generates a vector of index , and if , it reduces to a single (composite) index. For example, consider , then is simply a linear combination of individual study endpoints, with weights of . In other words, index is simply the average over all of the individual study endpoints.
Step 2 is to select the appropriate weight . In practice, there might be various ways to select the weights. For example, one may consider selecting appropriate weights based on variabilities associated with the individual study endpoints. Some researchers, on the other hand, may prefer selecting weights based on the observed p-values from individual study endpoints because the p-values could reflect the levels of substantial evidence regarding the safety and effectiveness of the test treatment under investigation, provided by the individual study endpoints. In this case, we may use the following hypotheses:
where are the treatment effects assessed by the endpoint , and are pre-specified margins of clinically important differences.
Under some appropriate assumptions, we can calculate the p-value under each based on the sample of , and the weights can be constructed based on and . Note that should be constructed such that a high value of is with a low value of . For example, we may consider selecting .
In practice, if we consider as linear function here, then (1) reduces to
In order to study the statistical properties of the developed therapeutic index described above, we need to specify sampling distribution of . In practice, for simplicity, we may assume follows the multi-dimensional normal distribution , where and with
3.3. Sample Size
Sample size plays an important role in clinical trials, providing substantial evidence about the safety and efficacy of the test treatment under study. Substantial evidence means that the intended trial can achieve the study objectives at a pre-specified level of significance with a desired (sufficient) power (i.e., the probability of correctly concluding the safety and efficacy of the test treatment under study). For this purpose, power analysis for sample size calculation (power calculation) is often performed in clinical trials.
For clinical trials, however, power calculation may not be feasible in some disease areas such as rare diseases, especially when there is an extremely low incidence rate. As an example, consider a real example concerning a safety study required by the FDA. A pharmaceutical company was asked to conduct a diabetes study with an extremely low incidence rate of glycated hemoglobin ( to demonstrate that there is no safety concern about the test treatment under investigation. The incidence rate of is extremely low at three per ten thousand. The power calculation indicated that a total sample size of 784,684 is required for detecting a clinically meaningful difference (1 per 10,000) at the 5% level of significance with a desired power of 80%, assuming that the incidence rate is 3 per 10,000. In this case, a power calculation is definitely not practical/feasible.
Alternatively, it is suggested that sample size justification based on probability statements be considered. We first select a sample size that is reasonable and workable based on resources/financial considerations, e.g., 800 subjects. Assuming that the incidence rate of is 0.0003, with the selected sample size of 800, we expect not to observe a single event during the conduct of the study. If we observe one single event, we can conclude that the test treatment under study is not safe. If we do not observe any event during the conduct of the study, we may conclude that there is insufficient evidence to demonstrate that the test treatment is not unsafe. The FDA accepted this approach for demonstrating that the test treatment is not unsafe, and more safety data are yet to be collected for the confirmation of safety.
It should be noted that the above example for sample size justification based on probability statements is to evaluate the hypothesis that “” rather than “”, where while . Clearly, not unsafe (not ) is not equivalent to safe (
In this section, for simplicity and for illustration purposes, we will focus on demonstrating not-ineffectiveness. The demonstration of not-unsafeness can be treated similarly. With a limited sample size available, following the idea of demonstrating not-unsafeness rather than safeness, we may demonstrate not-ineffectiveness rather than effectiveness. In other words, we will demonstrate not-ineffectiveness by testing the following hypotheses:
Testing the above hypotheses is similar to testing a non-inferiority hypothesis. Once the null hypothesis of (4) is rejected, i.e., the not-ineffectiveness of the test treatment has been established, more data may be collected to further test the following hypotheses for efficacy confirmation:
As mentioned above, the concept of not-ineffectiveness is not equivalent to that of effectiveness. There is a gap between not-ineffectiveness and effectiveness, which is referred to as the area of inconclusiveness. In practice, it is suggested that RWD be collected to rule out the probability of inconclusiveness. RWD are often obtained through the conduct of RWS for the purpose of confirming the effectiveness of the test treatment under study.
The method of composite likelihood, as a down-weighting approach, can be used to account for the impact of high variability in the RWD to rule out the probability of inconclusiveness and consequently confirm the efficacy of the test treatment under study. Let represent the observation of and present the observation of . For , can be viewed as a set of random variables following a binomial distribution: and . The composite likelihood of can be written as
where is the composite likelihood function, is the likelihood function, is the density function of , and is the density function of . If population variances and are known, the maximum composite likelihood estimator (MCLE) of is
where and are the sample means for the th arm of an RCT and RWS, respectively. Since
Define sample mean difference at the second stage as . The variance of can be approximately estimated as
Though MCLEs are asymptotically normally distributed, it is difficult to derive a valid test statistic under null hypothesis in Equation (4). Another way to test for effectiveness at the second stage is using the probability of inconclusiveness [5]. That is, if the inconclusiveness zone in Figure 1 is negligible, we may conclude effectiveness.
When variances are known, the inconclusiveness occurs if . Given significance level , the probability of inconclusiveness can be defined as
Thus, can be estimated as
If is small enough, we can reject and conclude effectiveness; otherwise, the test drug is ineffective.
3.5. Complex Innovative Design
As indicated in PDUFA VI (Prescription Drug User Fee Act VI) as a result of the 21st Century Cure Act enacted by the US Congress in December 2016, complex innovative trial designs are designs involving complex adaptations, Bayesian methods, or other features requiring simulations to determine operating characteristics. Thus, complex innovative designs (CIDs) include but are not limited to (i) n-of-1 trial designs; (ii) adaptive trial designs; (iii) master protocol (platform trial) designs; and (iv) Bayesian sequential designs. In this section, we will focus on the two commonly considered CIDs, i.e., adaptive designs and n-of-1 trial designs, which are briefly described below.
Adaptive Design—The concept of an adaptive trial design can be traced back to the early 1970s. An adaptive design clinical study is defined as a study that includes a prospectively planned opportunity for the modification of one or more specified aspects of the study design and hypotheses based on the analysis of data (usually interim data) from subjects in the study [13].
It has received much attention since the early 2000s. It gives the investigator(s) the flexibility to identify any signal, possible trend/pattern, and ideally optimal benefit regarding the safety/efficacy of the test treatment under investigation. It not only speeds up (shortens) the development process in a more efficient way but also increases the probability of success without undermining the integrity and validity of the development.
The following adaptive designs are commonly used in clinical development: (i) Adaptive randomization design; (ii) (Adaptive) group sequential design; (iii) Flexible sample size re-estimation design; (iv) Drop-the-losers (pick-the-winner) design; (v) Adaptive dose-finding design; (vi) Biomarker-adaptive design; (vii) Adaptive treatment-switching design; (viii) Adaptive-hypotheses design; (ix) Seamless adaptive design; and (x) Multiple adaptive design (any combinations of the above designs).
Complete n-of-1 Trial Design—An n-of-1 trial is defined as a clinical trial where a single patient is the entire trial or a single case study. In an n-of-1 trial, n is the number of treatments and 1 is the single patient. Random allocation can be used to determine the order in which an experimental and a control are given to a patient. An n-of-1 trial is a multiple crossover study in a single participant.
For comparing a test treatment with a reference product, a complete n-of-1 trial design depends on m—the number of dosing periods. As an example, when m = 3 (three dosing periods), the complete n-of-1 trial design consists of three periods. Each dosing period involves two choices (i.e., either R or T) and, thus, a total of = 8 sequences. This results in an 8 × 3 crossover design (see also Table 1).
A complete n-of-1 trial design is useful especially when there is a small patient population, such as in rare disease drug development, and there are no active treatments in the disease area, like COVID-19. Under a complete n-of-1 trial design, we will be able to obtain valuable information from limited number of subjects available for an efficient, accurate, and reliable assessment of the test treatments under investigation.
3.6. The Use of RWD/RWE
RWD are data related to patient health status and/or the delivery of health care routinely collected from a variety of sources. RWE is evidence derived from RWD through the application of research methods. For regulatory applications, RWE can further be defined as clinical or substantial evidence regarding the use and potential benefits or risks of a medical product derived from the analysis of RWD. For the approval of drug products, regardless of whether they are orphan drugs, the FDA requires that substantial evidence (SE) regarding the safety and efficacy be provided, and SE can only be obtained through the conduct of adequate randomized controlled clinical trials (RCTs). RWE can only be used in support of regulatory submission. To provide a better understanding, a comparison between SE and RWE is given in Table 2.
As seen in Table 2, there are some fundamental differences between data obtained from RCT and RWD, which have raised some concerns regarding the use of RWD in support of regulatory submission. These concerns include but are not limited to (i) representativeness; (ii) heterogeneity; (iii) confounding and interaction; (iv) missing data; (v) reproducibility and generalizability; and (vi) data quality and validity. These concerns have greatly impacted the assessment of the safety and efficacy of the test treatment under investigation. Once these concerns have been addressed, the use of RWD and RWE in support of regulatory submission can then be implemented following the steps: (i) gap analysis; (ii) data relevancy; (iii) data quality and reliability; (iv) and fit-for-regulatory purpose data.
3.7. Benefit–Risk Assessment
In rare disease drug development, despite the limited sample size available, one of the most difficult challenges is balancing the benefits and risks of the test treatment under investigation. This leads to the conduct of benefit–risk assessment (BRA).
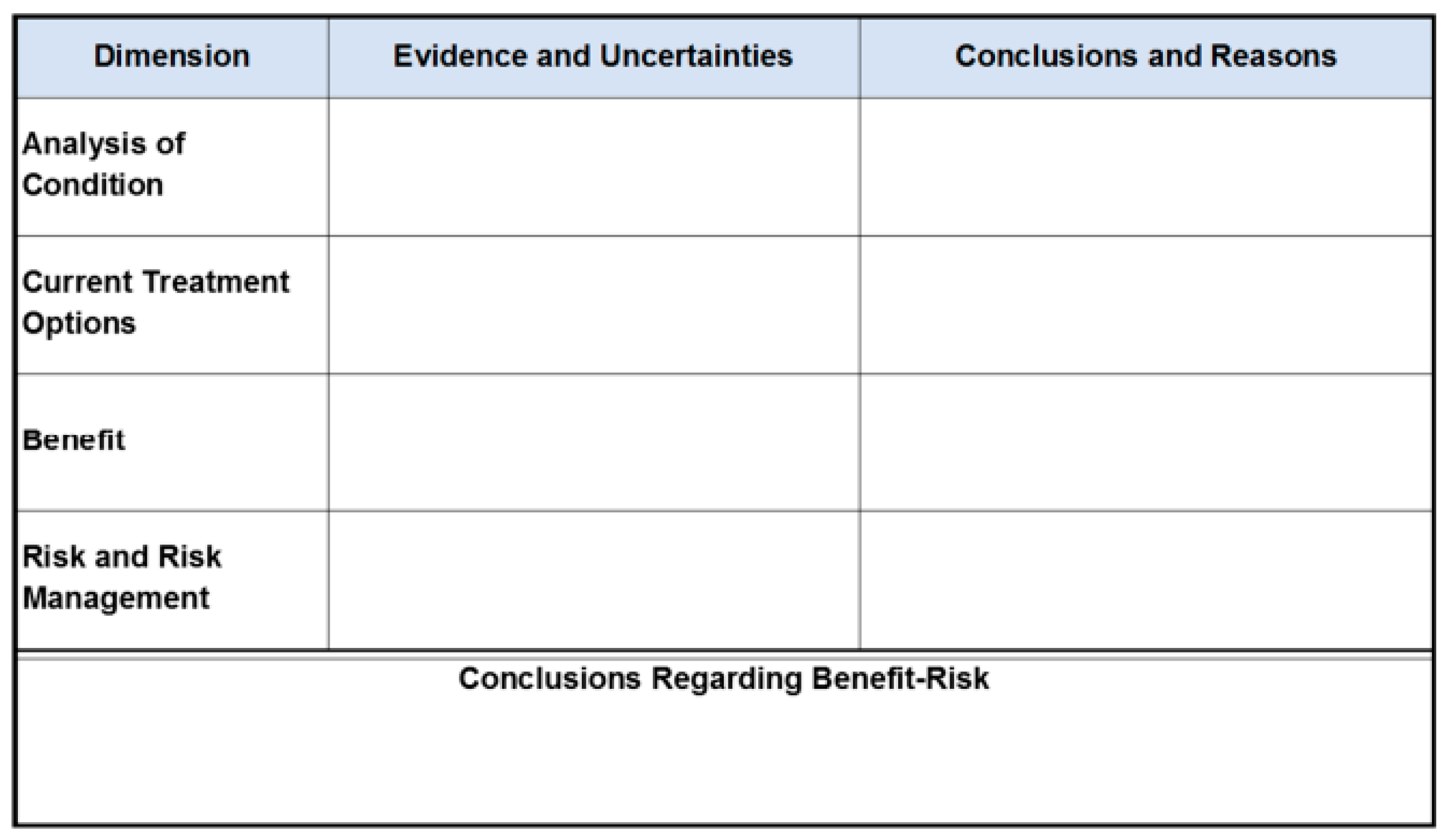
In 2023, the FDA published guidance to assist the sponsors in conducting benefit–risk assessments to support certain regulatory decisions about NDAs or BLAs, whether for pre-market approval or in the post-market setting. This involves decisions regarding regulatory requirements for approval, including the inclusion of a boxed warning in approved labeling, post-marketing study requirements and commitments, and risk evaluation and mitigation strategies [6]. The FDA’s guidance suggests the following BRA framework for new drug review.
Under the FDA’s BRA framework, no widely accepted quantitative methods for BRA exist. In practice, one of the most commonly used BRA metrics is the general benefit–risk (GBR) index proposed by [14], which has been generalized by several researchers in terms of determining weights for every endpoint, adding possible outcomes, generalizing to longitudinal data, and applying under a Bayesian framework ([15,16,17,18]).
In practice, although the GBR index and its generalizations are easy to compute, they suffer from the following disadvantages: (i) subjective weights and (ii) a lack of intuitive interpretation (e.g., the linear score from the GBR index is difficult to interpret). To construct a more comprehensive BRA, multi-criteria decision analysis (MCDA) was proposed by [19]. Subsequently, Bayesian MCDA was proposed to improve conventional MCDA to account for uncertainty in assessing benefit–risk balance ([20,21]). In addition, using different methods, stochastic multi-criteria acceptability analysis (SMAA) was developed to account for statistical uncertainty as well as provide a consensus weight [22]. However, the SMAA model is computationally complex, which can be time-consuming while incorporating Bayesian methods.
4. The Use of RWD/RWE in Support of Rare Disease Drug Development
As indicated earlier, in rare disease drug development, with a small patient population available, we can first use RCT data to test for the not-ineffectiveness (and/or not-unsafeness) of the test treatment under investigation. Once the not-ineffectiveness (and/or not-unsafeness) of the test treatment has been confirmed, we can then use RWD to support (confirm) the effectiveness (and/or safety) of the test treatment under study. In practice, the use of RWD/RWE as a complement to RCT data has been considered to support regulatory submission in rare disease drug development. However, there are some concerns regarding the use of RWD/RWE in support of rare disease drug development. For instance, (i) it is always a concern whether the RWD is representative of the target patient population with rare diseases under study; (ii) the heterogeneity across individual studies and/or resources contained in RWD; (iii) possible confounding/interaction with baseline demographics and/or patient characteristics; (iv) missing data/values in RWD; and (v) the reproducibility and generalizability of RWD. In addition, the validity of the RWD due to possible selection bias is also a concern. In this section, these concerns are briefly outlined. In addition, a proposal for the implementation of RWD/RWE in support of rare disease drug development is also discussed.
Representativeness of RWD—In practice, unlike clinical data collected from an RCT, which are obtained from a specific patient population under a controlled clinical environment, RWD contain data from different individual studies (with similar but different study protocols, study objectives, hypotheses, and/or study endpoints) or sources such as electronic health records (EHRs). They are collected with similar but different statistical procedures and/or structural/nonstructural formats, which may have resulted in a similar but different target patient population as compared to the original target population for the intended RCT. Thus, in evaluating a given disease under study, there is a concern if RWD represent the target patient population of the intended studies for regulatory submission in drug development. In practice, it is then suggested that the representativeness of RWD be carefully examined to avoid potential selection bias before they can be used in support of regulatory submission for regulatory review and approval.
Heterogeneity of RWD—Another challenging issue that needs to be addressed is heterogeneity across individual studies and/or resources of RDW, before RWD can be used for obtaining accurate and reliable RWE in support of regulatory submission. In practice, heterogeneity exists due to differences within and across individual studies and resources of RWD (e.g., with different means, variances, and sample sizes). It is suggested that statistical tests for the treatment-by-study (data resource) interaction (i.e., test for poolability) should be performed using either a random or mixed-effects model before RWD can be pooled for final analysis. Detailed information on evaluating the heterogeneity of RWD is found in [23].
Confounding/interaction of RWD—Confounding and interaction are commonly seen in RWD due to differences or imbalances in baseline demographics (e.g., age, gender, BMI or weight/height, race, etc.) and/or patient characteristics (e.g., disease severity, past medical history, concomitant medication(s), etc.) In practice, these possible confounding and interaction factors may not be observed. As a result, [24] proposed using the confounding function to summarize the impact of unobserved confounders on outcome variables to account for observed covariates to improve the inference based on RWD. To address interaction factors, data should not be pooled for overall analysis but can be pooled for final analysis if a significant qualitative interaction is observed.
Missing data in RWD—Missing or incomplete data are commonly encountered in clinical studies due to possible dropouts, loss to follow-up, withdrawal of informed consent, withdrawal by investigators, etc. Missingness may also occur due to non-medical reasons, such as health insurance policy and plan issues. In practice, it is necessary to develop a strategy to handle missing data in the RWD framework. One may first determine whether the missingness significantly alters the study conclusions. If there is no significant impact, then missingness is less critical [25]. Otherwise, a proper approach such as determining an estimand for handling missing data or incomplete data should be considered. For example, Ref. [26] proposed the following four-step procedure using an estimand to handle the missing data. The first step is to clarify the treatment estimand of interest with respect to the intercurrent event. The second step is to establish what data is missing for the chosen estimand. Then, the primary analysis is performed under the most plausible missing data assumptions, followed by a final sensitivity analysis based on alternative plausible assumptions. This four-step strategy will allow us to conduct an accurate and reliable assessment of the test treatment under investigation.
Reproducibility/generalizability of RWD—In clinical trials, reproducibility can be interpreted as the observed clinical result using RWD at one study center being reproducible at another study center, with the target population remaining the same. The reproducibility probability in a given clinical trial can be used to evaluate reproducibility based on the observed mean response and associated variability. Three approaches were proposed to assess reproducibility probability: the estimated power approach, the confidence-bound approach, and the Bayesian approach. To define generalizability, one target population’s clinical results (e.g., adults) can be generalized to another similar but different target patient population (e.g., children or elderly). In addition, the sensitivity index can be used to evaluate generalizability, as proposed by [27].
Validity of RWD—In practice, RWD may contain positive or negative studies and may be in a structured or unstructured format (see Figure 2). In this case, the validity of RWE derived from RWD is a concern for providing substantial evidence regarding the safety and efficacy of the test treatment under investigation. The validity of RWD/RWE is essential, especially when intended to support regulatory submission. In practice, studies with positive results are more likely accepted in RWD but may cause substantial selection bias. The selection bias in real-world data can be in structural and unstructured data settings. Based on this form of bias, three reproducibility probability-based approaches have been introduced to estimate the real proportion of positive studies in the structural and unstructured data. The reproducibility probability-based approach provides effective bias adjustment when the proportion of positive studies is different than the designed power. In most cases, the empirical power (EP) approach and the Bayesian approach provide robust and effective bias adjustment, and the confidence-bound (CB) approach provides an effective adjustment only when the bias is larger than 10%.
Implementation of RWD/RWE—Following the 21st Century Cures Act of 2016, the FDA established a program to evaluate the potential use of RWE in order to (i) support a new indication for a drug approved under Section 505(c) and (ii) satisfy post-approval requirements. In addition, the FDA published draft guidance on a framework for implementation, which describes courses of RWE, challenges, and pilot opportunities ([8,9]).
In order to use RWE to support regulatory review and approval, it is suggested that the difference between RWE and SE be carefully examined. For this purpose, the following process for implementation of RWD/RWE is proposed for regulatory consideration: (i) gap analysis between RWE and SE regarding safety and efficacy of the test treatment under investigation; (ii) data relevancy (e.g., whether RWD can be representative of the target patient population with the disease under study); (iii) data quality (e.g., accuracy, completeness, and transparency) and reliability; (iv) integrity and validity (e.g., information bias); and (v) fit-for-regulatory purpose data. The proposed process for the implementation of RWD/RWE in support of regulatory submission is useful in obtaining an efficient, accurate, and reliable assessment of the test treatment under investigation.
5. A Proposal for Regulatory Consideration
The current development process for drug products with normal conditions is first to test the null hypothesis that there is no clinical benefit (e.g., treatment effect) of the test treatment under study. Rejecting the null hypothesis leads to the conclusion of the alternative hypothesis—that there is a treatment effect of the test treatment under study. Thus, an appropriate sample size is selected to ensure that there is sufficient power for detecting such a treatment effect, assuming that the treatment effect truly exists. Note that the current process for drug development focuses on effectiveness (i.e., the study is often powered based on a primary efficacy endpoint) rather than both safety and efficacy simultaneously. This process may not be appropriate for direct application in rare disease drug development due to the small patient population available. Besides, the FDA emphasizes following the same standards for regulatory review and approval in developing rare disease drugs. In this case, the current development process needs to be modified to overcome the problem of a small patient population available in rare disease drug development and to meet the regulatory requirement of the same standards (e.g., sufficient power for correctly detecting clinical benefit if such clinical benefit truly exists). For this purpose, in this section, we propose an innovative approach by considering a two-stage design that includes a small scale of RCT and a large scale of real-world study (RWS), which is briefly described below.
By combining the collective innovative thoughts and approaches discussed in the previous sections regarding (i) the use of external control; (ii) the appropriate selection of study endpoints and/or the development of composite (therapeutic) index; (iii) probability-based sample size justification; (iv) the concept of demonstrating not-ineffectiveness and/or not-unsafeness; (v) the use of complex innovative design such as a two-stage seamless adaptive trial design and a complete n-of-1 trial design; (vi) the use of real-world data in support of regulatory submission; and (vii) individual benefit–risk assessment for providing a complete clinical picture of the performance of the test treatment under study, we would like to propose the following two-stage seamless adaptive trial design that combines an RCT with a limited number of patients with the rare disease under study and an RWS that collects RWD for the generation of RWE for regulatory consideration.
Under the proposed two-stage seamless adaptive trial design, the first stage is to demonstrate that the test treatment is not ineffective (and/or not unsafe) by testing the null hypothesis ()—that the test treatment is not effective (and/or not safe)—against the alternative hypothesis —that the test treatment is not ineffective (and/or not unsafe)—with a relatively small sample size. Once the null hypothesis of not-effectiveness (and/or not safe) is rejected, i.e., the not-ineffectiveness (and/or not-unsafeness) has been confirmed, we then test the null hypothesis ( not —that the test treatment is not ineffective (and/or not unsafe)—against the alternative hypothesis (—that the test treatment is effective (and/or safe)—using both data collected from the RCT at stage 1 and RWD collected from RWS at stage 2. The proposal is briefly summarized in the following steps.
Step 1.
Select a reasonable sample size based on resource/recruitment considerations. Then, justify a sample size based on a probability statement, such as the adaptive or non-adaptive version of the probability monitoring procedure proposed by [3];
Step 2.
Utilize a two-stage complex innovative design (CID) such as seamless adaptive design or complete n-of-1 design, that contains an RCT at the first stage and a real-world study (RWS) at the second stage (see, e.g., [27]);
Step 3.
Demonstrate that the test treatment is not ineffective (for efficacy evaluation) and/or not unsafe (for safety assessment) with a limited number of subjects selected at Stage 1. It should be noted that the null hypothesis to be tested at this step is a composite hypothesis for both efficacy and safety. Thus, the alternative hypothesis would be one of the following: (NN), (NS), (NE), (SN), (SS), (SE), (EN), (ES), or (EE), where N = non-inferiority, S = superiority, and E = therapeutic equivalence (see Table 3 below);
Step 4.
Provide clinical evidence to eliminate the probability of inconclusiveness regarding safety and efficacy based on RCT data from Stage 1 and RWE, which are generated from RWD collected from RWS at Stage 2. The implementation of RWD for the generation of RWE should follow the process discussed in the previous section;
Step 5.
Conduct individual benefit–risk assessments to provide a complete clinical picture in terms of the benefits and risks of the test treatment under investigation. In practice, it is very likely that one study endpoint (either the safety endpoint or efficacy endpoint) meets the study objective, while the other study endpoints will fail to achieve the study objective. In this case, benefit–risk assessments may provide useful information in making critical decisions regarding the approval of the test treatment under investigation.
Under the proposed two-stage seamless adaptive trial design that combines an RCT and an RWS, the standard methods based on individual p-values (MIP), the sum of individual p-values (MSP), the product of individual p-values (MPP), and a normal-inverse combination test with different weights could be applied. If there is a planned interim analysis to stop the trial early due to safety concerns, futility, and/or efficacy, appropriate stopping boundaries can be determined based on individual p-values to control the overall type I error rate at a pre-specified level of significance.
For rare disease drug development, under the proposed two-stage seamless adaptive design that combines an RCT at the first stage and an RWS at the second stage, it is suggested that regulatory agencies may initiate an approvable letter once not-ineffectiveness and/or not-unsafeness have been confirmed at the first stage, based on the small-scale RCT study. Then, relevant clinical data may be collected from RWS at the second stage in support of regulatory evaluation and approval for the effectiveness and safety of the test treatment under investigation.
6. Concluding Remarks
In recent years, rare disease drug development has attracted much attention due to unmet medical needs. To encourage sponsors to develop efficient treatments, the FDA has kicked off some incentive programs. However, these incentive programs may not help in achieving the study objectives due to limited subjects with the disease. To overcome this problem, some out-of-the-box innovative thoughts/approaches are necessarily considered for maintaining the integrity, quality, and scientific validity of rare disease drug development. These innovative thoughts and/or approaches include (i) the use of external control; (ii) the selection of appropriate study endpoints; (iii) sample size justification based on probability statements; (iv) the concept of demonstrating the not-ineffectiveness and not-unsafeness of the test treatment under study; (v) the use of a complex innovative design such as a two-stage seamless adaptive trial design or a complete n-of-1 trial design for flexibility and the efficient assessment of the test treatment under study; (vi) the use of RWD/RWE in support of regulatory submission; and (vii) the conduct of benefit–risk assessments for decision-making in regulatory review and approval process.
Although RWD provide robust evidence about the real-world performance of the test drug, they are less powerful in producing causal-effect estimates compared with RCT data. In other words, RWE generated by RWD could be biased due to a lack of randomization and blinding. To map RWE to substantial evidence, more detailed research in terms of evaluating the relevancy/quality of RWD is necessary. A gap analysis to test whether the gap between RWE and substantial evidence is acceptable is helpful in assessing the quality of RWE. Thus, in practice, the use of RWD/RWE in support of regulatory submission is possible provided that (i) RWD is representative of the target patient population under study; (ii) there are no issues of selection bias, heterogeneity, confounding/interaction, and missing data regarding RWD; (iii) the gap analysis between RWE and substantial evidence in terms of the sensitivity index between RCTs and RWS is acceptable to the regulatory agency, such as the FDA; and (iv) the fit-for-regulatory purpose data derived from RWD are acceptable to the regulatory agency. It should be noted that in the proposed two-stage seamless adaptive design, we simply focus on hypotheses testing and use individual p-values to assess the safety and efficacy of the test treatment under investigation. Alternatively, one may consider a confidence interval approach or a probability-based confidence interval approach for the assessment of the test treatment under study.
Note that simulation studies demonstrate that the hybrid two-stage adaptive design yields a maximum sample size and expected sample size smaller than the sample size derived for traditional superiority tests in most cases. The procedure to down-weigh information from RWS can effectively mitigate the impact of the high variability of the RWS population on statistical inference for effectiveness. Across various settings for population means and population variances, the proposed design has good performance in controlling the type I error rate and maintaining statistical power. Specifically, the statistical power is consistently higher than the desired level, and the type I error rate is well-controlled when the sample size is large. In practice, when the RCT sample size is too small, the capability of controlling type I error-rate inflation decreases. The probability of correctly identifying inconclusiveness between not-ineffectiveness (not-unsafeness) and effectiveness (safety) is always over 0.60. In general, this design serves as a valuable reference for trials aiming to generate RWE and evaluate effectiveness, particularly in scenarios with extensive available RWD, such as in rare disease drug development and oncology drug development.
Author Contributions
Validation, S.S.C.; Writing—original draft, S.C.C.; Writing—review & editing, A.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Data Availability Statement
Data are contained within the article
Conflicts of Interest
Annpey Pong was employed by Merck & Co., Inc. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The Merck & Co., Inc. had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
FDA. Guidance for Industry—Framework for FDA’s Real-World Evidence Program; The United States Food and Drug Administration: Silver Spring, MD, USA, 2018.
FDA. Guidance for Industry—Rare Diseases: Common Issues in Drug Development; The United States Food and Drug Administration: Silver Spring, MD, USA, 2019.
Huang, Z.; Chow, S.C. Probability monitoring procedure for sample size determination. J. Biopharm. Stat.2019, 29, 887–896. [Google Scholar] [CrossRef] [PubMed]
Filozof, C.; Chow, S.C.; Dimick-Santos, L.; Chen, Y.-F.; Williams, R.N.; Goldstein, B.J.; Sanyal, A. Clinical endpoints and adaptive clinical trials in precirrhotic nonalcoholic steatohapitis: Facilitating development approaches for an emerging epidemic. Hepatol. Commun.2017, 1, 577–585. [Google Scholar] [CrossRef] [PubMed]
Chow, S.C.; Huang, Z. Demonstrating effectiveness or demonstrating not ineffectiveness—A potential solution for rare disease drug development. J. Biopharm. Stat.2019, 29, 897–907. [Google Scholar] [CrossRef] [PubMed]
FDA. Guidance for Industry—Benefit-Risk Assessment for New Drug and Biological Products; The United States Food and Drug Administration: Silver Spring, MD, USA, 2023.
Fan, M.; Chan, A.Y.; Yan, V.K.; Tong, X.; Lau, L.K.; Wan, E.Y.; Tam, E.Y.; Ip, P.; Lum, T.Y.; Wong, I.C.; et al. Postmarketing safety of orphan drugs: A longitudinal analysis of the US Food and Drug Administration database between 1999 and 2018. Orphanet J. Rare Dis.2022, 17, 3. [Google Scholar] [CrossRef] [PubMed]
FDA. Guidance for Industry—Submitting Documents Using Real-World Data and Real-World Evidence to FDA for Drugs and Biologics; The United States Food and Drug Administration: Silver Spring, MD, USA, 2019.
FDA. Guidance for Industry—Real-World Data: Assessing Electronic Health Records and Medical Claims Data to Support Regulatory Decision-Making for Drug and Biological Products; The United States Food and Drug Administration: Silver Spring, MD, USA, 2021. Available online: https://www.fda.gov/media/152503/download (accessed on 18 October 2021).
Roy, A. Some Thoughts on the Distribution of Earnings. Oxf. Econ. Pap.1951, 3, 135–145. [Google Scholar] [CrossRef]
Rubin, D. Estimating Causal Effects to Treatments in Randomised and Nonrandomised Studies. J. Educ. Psychol.1974, 66, 688–701. [Google Scholar] [CrossRef]
Caliendo, M.; Kopeinig, S. Some Practical Guidance for the Implementation of Propensity Score Matching; DIW Discussion Papers, No. 485; Deutsches Institut für Wirtschaftsforschung (DIW): Berlin, Germany, 2005. [Google Scholar]
FDA. Draft Guidance for Industry—Adaptive Design Clinical Trials for Drugs and Biologics; The United States Food and Drug Administration: Rockville, MD, USA, 2010.
Chuang-Stein, C.; Mohberg, N.R.; Sinkula, M.S. Three measures for simultaneously evaluating benefits and risks using categorical data from clinical trials. Stat. Med.1991, 10, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
Pritchett, Y.L.; Tamura, R. Global benefit–risk assessment in designing clinical trials and some statistical considerations of the method. Pharm. Stat. J. Appl. Stat. Pharm. Ind.2008, 7, 170–178. [Google Scholar] [CrossRef] [PubMed]
Yan, D.; Ahn, C.; Azadeh, S.; Atlas, M.; Tiwari, R. A Bayesian approach to benefit-risk assessment in clinical studies with longitudinal data. J. Biopharm. Stat.2020, 30, 574–591. [Google Scholar] [CrossRef] [PubMed]
Mussen, F.; Salek, S.; Walker, S. A quantitative approach to benefit-risk assessment of medicines—Part 1: The development of a new model using multi-criteria decision analysis. Pharmacoepidemiol. Drug Saf.2007, 16, S2–S15. [Google Scholar] [CrossRef] [PubMed]
Menzies, T.; Saint-Hilary, G.; Mozgunov, P. A comparison of various aggregation functions in multi-criteria decision analysis for drug benefit–risk assessment. Stat. Methods Med. Res.2022, 31, 899–916. [Google Scholar] [CrossRef] [PubMed]
Waddingham, E.; Mt-Isa, S.; Nixon, R.; Ashby, D. A Bayesian approach to probabilistic sensitivity analysis in structured benefit-risk assessment. Biom. J.2016, 58, 28–42. [Google Scholar] [CrossRef] [PubMed]
Tervon, T.; Van Valkenhoef, G.; Buskens, E.; Hillege, H.L.; Postmus, D. A stochastic multicriteria model for evidence-based decision making in drug benefit-risk analysis. Stat. Med.2011, 30, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
Moran, M.; Nickens, D.; Adcock, K.; Bennetts, M.; Desscan, A.; Charnley, N.; Fife, K. Sunitinib for metastatic renal cell carcinoma: A systematic review and meta-analysis of real-world and clinical trials data. Target. Oncol.2019, 14, 405–416. [Google Scholar] [CrossRef] [PubMed]
Yang, S.; Zeng, D.; Wang, X. Improved inference for heterogeneous treatment effects using real-world data subject to hidden confounding. arXiv2020, arXiv:2007.12922. [Google Scholar]
Girman, C.J.; Ritchey, M.E.; Zhou, W.; Dreyer, N.A. Considerations in characterizing real-world data relevance and quality for regulatory purposes: A commentary. Pharmacoepidemiol. Drug Saf.2019, 28, 439–442. [Google Scholar] [CrossRef] [PubMed]
Cro, S.; Morris, T.P.; Kahan, B.C.; Cornelius, V.R.; Carpenter, J.R. A four-step strategy for handling missing outcome data in randomised trials affected by a pandemic. BMC Med. Res. Methodol.2020, 20, 208. [Google Scholar] [CrossRef] [PubMed]
Chow, S.C. Innovative Methods for Rare Disease Drug Development; CRC Press: New York, NY, USA, 2020. [Google Scholar]
Figure 1.
FDA’s Benefit–Risk Framework for New Drug Review. Source: [6].
Figure 1.
FDA’s Benefit–Risk Framework for New Drug Review. Source: [6].
Figure 2.
RWD that contains positive/negative and structured/unstructured data.
Figure 2.
RWD that contains positive/negative and structured/unstructured data.
Table 1.
Examples of Complete n-of-1 Designs with p = 4.
Table 1.
Examples of Complete n-of-1 Designs with p = 4.
Group
Period 1
Period 2
Period 3
Period 4
1
R
R
R
R
2
R
T
R
R
3
T
T
R
R
4
T
R
R
R
5
R
R
T
R
6
R
T
T
T
7
T
R
T
R
8
T
T
T
T
9
R
R
R
T
10
R
R
T
T
11
R
T
R
T
12
R
T
T
R
13
T
R
R
T
14
T
R
T
T
15
T
T
R
T
16
T
T
T
R
Note: The first block (a 4 × 2 crossover design) is a complete n-of-1 design with 2 periods, while the second block is a complete n-of-1 design with 3 periods. T = test drug product and R = reference drug product.
Table 2.
Fundamental Differences between Substantial Evidence and Real-World Evidence.
Table 2.
Fundamental Differences between Substantial Evidence and Real-World Evidence.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
Chow, S.C.; Pong, A.; Chow, S.S.
Novel Design and Analysis for Rare Disease Drug Development. Mathematics2024, 12, 631.
https://doi.org/10.3390/math12050631
AMA Style
Chow SC, Pong A, Chow SS.
Novel Design and Analysis for Rare Disease Drug Development. Mathematics. 2024; 12(5):631.
https://doi.org/10.3390/math12050631
Chicago/Turabian Style
Chow, Shein Chung, Annpey Pong, and Susan S. Chow.
2024. "Novel Design and Analysis for Rare Disease Drug Development" Mathematics 12, no. 5: 631.
https://doi.org/10.3390/math12050631
APA Style
Chow, S. C., Pong, A., & Chow, S. S.
(2024). Novel Design and Analysis for Rare Disease Drug Development. Mathematics, 12(5), 631.
https://doi.org/10.3390/math12050631
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.
Article Metrics
No
No
Article Access Statistics
For more information on the journal statistics, click here.
Multiple requests from the same IP address are counted as one view.
Chow, S.C.; Pong, A.; Chow, S.S.
Novel Design and Analysis for Rare Disease Drug Development. Mathematics2024, 12, 631.
https://doi.org/10.3390/math12050631
AMA Style
Chow SC, Pong A, Chow SS.
Novel Design and Analysis for Rare Disease Drug Development. Mathematics. 2024; 12(5):631.
https://doi.org/10.3390/math12050631
Chicago/Turabian Style
Chow, Shein Chung, Annpey Pong, and Susan S. Chow.
2024. "Novel Design and Analysis for Rare Disease Drug Development" Mathematics 12, no. 5: 631.
https://doi.org/10.3390/math12050631
APA Style
Chow, S. C., Pong, A., & Chow, S. S.
(2024). Novel Design and Analysis for Rare Disease Drug Development. Mathematics, 12(5), 631.
https://doi.org/10.3390/math12050631
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.






{kind=link}
{kind=link}