3.1. Selection of Optimal Parameters , , and
The selection of parameters is the core of the prediction model. Extracting effective feature information from protein sequences is a key step in constructing an ECM protein prediction model. In order to obtain better feature information, the parameters need to be continuously adjusted. In this paper, the training dataset was used as the research object to perform feature extraction on protein sequences. The selection of parameters
in the PseAAC,
in the PsePSSM, and
in the AD play an important role in model construction. If
,
, and
are selected too small, the extracted sequence information is reduced, and it cannot effectively describe the characteristics of protein sequence. If the values of parameters
,
, and
are too large, it causes noise interference and affects the prediction results. In order to find the best parameters, the
value was set to 10, 20, 30, 40, and 50, the
value was set to 10, 20, 30, 40, and 49, and the
value was set to 10, 20, 30, 40, and 50. The RF classifier was used to classify the dataset. ACC, Sn, Sp, and BACC were used as the evaluation indicators. The results were tested by the method of LOOCV. The evaluation values obtained by selecting different parameters in the PseAAC algorithm, PsePSSM algorithm, and AD algorithm are shown in
Table 3,
Table 4 and
Table 5, respectively. The change in BACC value obtained by the feature extraction algorithm upon selecting different parameters is shown in
Figure S1 (Supplementary Materials).
The
value in PseAAC encoding reflects sequence correlation factors at different levels of amino-acid sequence information. Different values of
have different effects on the prediction performance of the model. Considering that the sample sequence length was 50, the
values in PseAAC encoding were set to 10, 20, 30, 40, and 50. As can be seen from
Table 3, the evaluation indicators changed with
. ACC, Sp, and BACC reached the highest when the value of
was 20. At this time, the value of ACC was 92.63%, the value of Sn was 12.71%, and the value of BACC was 56.33%. In summary,
was selected as the best parameter of PseAAC coding. At this time, the performance of the model was best. When
was 20, each protein sequence generated a 40-dimensional feature vector.
Different values of the parameter
of the PsePSSM encoding have a certain impact on the prediction performance of the model. Considering that the minimum sample sequence length was 50, the value of parameter
in PsePSSM encoding was set to 10, 20, 30, 40, and 49 in this order. It can be seen from
Table 4 that the evaluation index changed with the value of
. When the value of
in the training dataset was 30, the maximum value of BACC was 60.44%, which means that PsePSSM had the best influence on the model performance. Therefore, the value of
= 30 was the best parameter in PsePSSM. At this time, each protein sequence generated a 620-dimensional feature vector.
Different
values in AD have different effects on the prediction performance of the model. The
values in AD were set to 10, 20, 30, 40 and 50 in turn.
Table 5 shows that ACC, Sn, Sp, and BACC changed with the change in
. When
was 20 and 40, the values of ACC, Sp, and BACC in the training dataset slightly decreased, and when
was 30, BACC was the highest, reaching 57.63%. When
was 30, each protein sequence generated a 630-dimensional feature vector.
3.2. The Effect of Feature Extraction Algorithm on Prediction Results
Extracting protein sequence information using the feature extraction algorithm is an important stage in the construction of ECM protein prediction models. In this paper, five feature extraction algorithms were selected: PseAAC, PsePSSM, EBGW, AD, and LD. In order to better learn the performance of the model, this article made comparisons using five separate feature coding methods and a hybrid feature coding method named ALL (EBGW + PseAAC + PsePSSM + LD + AD). The feature vectors extracted by the six feature extraction methods were input into the RF classifier to predict them respectively. ACC, BACC, Sn, and Sp were used as evaluation indicators to assess the performance of ECMP-RF. LOOCV was used for evaluation. The results predicted by different feature extraction methods are shown in
Table 6.
In the training dataset, the ACC maximum and BACC maximum were 93.62% and 62.76% respectively, which were obtained by the LD algorithm. The ALL feature extraction method obtained an ACC of 95.03%, which was 1.41% to 2.60% higher than the result obtained by a single feature prediction. The BACC was 71.53%, which is 8.77%–15.61% higher than the result obtained by a single feature prediction. The difference in prediction results reflects the effect of feature extraction algorithms on the robustness of the prediction model. The ALL feature extraction method can not only express protein evolutionary information but also effectively use protein physicochemical properties, sequence information, and structure information. Compared with the single feature extraction method, it has better prediction performance.
3.3. Influence of Class Imbalance on Prediction Results
The two datasets used in this article, i.e., the training dataset and testing dataset, were highly imbalanced, with the ratio of positive and negative samples reaching 1:11 and 1:1.5 respectively. In this case, there was a serious imbalance between the dataset categories, and the prediction results of the classifier would be biased toward a large number of categories, which would adversely affect the prediction performance of the model. Therefore, we used SMOTE to improve the generalization ability of model. SMOTE achieves sample balance by deleting or adding some samples. LOOCV and RF classification algorithms were used to test balanced and unbalanced data. The prediction results are shown in
Table 7.
Because of the imbalance of datasets, the evaluation of ACC index was biased. At the same time, the number of non-ECM proteins was much larger than the number of ECM proteins; thus, Sn and SP indexes could not accurately express the performance of the ECMP-RF, while the BACC could reasonably measure the performance of the model in all the above indicators. It can be seen in
Table 7 that the dataset processed by SMOTE significantly improved the BACC. On the imbalanced training dataset, the BACC was 71.53%. After the dataset was balanced, the BACC was 97.04%, which is 25.51% higher than before. As a result, we think that, after SMOTE processing, the prediction performance of ECMP-RF was greatly improved.
3.4. The Effect of Feature Selection Algorithm on Prediction Results
Protein sequence features were extracted by EBGW, PseAAC, PsePSSM, LD, and AD feature extraction methods, and a 1950-dimensional feature vector was obtained after fusion. Since the fused feature vector had a large dimension and redundancy, it was very important to eliminate redundant information and keep the effective information of original data. Choosing different feature selection methods had a certain impact on the ACC of ECM protein prediction. To this end, this paper chose seven feature selection methods, namely, PCA, FA, MI, LASSO, KPCA, LR, and EN. RF was used as a classifier, and its results were tested using LOOCV. The optimal feature extraction method was selected by comparing the evaluation indicators. The results obtained by different feature selection methods are shown in
Table 8.
Table 8 shows that different feature selection methods had a certain impact on the prediction performance of the model. On the training dataset, the BACC of the EN algorithm was 97.28%, which is 0.11%, 0.16%, 8.14%, 0.12%, 0.10%, and 0.52% higher than that of PCA, FA, MI, LASSO, KPCA, and LR. In addition, ACC, Sn, and SP were 97.28%, 98.78%, and 95.78%, respectively. This shows that, compared with PCA, FA, MI, LASSO, KPCA, and LR, the EN algorithm could effectively select a subset of features, and the prediction effect in the model was better.
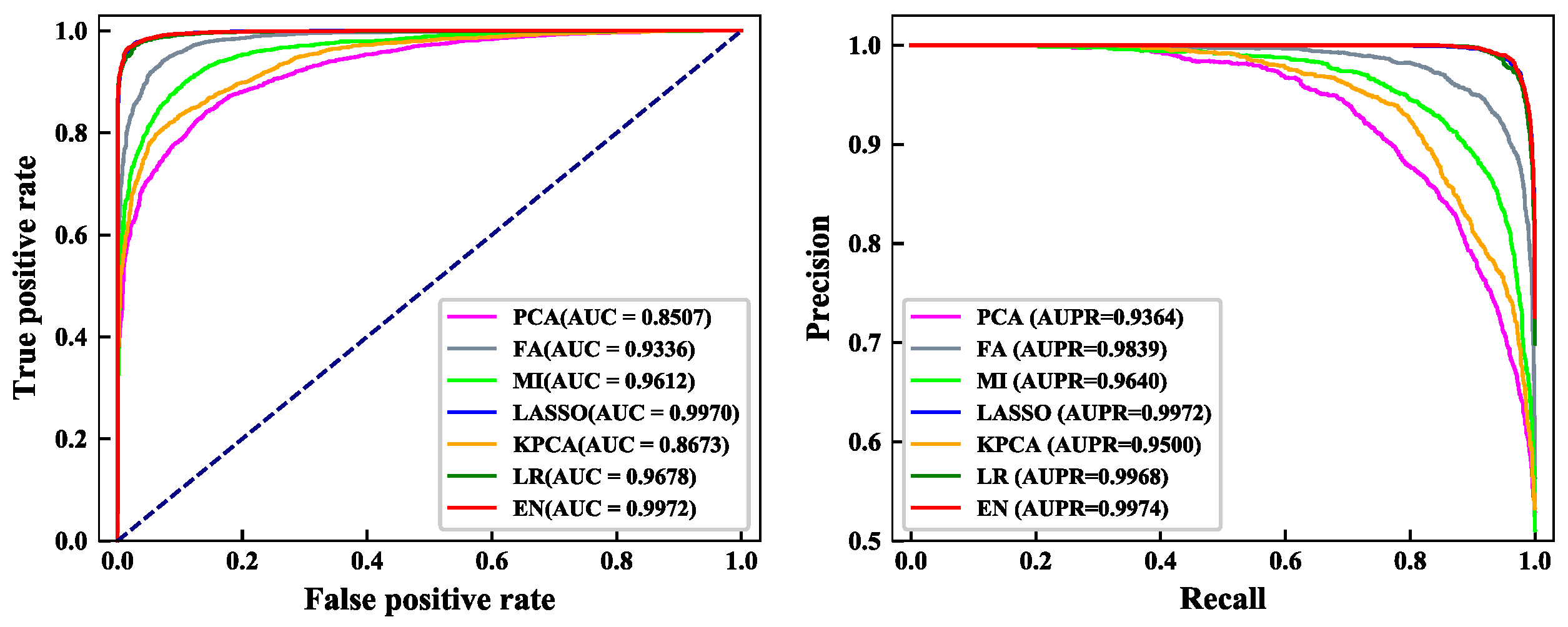
From
Figure 2, we can see the details of the ROC curve and PR curve of the training dataset under the seven feature selection methods. AUC and AUPR are important evaluation indicators that indicate the classification performance of the model. Higher values of AUC and AUPR denote the better the performance of the model. Results show that the area under the ROC and PR curves predicted by the EN algorithm was the largest. The AUC value was 0.9972, which is 0.02%–14.65% higher than the AUC value obtained by other feature selection methods. The AUPR value was 0.9974, which is 0.02%–6.10% higher than the AUPR value obtained by other feature selection methods. In conclusion, the feature selection result of the EN algorithm was the highest.
3.5. The Effect of The Classifier Algorithm on the Prediction Results
Classification algorithms play an important role in building the ECM protein prediction model. In this paper, six common classification algorithms were selected for comparison, namely GBDT, AdaBoost, naïve Bayes, SVM, LR, and RF. The feature subset constructed by EN was input into the six classifiers, and the results were tested by LOOCV. The prediction results of the training dataset on the six classifiers are shown in
Table 9. The ROC and PR curves of the six classification algorithms are shown in
Figure 3.
Table 9 shows that, on the training dataset, the BACC obtained by GBDT, AdaBoost, naïve Bayes, SVM, LR, and RF was 83.11%, 93.68%, 83.38%, 84.28%, 91.18%, and 97.28%. Among them, RF’s BACC was 14.17%, 3.60%, 13.90%, 13.00%, and 6.10% higher than that of GBDT, AdaBoost, naïve Bayes, SVM, and LR algorithms. This shows that the classification performance of RF was superior.
It can be seen from
Figure 3 that the AUC value of GBDT was 0.9071, the AUC value of AdaBoost was 0.9857, the AUC value of naïve Bayes was the lowest at 0.8822, the AUC value of SVM was 0.9262, the AUC value of LR was 0.9701, and the highest AUC value was achieved by RF at 0.9972. At the same time, the AUPR value of RF also reached 0.9974, which is 1.15%–14.62% higher than that of the other classifiers. Therefore, this paper chose RF as the classifier.
3.6. Comparison with Other Methods
At present, many researchers conducted prediction research on ECM proteins. In order to prove the validity and discrimination of the model, this section compares the ECMP-RF prediction model with other prediction models using the same datasets. To be fair, we use the LOOCV test to compare the expected method with other latest methods on the training dataset and testing dataset.
Figure 4 visually represents the prediction results of ECMP-RF and existing methods on the training dataset. Obviously, of all the methods, ECMPRED had the lowest ACC, Sp, and BACC, while PECMP [
24] had the lowest Sn. It is worth noting that, although ECMPP [
23] had the highest Sp, its Sn was poor, resulting in poor BACC results. At the same time, the proposed method ECMP-RF could improve the prediction performance of the model and get better results. As the most important evaluation index of the model, BACC was 97.3% in the training dataset. The BACC value of this paper was 19.5% higher than ECMPP, 24.2% higher than PECMP, 26.3% higher than ECMPERD [
25], 10.9% higher than IECMP [
27], 6.4% higher than ECMP-HybKNN [
28], and 3.6% higher than TargetECMP [
29]. In addition, the ACC and Sn of ECMP-RF had certain advantages. The comparison results of the training datasets are shown in
Table S1 (Supplementary Materials). In short, we conclude that the method we propose is superior to existing models.
In order to objectively evaluate the methods proposed in this paper, we used the same testing dataset to test the prediction performance of the above methods. The specific results are shown in
Figure 5. The BACC of the testing dataset on ECMP-RF was 97.9%, which is 33.9%, 29.2%, 42.9%, 20.4%, and 11.4% higher than ECMPP [
23], PECMP [
24], ECMPRED [
25], IECMP [
27], and TargetECMP [
29], respectively. The prediction results show that, compared with other ECM protein prediction methods, the ACC of the ECMP-RF model prediction was much higher than that of other methods, which verifies the excellent performance of ECMP-RF. The comparison results on the testing dataset are presented in
Table S2 (Supplementary Materials).

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}