
Iterative Variable Selection for High-Dimensional Data: Prediction of Pathological Response in Triple-Negative Breast Cancer

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Methodology and Algorithms
2.1. The Sparse-Group Lasso
2.2. Selection of the Optimal Regularization Parameter
| Algorithm 1: Two-parameter iterative sparse-group lasso (isgl) |
 |
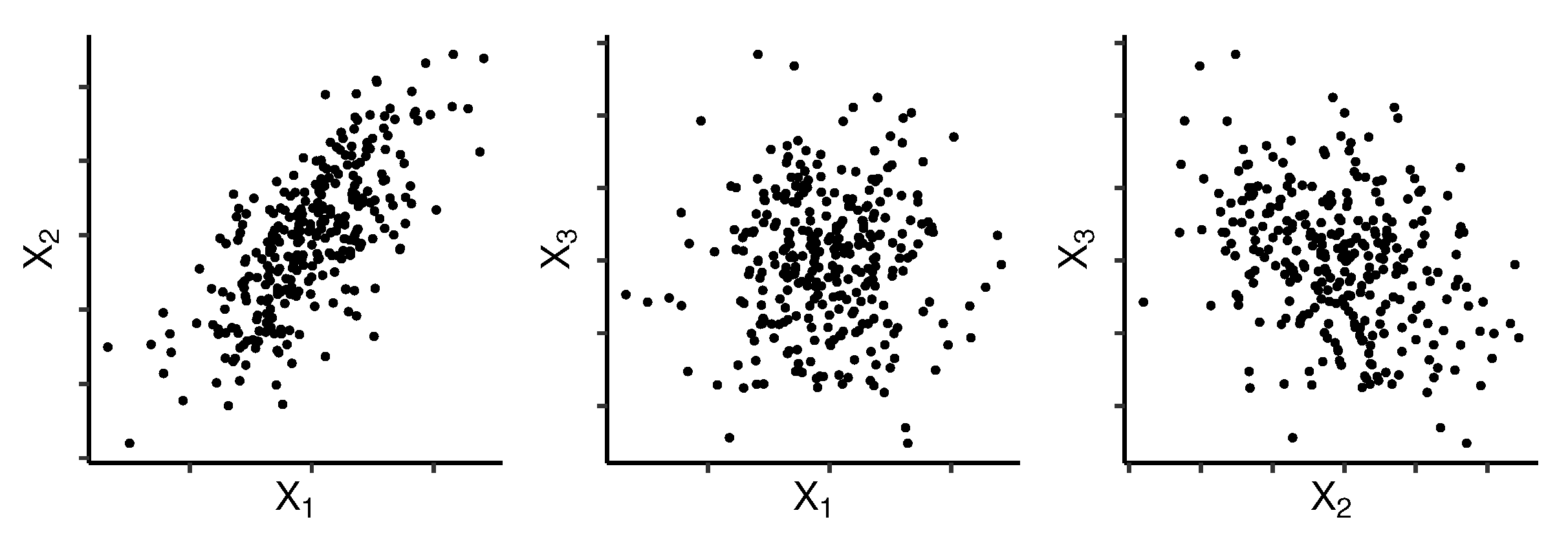
2.3. Grouping Variables Using Principal Component Analysis
2.4. Mining Influent Variables under a Cross-Validation Approach
| Algorithm 2: |
 |
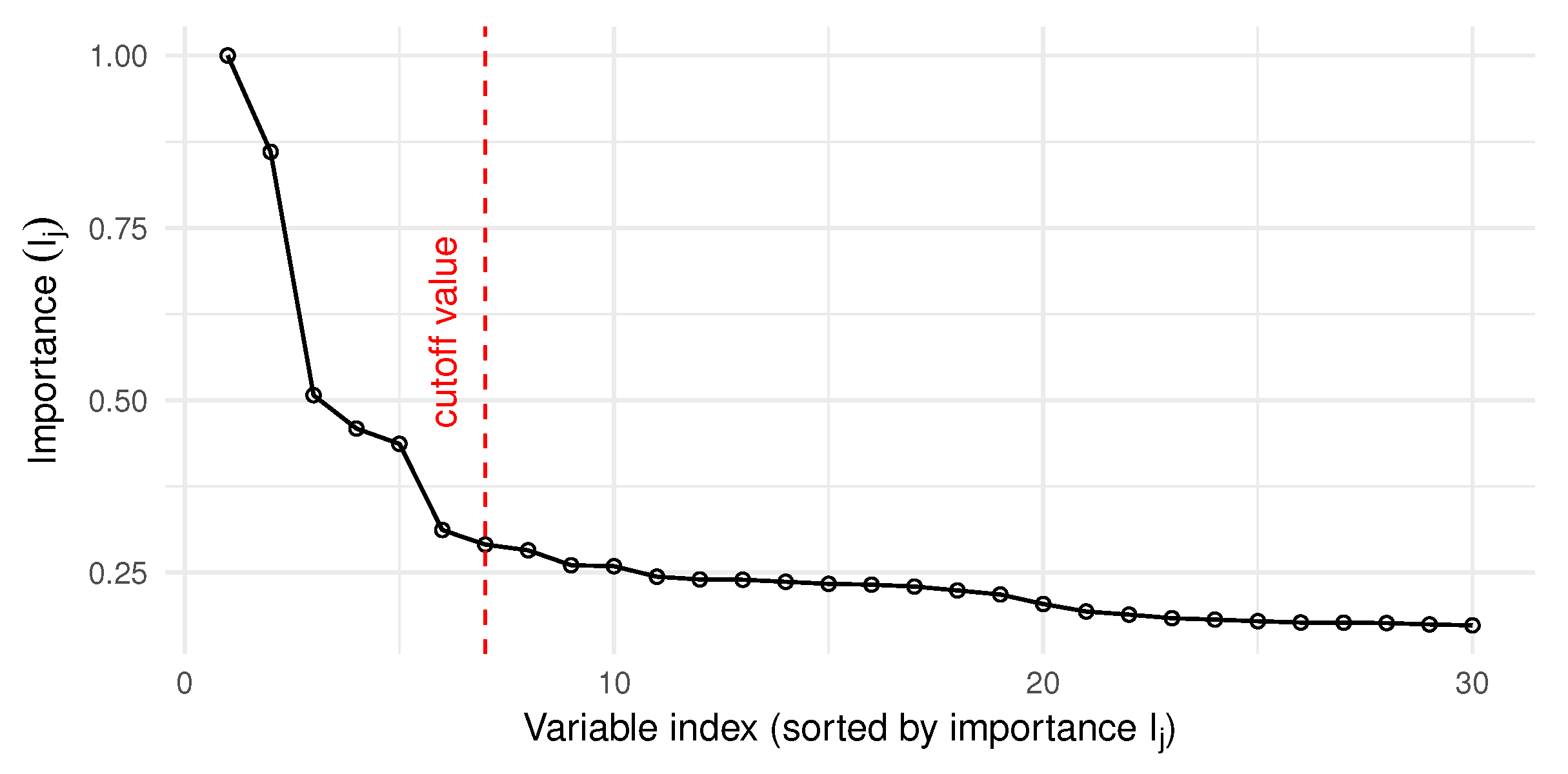
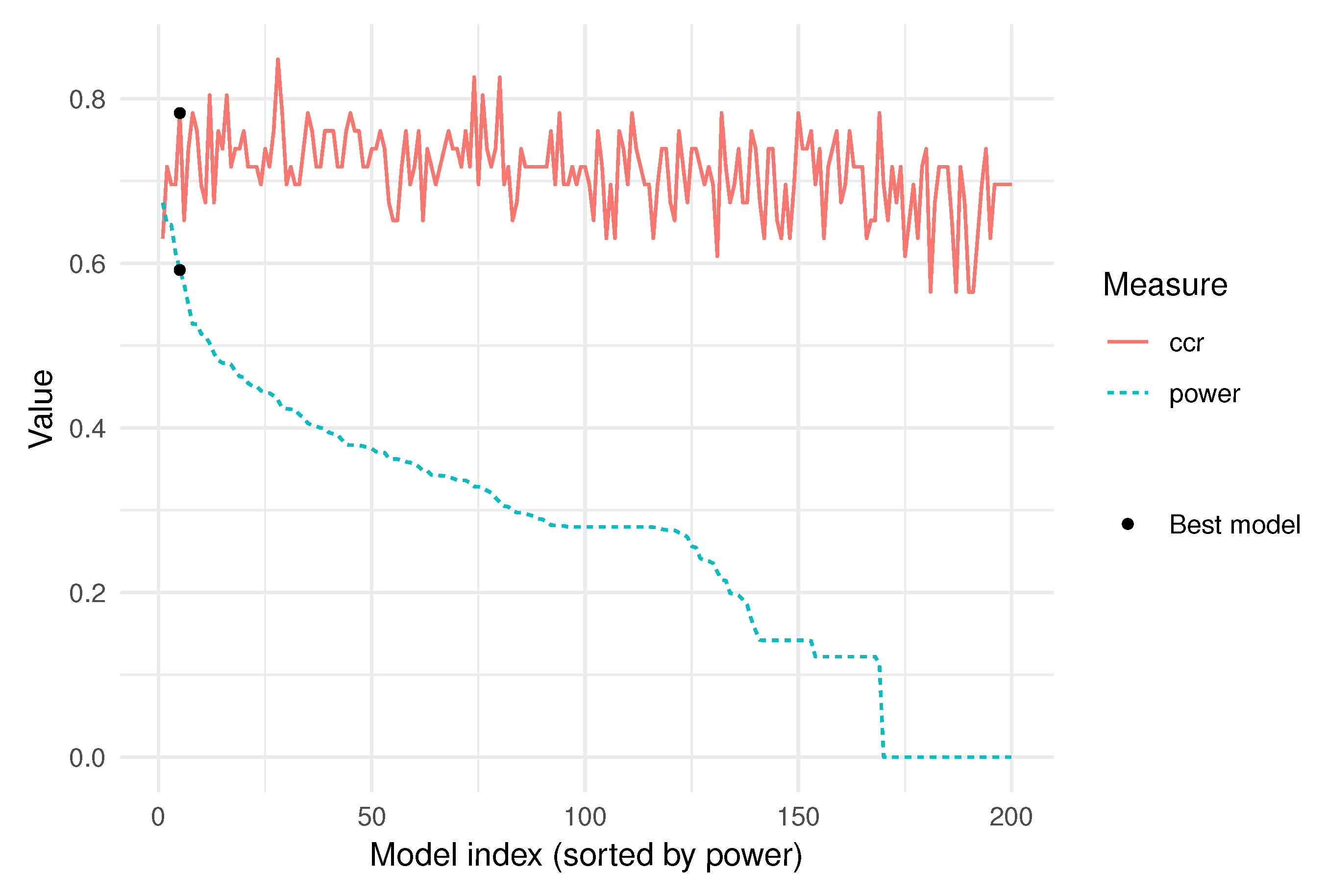
2.5. Selection of the Best Model
3. A Simulation Study
- (SFHT_1)
- and are i.i.d , for .
- (SFHT_2)
- In this example, is generated as in SFHT_1, but the rows of the model matrix are i.i.d. generated from a multivariate gaussian distribution with .
- (Tibs_1)
- and the rows of are i.i.d. generated from a multivariate gaussian distribution with .
- (Tibs_4)
- and the rows of are i.i.d. generated from a multivariate gaussian distribution with , and .
- (ZH_d)
- and the rows of were generated as follows:
- , i.i.d. for
where are i.i.d. , for .
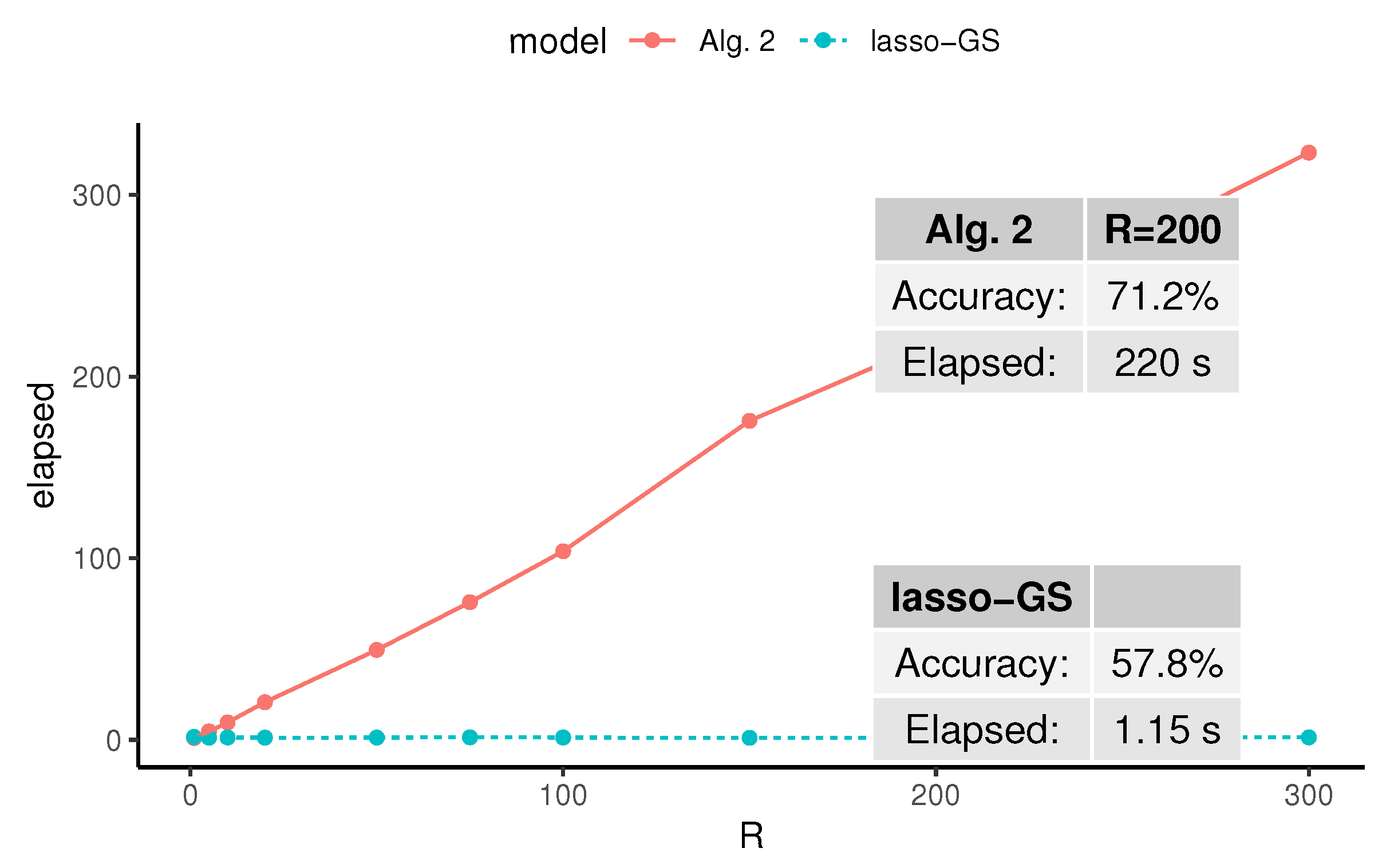
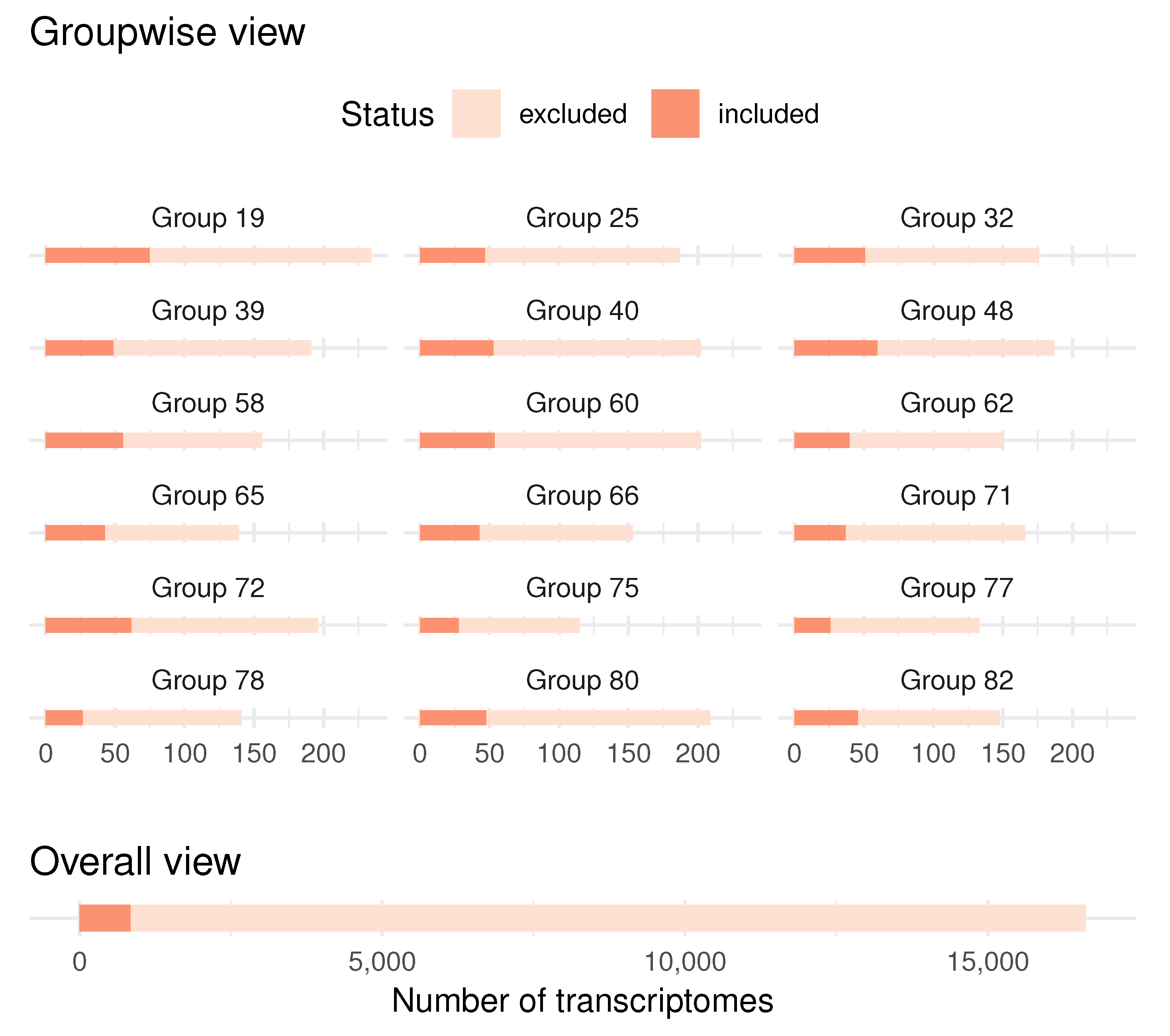
4. Application to Biomedical Data
5. Conclusions
- A clustering of the variables, based on PCA, makes it possible to work with an arbitrarily large number of variables without specifying groups a priori.
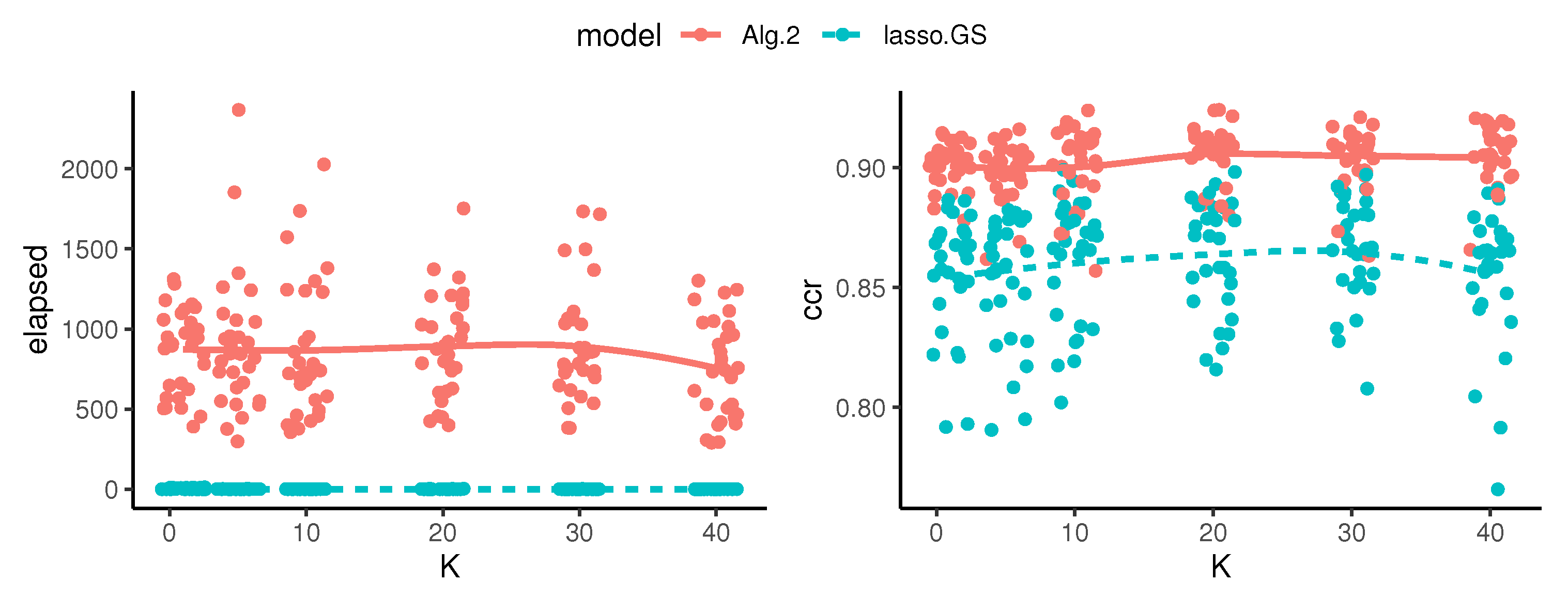
- The iterative sparse group lasso removes the dependence on the hyper-parameters of the sparse group lasso, but is sensible to the train–validate sample partitions. This problem has been solved running the algorithm for a large number of different train–validate sample partitions (Algorithm 2).
- The correct classification rate of each model in its respective validation sample is stored. Notice that this is an overestimation of the true correct classification rate on future observations, and the highest validation rate does not imply the best model.
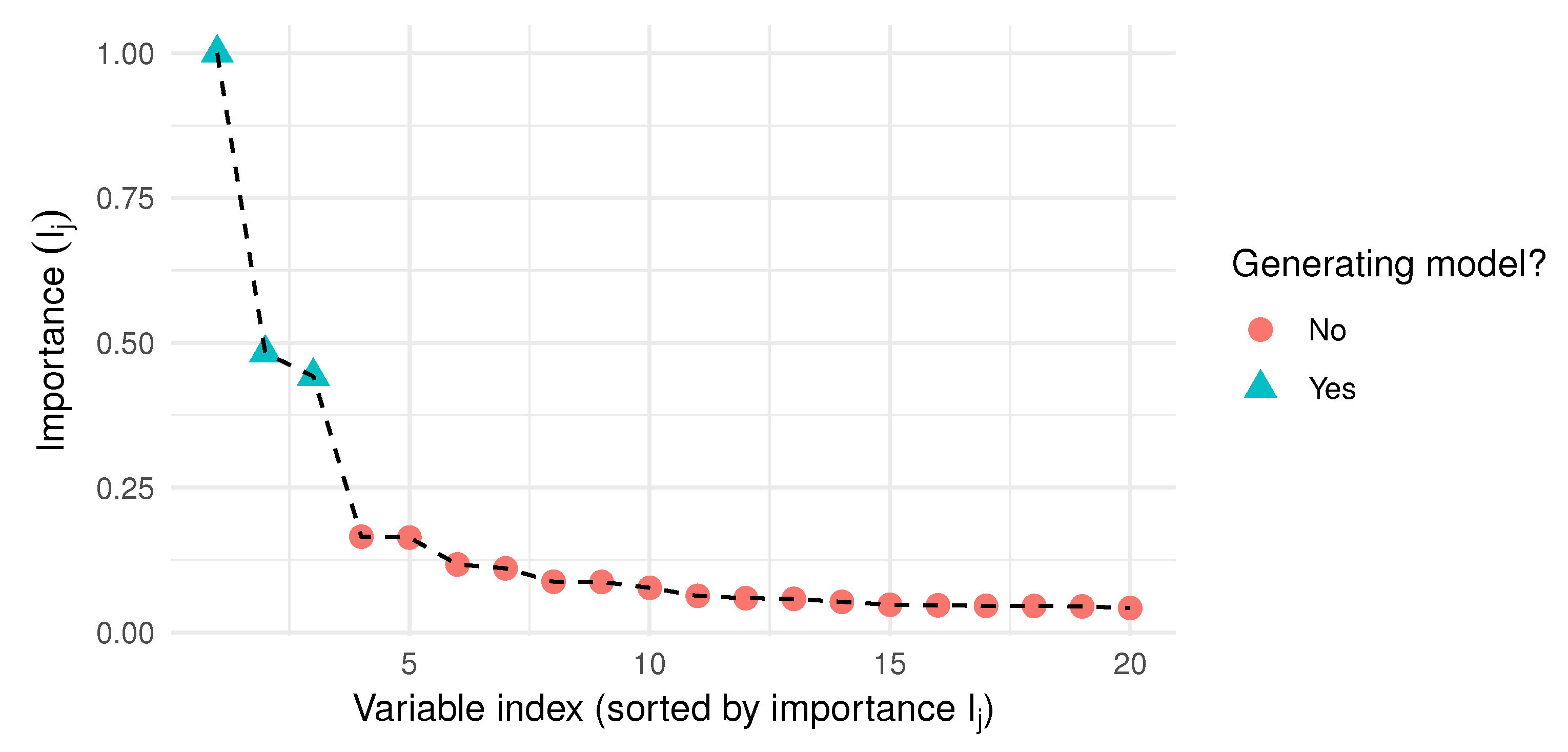
- The importance index weighs the variables, based on the correct classification rate of the models that include them.
- The power index weighs the models, based on the importance of the variables they include.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.; Forman, D.; Bray, F. GLOBOCAN 2012 v1. 0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11; International Agency for Research on Cancer: Lyon, France, 2013. [Google Scholar]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [Green Version]
- Cortazar, P.; Zhang, L.; Untch, M.; Mehta, K.; Costantino, J.P.; Wolmark, N.; Bonnefoi, H.; Cameron, D.; Gianni, L.; Valagussa, P.; et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 2014, 384, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Symmans, W.F.; Wei, C.; Gould, R.; Yu, X.; Zhang, Y.; Liu, M.; Walls, A.; Bousamra, A.; Ramineni, M.; Sinn, B.; et al. Long-Term Prognostic Risk After Neoadjuvant Chemotherapy Associated With Residual Cancer Burden and Breast Cancer Subtype. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 1049–1060. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; López-Tarruella, S.; Garcia-Saenz, J.A.; Khan, Q.J.; Gomez, H.; Prat, A.; Moreno, F.; Jerez-Gilarranz, Y.; Barnadas, A.; Picornell, A.C.; et al. Pathological response and survival in triple-negative breast cancer following neoadjuvant carboplatin plus docetaxel. Clin. Cancer Res. 2018, 24, 5820–5829. [Google Scholar] [CrossRef] [Green Version]
- Tabchy, A.; Valero, V.; Vidaurre, T.; Lluch, A.; Gomez, H.L.; Martin, M.; Qi, Y.; Barajas-Figueroa, L.J.; Souchon, E.A.; Coutant, C.; et al. Evaluation of a 30-gene paclitaxel, fluorouracil, doxorubicin and cyclophosphamide chemotherapy response predictor in a multicenter randomized trial in breast cancer. Clin. Cancer Res. 2010, 16, 5351–5361. [Google Scholar] [CrossRef] [Green Version]
- Hatzis, C.; Pusztai, L.; Valero, V.; Booser, D.J.; Esserman, L.; Lluch, A.; Vidaurre, T.; Holmes, F.; Souchon, E.; Wang, H.; et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 2011, 305, 1873–1881. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.C.; Wooten, E.C.; Tsimelzon, A.; Hilsenbeck, S.G.; Gutierrez, M.C.; Elledge, R.; Mohsin, S.; Osborne, C.K.; Chamness, G.C.; Allred, D.C.; et al. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet 2003, 362, 362–369. [Google Scholar] [CrossRef]
- De Los Campos, G.; Gianola, D.; Allison, D.B. Predicting genetic predisposition in humans: the promise of whole-genome markers. Nat. Rev. Genet. 2010, 11, 880. [Google Scholar] [CrossRef]
- Lupski, J.R.; Belmont, J.W.; Boerwinkle, E.; Gibbs, R.A. Clan genomics and the complex architecture of human disease. Cell 2011, 147, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Offit, K. Personalized medicine: New genomics, old lessons. Hum. Genet. 2011, 130, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Szymczak, S.; Biernacka, J.M.; Cordell, H.J.; González-Recio, O.; König, I.R.; Zhang, H.; Sun, Y.V. Machine learning in genome-wide association studies. Genet. Epidemiol. 2009, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, N.; Friedman, J.; Hastie, T.; Tibshirani, R. A sparse-group lasso. J. Comput. Graph. Stat. 2013, 22, 231–245. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Yuan, M.; Lin, Y. Model selection and estimation in regression with grouped variables. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 2006, 68, 49–67. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regression shrinkage and selection via the elastic net, with applications to microarrays. J. R. Stat. Soc. Ser. B 2003, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Laria, J.C.; Carmen Aguilera-Morillo, M.; Lillo, R.E. An iterative sparse-group lasso. J. Comput. Graph. Stat. 2019, 28, 722–731. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Azevedo Costa, M.; de Souza Rodrigues, T.; da Costa, A.G.F.; Natowicz, R.; Pádua Braga, A. Sequential selection of variables using short permutation procedures and multiple adjustments: An application to genomic data. Stat. Methods Med Res. 2017, 26, 997–1020. [Google Scholar] [CrossRef]
- Sharma, P.; López-Tarruella, S.; García-Saenz, J.A.; Ward, C.; Connor, C.; Gómez, H.L.; Prat, A.; Moreno, F.; Jerez-Gilarranz, Y.; Barnadas, A.; et al. Efficacy of neoadjuvant carboplatin plus docetaxel in triple negative breast cancer: Combined analysis of two cohorts. Clin. Cancer Res. 2017, 23, 649–657. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, Y.; Gao, Y.; Cui, Y.; Liu, H.; Li, M.; Tian, Y. Downregulation of HNF1 homeobox B is associated with drug resistance in ovarian cancer. Oncol. Rep. 2014, 32, 979–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanrahan, K.; O’neill, A.; Prencipe, M.; Bugler, J.; Murphy, L.; Fabre, A.; Puhr, M.; Culig, Z.; Murphy, K.; Watson, R.W. The role of epithelial–mesenchymal transition drivers ZEB1 and ZEB2 in mediating docetaxel-resistant prostate cancer. Mol. Oncol. 2017, 11, 251–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín-Aguilera, M.; Codony-Servat, J.; Reig, Ò.; Lozano, J.J.; Fernández, P.L.; Pereira, M.V.; Jiménez, N.; Donovan, M.; Puig, P.; Mengual, L.; et al. Epithelial-to-mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer. Mol. Cancer Ther. 2014, 13, 1270–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puhr, M.; Hoefer, J.; Schäfer, G.; Erb, H.H.; Oh, S.J.; Klocker, H.; Heidegger, I.; Neuwirt, H.; Culig, Z. Epithelial-to-mesenchymal transition leads to docetaxel resistance in prostate cancer and is mediated by reduced expression of miR-200c and miR-205. Am. J. Pathol. 2012, 181, 2188–2201. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Matassa, D.; Amoroso, M.; Lu, H.; Avolio, R.; Arzeni, D.; Procaccini, C.; Faicchia, D.; Maddalena, F.; Simeon, V.; Agliarulo, I.; et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. 2016, 23, 1542. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.; Yin, J.; He, H.; Li, W.; Hou, C.; Qian, X.; Mao, N.; Pan, L. Mitochondrial comparative proteomics of human ovarian cancer cells and their platinum-resistant sublines. Proteomics 2010, 10, 3789–3799. [Google Scholar] [CrossRef]
- Chappell, N.P.; Teng, P.n.; Hood, B.L.; Wang, G.; Darcy, K.M.; Hamilton, C.A.; Maxwell, G.L.; Conrads, T.P. Mitochondrial proteomic analysis of cisplatin resistance in ovarian cancer. J. Proteome Res. 2012, 11, 4605–4614. [Google Scholar] [CrossRef]
- Marrache, S.; Pathak, R.K.; Dhar, S. Detouring of cisplatin to access mitochondrial genome for overcoming resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 10444–10449. [Google Scholar] [CrossRef] [Green Version]
- Belotte, J.; Fletcher, N.M.; Awonuga, A.O.; Alexis, M.; Abu-Soud, H.M.; Saed, M.G.; Diamond, M.P.; Saed, G.M. The role of oxidative stress in the development of cisplatin resistance in epithelial ovarian cancer. Reprod. Sci. 2014, 21, 503–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAdam, E.; Brem, R.; Karran, P. Oxidative Stress–Induced Protein Damage Inhibits DNA Repair and Determines Mutation Risk and Therapeutic Efficacy. Mol. Cancer Res. 2016, 14, 612–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PC1 | PC2 | |
|---|---|---|
| −0.67 | 0.40 | |
| −0.70 | −0.08 | |
| 0.23 | 0.91 |
| Simulation A | |||||
|---|---|---|---|---|---|
| p = 40 | p = 100 | p = 400 | p = 1000 | p = 4000 | |
| Alg. 2 | 77.82 (0.88) | 73.97 (1.04) | 65.42 (1.23) | 62.1 (1.09) | 56.95 (1.19) |
| Lasso (Grid Search) | 82.61 (0.62) | 81.1 (0.95) | 83.08 (0.7) | 83.48 (0.56) | 84.27 (0.86) |
| Alg. 2 (Known Groups) | 74.66 (1.24) | 67.01 (1.8) | 61.38 (1.8) | 59.07 (1.49) | 54.54 (1.19) |
| Simulation B | |||||
| p = 40 | p = 100 | p = 400 | p = 1000 | p = 4000 | |
| Alg. 2 | 84.72 (0.65) | 82.28 (0.99) | 75.63 (1.14) | 73.33 (1.55) | 68.87 (1.46) |
| Lasso (Grid Search) | 87.82 (0.57) | 88.01 (0.67) | 88.16 (0.65) | 89.25 (0.43) | 88.5 (0.49) |
| Alg. 2 (Known Groups) | 80.36 (0.93) | 81.57 (0.82) | 75.72 (1.18) | 74.98 (0.96) | 69.48 (2.04) |
| Simulation ZHa | |||||
| p = 40 | p = 100 | p = 400 | p = 1000 | p = 4000 | |
| Alg. 2 | 79.44 (0.76) | 76.76 (0.87) | 71.2 (0.86) | 68.52 (1.08) | 66.93 (1.46) |
| Lasso (Grid Search) | 82.75 (0.79) | 83.39 (0.6) | 83.54 (0.41) | 85.68 (0.42) | 84.68 (0.45) |
| Alg. 2 (Known Groups) | 78.72 (1.12) | 75.75 (1.22) | 70.81 (1.51) | 69.54 (1.95) | 61.98 (1.84) |
| Simulation ZHc | |||||
| p = 40 | p = 100 | p = 400 | p = 1000 | p = 4000 | |
| Alg. 2 | 89.11 (0.3) | 87.95 (0.38) | 88 (0.52) | 88.96 (0.54) | 89.68 (0.4) |
| Lasso (Grid Search) | 92.1 (0.43) | 92.52 (0.55) | 93.27 (0.34) | 93.21 (0.35) | 92.7 (0.38) |
| Alg. 2 (Known Groups) | 83.98 (0.86) | 83.11 (0.64) | 81.12 (0.8) | 81.81 (0.67) | 81.52 (0.97) |
| Simulation ZHd | |||||
| p = 40 | p = 100 | p = 400 | p = 1000 | p = 4000 | |
| Alg. 2 | 85.91 (0.81) | 83.44 (0.87) | 83.19 (0.81) | 80.41 (1.16) | 71.89 (1.51) |
| Lasso (Grid Search) | 89.98 (0.63) | 89.96 (0.86) | 90.7 (0.5) | 91.87 (0.6) | 89.94 (0.98) |
| Alg. 2 (Known Groups) | 79.79 (1.59) | 75.55 (1.74) | 68.4 (1.71) | 62.58 (1.62) | 55.93 (1.29) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laria, J.C.; Aguilera-Morillo, M.C.; Álvarez, E.; Lillo, R.E.; López-Taruella, S.; del Monte-Millán, M.; Picornell, A.C.; Martín, M.; Romo, J. Iterative Variable Selection for High-Dimensional Data: Prediction of Pathological Response in Triple-Negative Breast Cancer. Mathematics 2021, 9, 222. https://doi.org/10.3390/math9030222
Laria JC, Aguilera-Morillo MC, Álvarez E, Lillo RE, López-Taruella S, del Monte-Millán M, Picornell AC, Martín M, Romo J. Iterative Variable Selection for High-Dimensional Data: Prediction of Pathological Response in Triple-Negative Breast Cancer. Mathematics. 2021; 9(3):222. https://doi.org/10.3390/math9030222
Chicago/Turabian StyleLaria, Juan C., M. Carmen Aguilera-Morillo, Enrique Álvarez, Rosa E. Lillo, Sara López-Taruella, María del Monte-Millán, Antonio C. Picornell, Miguel Martín, and Juan Romo. 2021. "Iterative Variable Selection for High-Dimensional Data: Prediction of Pathological Response in Triple-Negative Breast Cancer" Mathematics 9, no. 3: 222. https://doi.org/10.3390/math9030222
APA StyleLaria, J. C., Aguilera-Morillo, M. C., Álvarez, E., Lillo, R. E., López-Taruella, S., del Monte-Millán, M., Picornell, A. C., Martín, M., & Romo, J. (2021). Iterative Variable Selection for High-Dimensional Data: Prediction of Pathological Response in Triple-Negative Breast Cancer. Mathematics, 9(3), 222. https://doi.org/10.3390/math9030222