
Feature Selection for Colon Cancer Detection Using K-Means Clustering and Modified Harmony Search Algorithm




Abstract
:1. Introduction
2. Related Work
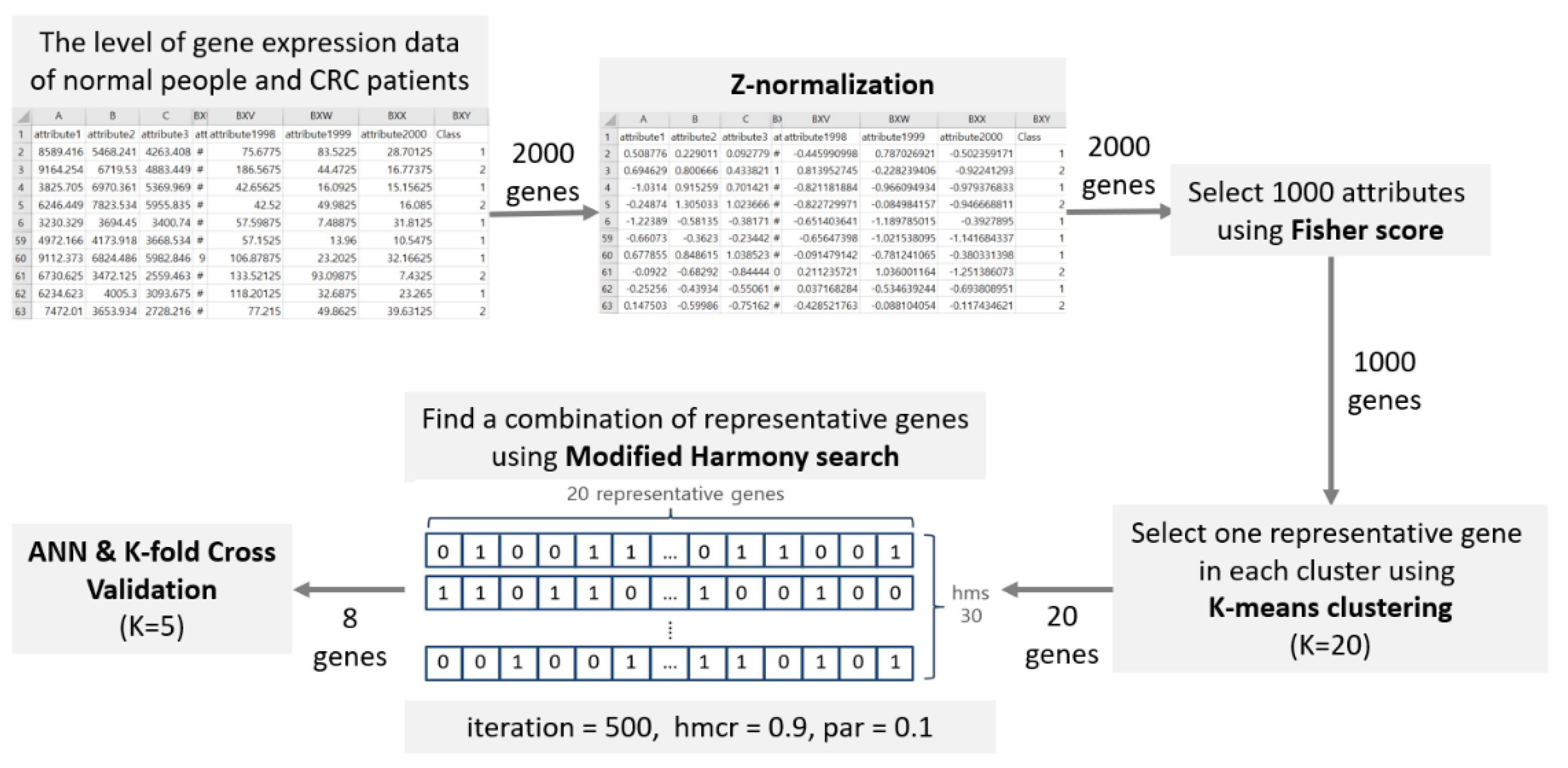
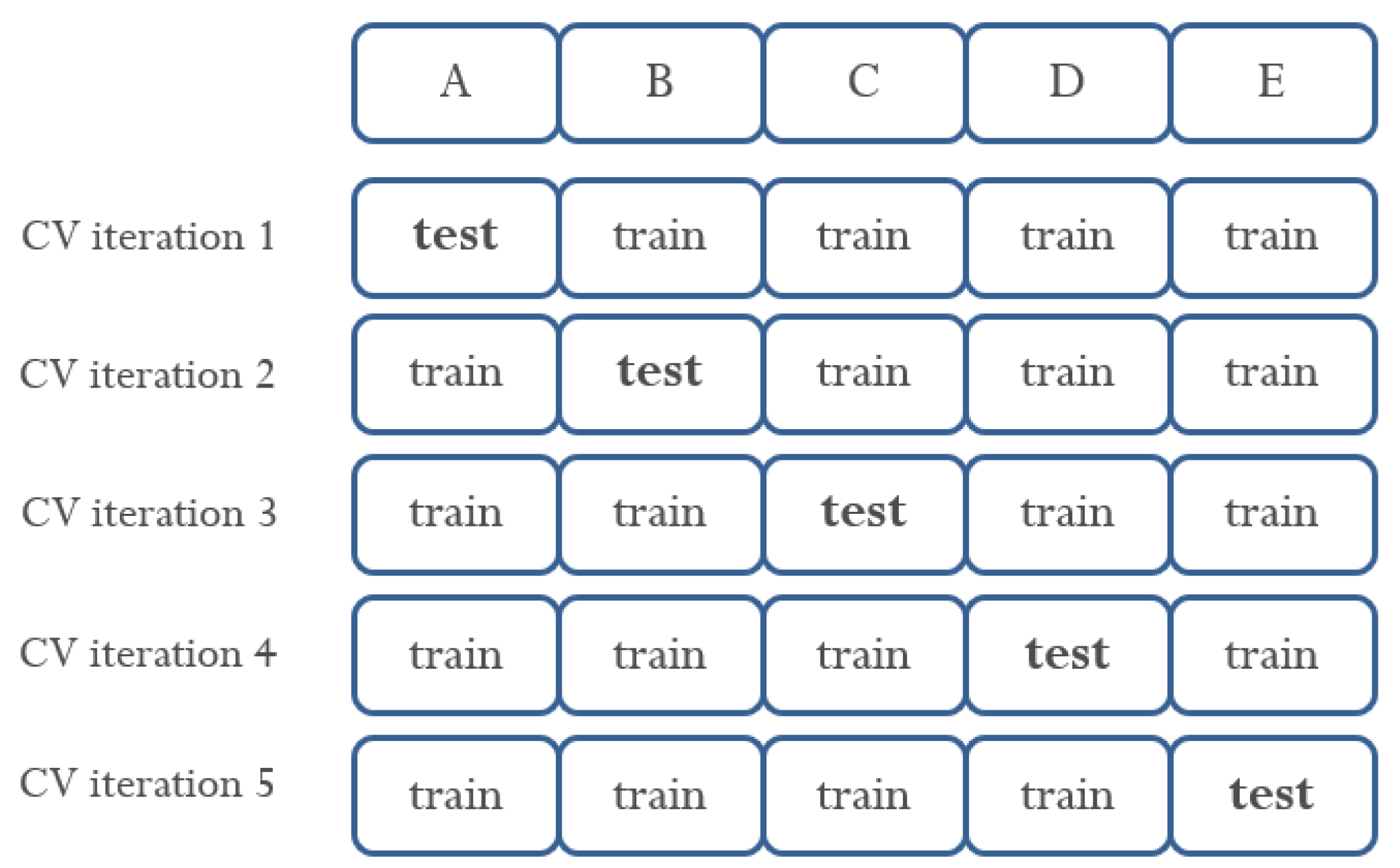
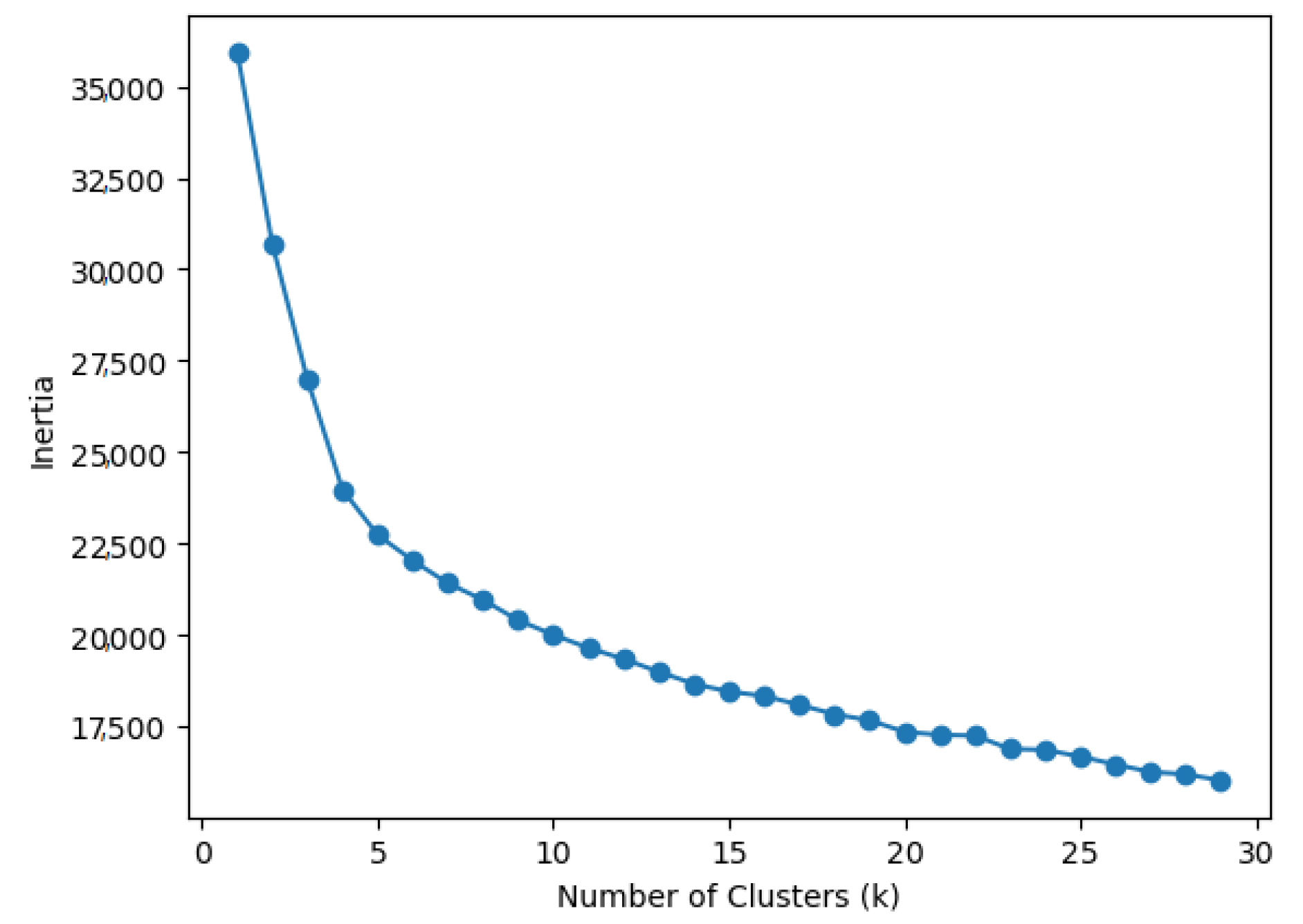
3. Materials and Methods
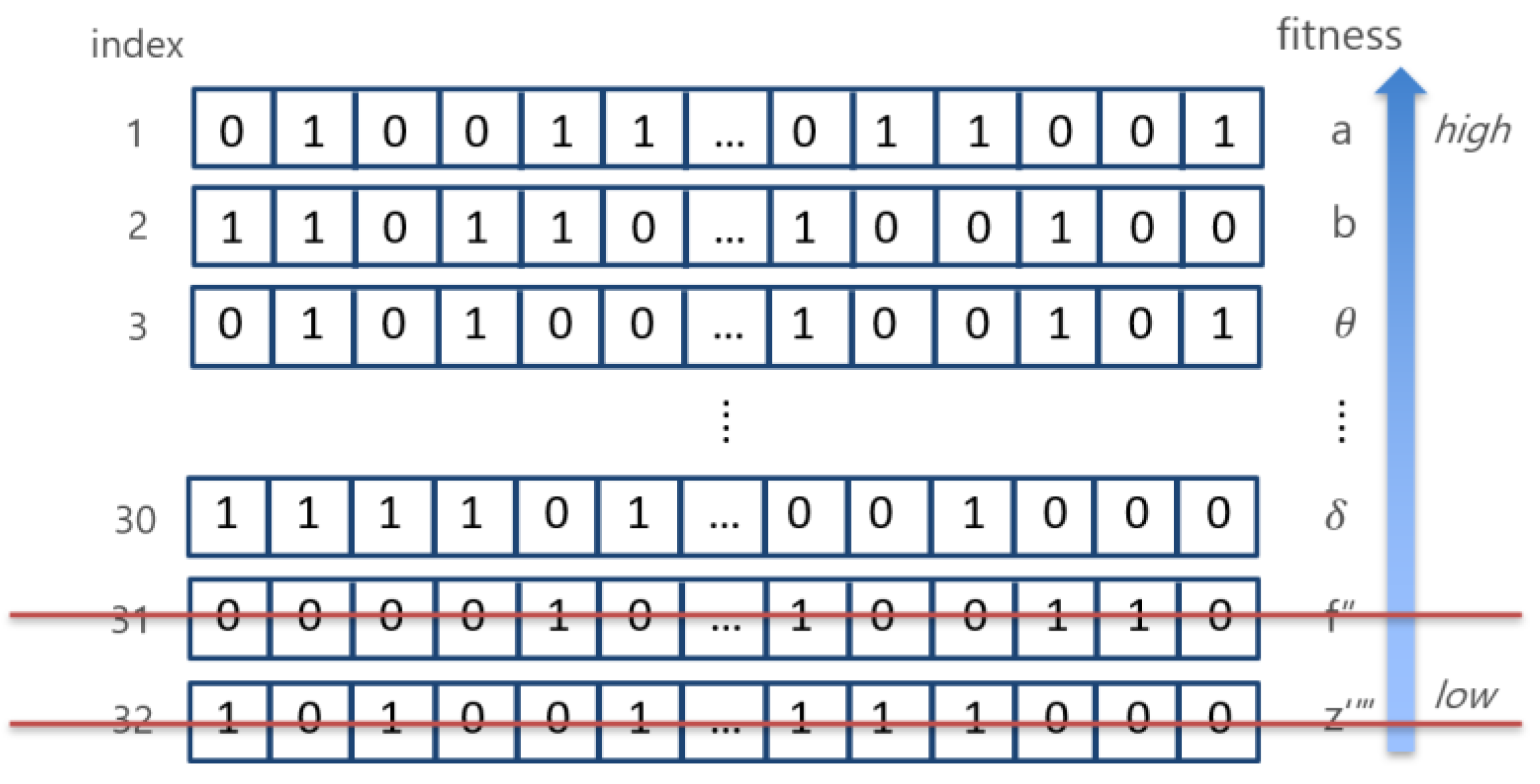
| Algorithm 1. Pseudocode of Modified Harmony Search Algorithm |
| 1. Set the parameters BDR, HMS, HMCR, PAR 2. Set itr: =0 {iteration initialization} 3. Initialize Harmony with 0 and 1 (binary value) 4. BDR = HMS*0.2 //set the upper and lower area boundary 5. For (i = 1: i ≤ HMS) then 6. generate initial Harmony 7. End for 8. Repeat 9. For ( = 1: N) then //Harmony search in upper area 10. = Randomly select from to 11. end for 12. generate new Harmony ( 13. If (Rand(0,1) < HMCR) then //Harmony search in lower area 14. For ( = 1: N) then 15. Randomly select from to 16. If (Rand(0,1) < PAR) then 17. 18. end if 19. end for 20. generate new Harmony () 21. else 22. generate new Harmony randomly 23. end if 24. if (fit() < fit()) 25. update harmony memory 26. end if 27. set itr+=1 28. until (itr < maxit) 29. Get the best harmony |
4. Results
5. Comparisons with Other Method Surveys
6. Conclusions and Future Works
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2018. [Google Scholar]
- Center, M.M.; Jemal, A.; Smith, R.A.; Ward, E. Worldwide Variations in Colorectal Cancer. CA A Cancer J. Clin. 2009, 59, 366–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Fedewa, S.A.; Anderson, W.F.; Miller, K.D.; Ma, J.; Rosenberg, P.S.; Jemal, A. Colorectal Cancer Incidence Patterns in the United States, 1974–2013. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [Green Version]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Gastroenterol. Rev. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Soravia, C.; Bapat, B.; Cohen, Z. Familial adenomatous polyposis (FAP) and hereditary nonpolyposis colorectal cancer (HNPCC): A review of clinical, genetic and therapeutic aspects. Schweiz. Med. Wochenschr. 1997, 127, 682. [Google Scholar] [PubMed]
- National Center for Biotechnology Information (US). Genes and Disease. Bethesda (MD): National Center for Biotechnology Information (US). Colon Cancer. 1998. Available online: https://www.ncbi.nlm.nih.gov/books/NBK22218/ (accessed on 15 January 2021).
- Burt, R.; Neklason, D.W. Genetic Testing for Inherited Colon Cancer. Gastroenterology 2005, 128, 1696–1716. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Li, Z.; Han, J. Generalized fisher score for feature selection. arXiv 2012, arXiv:1202.3725. [Google Scholar]
- Coates, A.; Ng, A.Y. Learning Feature Representations with K-Means. In Pattern Recognition. ICPR International Workshops and Challenges; Springer: New York, NY, USA, 2012; pp. 561–580. [Google Scholar]
- Lee, K.S.; Geem, Z.W. A new meta-heuristic algorithm for continuous engineering optimization: Harmony search theory and practice. Comput. Methods Appl. Mech. Eng. 2005, 194, 3902–3933. [Google Scholar] [CrossRef]
- Bertoni, A.; Folgieri, R.; Valentini, G. Bio-molecular cancer prediction with random subspace ensembles of support vector machines. Neurocomputing 2005, 63, 535–539. [Google Scholar] [CrossRef] [Green Version]
- Valentini, G.; Masulli, F. NEURObjects: An object-oriented library for neural network development. Neurocomputing 2002, 48, 623–646. [Google Scholar] [CrossRef] [Green Version]
- Marvi-Khorasani, H.; Usefi, H. Feature Clustering Towards Gene Selection. In Proceedings of the 2019 18th IEEE International Conference on Machine Learning And Applications (ICMLA), Boca Raton, FL, USA, 16–19 December 2019; pp. 1466–1469. [Google Scholar]
- Princeton University Gene Expression Project. Available online: http://microarray.princeton.edu/oncology/ (accessed on 15 January 2021).
- Misquitta, C.M.; Iyer, V.R.; Werstiuk, E.S.; Grover, A.K. The role of 3′-untranslated region (3′-UTR) mediated mRNA stability in cardiovascular pathophysiology. Mol. Cell. Biochem. 2001, 224, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Cheadle, C.; Vawter, M.P.; Freed, W.J.; Becker, K.G. Analysis of Microarray Data Using Z Score Transformation. J. Mol. Diagn. 2003, 5, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Bry, X.; Trottier, C.; Verron, T.; Mortier, F. Supervised component generalized linear regression using a PLS-extension of the Fisher scoring algorithm. J. Multivar. Anal. 2013, 119, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Hickmann, K.S.; Fairchild, G.; Priedhorsky, R.; Generous, N.; Hyman, J.M.; Deshpande, A.; Del Valle, S.Y. Forecasting the 2013–2014 Influenza Season Using Wikipedia. Plos Comput. Biol. 2015, 11, e1004239. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, B.; Lv, H.; Yin, Q.; Zhang, Q.; Wei, X. Constraining DNA Sequences with a Triplet-Bases Unpaired. IEEE Trans. Nanobioscience 2020, 19, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-Y.; Lee, I.-H.; Kim, D.; Zhang, B.-T. Multiobjective Evolutionary Optimization of DNA Sequences for Reliable DNA Computing. IEEE Trans. Evol. Comput. 2005, 9, 143–158. [Google Scholar] [CrossRef] [Green Version]
- Chaves-González, J.M.; Vega-Rodríguez, M.A. DNA strand generation for DNA computing by using a multi-objective differential evolution algorithm. Biosystems 2014, 116, 49–64. [Google Scholar] [CrossRef]
- Chaves-González, J.M. Hybrid multiobjective metaheuristics for the design of reliable DNA libraries. J. Heuristics 2015, 21, 751–788. [Google Scholar] [CrossRef]
- Chaves-González, J.M.; Vega-Rodríguez, M.A.; Granado-Criado, J.M. A multiobjective swarm intelligence approach based on artificial bee colony for reliable DNA sequence design. Eng. Appl. Artif. Intell. 2013, 26, 2045–2057. [Google Scholar] [CrossRef]
- Elyasigomari, V.; Lee, D.; Screen, H.; Shaheed, M. Development of a two-stage gene selection method that incorporates a novel hybrid approach using the cuckoo optimization algorithm and harmony search for cancer classification. J. Biomed. Inform. 2017, 67, 11–20. [Google Scholar] [CrossRef]
- Mohsen, A.M.; Khader, A.T.; Ramachandram, D. HSRNAFold: A harmony search algorithm for RNA secondary structure prediction based on minimum free energy. In Proceedings of the 2008 International Conference on Innovations in Information Technology, Al Ain, United Arab Emirates, 16–18 December 2008; pp. 11–15. [Google Scholar]
- Faraji, B.; Esfahani, Z.; Rouhollahi, K.; Khezri, D. Optimal Canceling of the Physiological Tremor for Rehabilitation in Parkinson’s disease. J. Exerc. Sci. Med. 2020, 11. [Google Scholar]
- Jain, A.K.; Mao, J.; Mohiuddin, M. Neural networks: A tutorial. IEEE Comput. 1996, 29, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Elad, A.; Kimmel, R. On bending invariant signatures for surfaces. IEEE Trans. Pattern Anal. Mach. Intell. 2003, 25, 1285–1295. [Google Scholar] [CrossRef]
- Grasso, S.; Tristante, E.; Saceda, M.; Carbonell, P.; Mayor-López, L.; Carballo-Santana, M.; Martínez-Lacaci, I. Resistance to Selumetinib (AZD6244) in colorectal cancer cell lines is mediated by p70S6K and RPS6 activation. Neoplasia 2014, 16, 845–860. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.C.; Chien, M.H.; Lai, T.C.; Su, C.Y.; Jan, Y.H.; Hsiao, M.; Chen, C.L. Monoamine Oxidase B Expression Correlates with a Poor Prognosis in Colorectal Cancer Patients and Is Significantly Associated with Epitheli-al-to-Mesenchymal Transition-Related Gene Signatures. Int. J. Mol. Sci. 2020, 21, 2813. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Roehrl, M.H.; Wang, J.Y. Proteomic profiling of antibody-inducing immunogens in tumor tissue identifies PSMA1, LAP3, ANXA3, and maspin as colon cancer markers. Oncotarget 2018, 9, 3996–4019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves Martins, B.A.; De Bulhões, G.F.; Cavalcanti, I.N.; Martins, M.M.; de Oliveira, P.G.; Martins, A.M.A. Biomarkers in colorectal cancer: The role of translational proteomics research. Front. Oncol. 2019, 9, 1284. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Wu, T.; Xu, L.; Liu, A.; Ji, Y.; Hu, G. Upstream binding factor up-regulated in hepatocellular carcinoma is related to the survival and cisplatin-sensitivity of cancer cells. FASEB J. 2002, 16, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Korman, L.Y.; Sayadi, H.; Bass, B.; Moody, T.W.; Harmon, J.W. Distribution of vasoactive intestinal polypeptide and substance P receptors in human colon and small intestine. Dig. Dis. Sci. 1989, 34, 1100–1108. [Google Scholar] [CrossRef]
- Wong, T.-S.; Gao, W.; Chan, J.Y.-W. Transcription Regulation of E-Cadherin by Zinc Finger E-Box Binding Homeobox Proteins in Solid Tumors. BioMed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Uriarte, R.; De Andrés, S.A. Gene selection and classification of microarray data using random forest. BMC Bioinform. 2006, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Alon, U.; Barkai, N.; Notterman, D.A.; Gish, K.; Ybarra, S.; Mack, D.; Levine, A.J. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1999, 96, 6745–6750. [Google Scholar] [CrossRef] [Green Version]
- Dettling, M.; Bühlmann, P. Boosting for tumor classification with gene expression data. Bioinformatics 2003, 19, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, Y.; Wu, Z. Colon cancer data analysis by chameleon algorithm. Health Inf. Sci. Syst. 2019, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Song, X.; Huang, J. Supervised group Lasso with applications to microarray data analysis. BMC Bioinform. 2007, 8, 60. [Google Scholar] [CrossRef] [Green Version]
- Hedenfalk, I.; Duggan, D.; Chen, Y.; Radmacher, M.; Bittner, M.; Simon, R.; Trent, J. Gene-expression profiles in he-reditary breast cancer. N. Eng. J. Med. 2001, 344, 539–548. [Google Scholar] [CrossRef]
- Prat, J.; Ribé, A.; Gallardo, A. Hereditary ovarian cancer. Hum. Pathol. 2005, 36, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Thapa, S.; Lomholt, M.A.; Krog, J.; Cherstvy, A.G.; Metzler, R. Bayesian analysis of single-particle tracking data using the nested-sampling algorithm: Maximum-likelihood model selection applied to stochastic-diffusivity data. Phys. Chem. Chem. Phys. 2018, 20, 29018–29037. [Google Scholar] [CrossRef]
- Muñoz-Gil, G.; Garcia-March, M.A.; Manzo, C.; Martín-Guerrero, J.D.; Lewenstein, M. Single trajectory characterization via machine learning. New J. Phys. 2019, 22, 013010. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Number | Attribute 1 | After Z-Normalization |
|---|---|---|
| 1 | 8589.4163 | 0.508776126 |
| 2 | 9164.2537 | 0.694628976 |
| 3 | 3825.705 | −1.031397365 |
| 4 | 6246.4487 | −0.248737577 |
| 5 | 3230.3287 | −1.223890725 |
| 6 | 2510.325 | −1.45667784 |
| 57 | 4653.2375 | −0.763844707 |
| 58 | 4972.1662 | −0.660730665 |
| 59 | 9112.3725 | 0.67785507 |
| 60 | 6730.625 | −0.092196709 |
| 61 | 6234.6225 | −0.252561151 |
| 62 | 7472.01 | 0.147503275 |
| 357 | 457 | 750 | 722 | 1635 | 982 | 936 | 1897 | 1515 | 316 | 1069 | 1170 | 158 | 737 | 640 | 482 | 109 | 980 | 43 | 1244 | AA | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −0.1992 | 0.5071 | 0.1017 | −0.0684 | −0.6675 | −0.0300 | −0.5490 | −0.6626 | 0.1372 | −0.0531 | −0.3638 | −0.6533 | −0.5021 | −0.3102 | −0.2184 | −0.8842 | −0.8417 | 0.4786 | −0.2234 | −0.2627 | 0 |
| 2 | −0.7666 | 0.4399 | 0.5756 | 0.3717 | 0.9967 | 0.4240 | −0.2509 | 0.4489 | −0.0195 | −0.6426 | 1.8825 | −0.3336 | −0.1146 | 1.2792 | 0.3798 | −0.4430 | 0.1098 | 0.7575 | −0.7258 | 0.0056 | 1 |
| 3 | 3.0568 | −0.1159 | 1.2351 | −0.5044 | −0.8140 | −0.9917 | −0.7655 | −0.8108 | −1.2720 | −1.2896 | 0.7242 | −1.1906 | −0.5315 | −0.8249 | −0.8806 | −1.1246 | −1.1643 | 0.2576 | −1.3183 | −1.0884 | 0 |
| 4 | 1.2695 | −0.0903 | 0.8739 | −0.2384 | −0.5939 | −0.8132 | −0.7175 | −0.5897 | −0.5017 | −0.9306 | −0.3337 | −0.9889 | −0.6994 | 0.1333 | −0.4219 | −0.8763 | −1.1293 | 0.3639 | −1.0305 | −0.7914 | 1 |
| 5 | −0.0648 | −0.3319 | −0.6814 | −0.8450 | −0.6507 | −0.9795 | −0.5607 | −0.4224 | −1.1695 | −0.6220 | −0.3618 | −0.5248 | −0.3469 | −1.0208 | −0.3840 | −0.3813 | −0.3577 | 0.3520 | 0.1902 | −0.6380 | 0 |
| 6 | −0.2609 | −0.4963 | −1.1186 | −1.2516 | −0.2586 | −1.0143 | −0.7658 | 0.0749 | −1.2676 | −0.6466 | −0.7588 | −0.9889 | −0.5115 | −0.7993 | −0.9140 | −0.7159 | −1.1862 | −0.0364 | −0.9349 | −0.6244 | 1 |
| 7 | −0.6730 | 0.9887 | −0.1339 | −0.0290 | −0.7465 | −0.3074 | −0.3726 | −0.6688 | −0.8856 | −0.6814 | −0.8619 | −0.8984 | −0.3543 | −1.0381 | −0.2699 | −0.5531 | −0.5336 | −0.0188 | −0.5576 | −0.7322 | 0 |
| 8 | −0.6308 | −0.1363 | −0.8422 | −0.4778 | −0.3815 | −0.7124 | −0.3125 | −0.6811 | −1.2536 | −0.3774 | −0.3012 | −1.0148 | −0.1874 | −0.3649 | −0.6253 | −0.4677 | −1.0593 | 1.0362 | −1.0438 | −0.3638 | 1 |
| 9 | −1.0475 | 2.2475 | 1.6391 | 0.1700 | −0.8709 | 0.5576 | 0.8973 | −0.5581 | 1.4764 | 0.8136 | −0.5818 | −0.8986 | −0.2457 | −0.3360 | 0.9101 | 0.9838 | −1.0531 | 0.2195 | 1.7120 | −1.6739 | 0 |
| 10 | −0.2550 | 0.3901 | −0.1093 | −0.4833 | 0.5957 | −0.7036 | −0.5306 | 0.5286 | −1.0627 | −0.5627 | −0.3806 | 0.1106 | 0.9938 | −0.5130 | −0.1034 | −0.2700 | 0.5369 | 0.2222 | −0.5102 | 0.4171 | 1 |
| 11 | 0.4643 | 3.1825 | −0.3754 | 2.5536 | 0.3393 | 3.0958 | 0.8291 | −0.2470 | −0.7839 | 1.0607 | −0.1222 | 3.4112 | 3.1890 | −0.8244 | 5.5061 | 2.0606 | 3.0506 | 1.6047 | 1.6041 | −0.0807 | 0 |
| 12 | 1.0635 | 0.8152 | 0.8134 | 0.7939 | 0.4041 | 0.7287 | −0.3338 | 0.6967 | 0.2758 | 0.0028 | 1.3968 | −0.2761 | 0.2502 | 1.4674 | 0.5655 | −0.3176 | 0.1029 | 0.0466 | 0.0120 | −0.3680 | 1 |
| 13 | −0.3604 | −0.3781 | −0.5081 | −0.0171 | −0.2191 | −0.6880 | 0.0041 | −0.3768 | −0.9954 | −0.2205 | −0.5420 | −0.2176 | −0.5044 | −0.7372 | −0.5431 | −0.4008 | 0.2132 | 0.3524 | −0.0848 | −0.2478 | 0 |
| 14 | 0.6732 | −0.6340 | −0.5889 | −0.7660 | −0.0504 | −0.9001 | −0.5282 | −0.1615 | −0.7677 | −0.4724 | −0.5403 | −0.8730 | −0.8915 | −0.5668 | −0.7117 | −0.7386 | −0.4462 | 0.0252 | −0.4674 | −0.5418 | 1 |
| 15 | 0.1503 | −0.4892 | −0.0644 | −0.6390 | −0.4183 | −0.6987 | −0.1476 | −0.4740 | −0.3908 | 0.3067 | −0.1940 | −0.4529 | −0.2461 | −0.2099 | −0.6133 | −0.1809 | −0.3371 | 2.8237 | 0.4598 | 0.3234 | 0 |
| 16 | 0.1676 | −0.7636 | −0.6108 | −1.2205 | −0.5852 | −1.1287 | −0.2759 | −0.8298 | −0.8891 | 0.1060 | −0.8727 | −0.8725 | −0.8945 | −1.0600 | −0.9146 | −0.3240 | −0.0326 | 0.4168 | 0.0068 | −1.1281 | 1 |
| 17 | −0.5718 | −0.8460 | −1.2704 | −0.3834 | −0.8923 | −0.3836 | −0.5605 | −0.6439 | −1.2231 | −0.7155 | −0.8652 | −0.9879 | −1.1323 | −0.8553 | −0.4576 | −0.6124 | −0.9954 | −1.0213 | 0.5998 | −0.9727 | 0 |
| 18 | −0.9664 | −0.6742 | −1.2115 | −0.7833 | −0.0885 | −0.4719 | −0.8443 | −0.5524 | −1.2840 | −1.1437 | −0.9806 | −1.0695 | −1.2193 | −0.9612 | −0.4430 | −0.8493 | −1.1887 | −0.3629 | −1.0256 | −0.6510 | 1 |
| 19 | −1.0436 | −0.2089 | −0.6123 | −0.2747 | −0.9536 | −0.6383 | −0.2481 | −0.6895 | −0.7374 | −0.1340 | −0.7427 | −0.7266 | −0.9080 | −0.8854 | −0.0453 | −0.1587 | −1.2486 | −0.3400 | 0.2375 | −0.9134 | 0 |
| 20 | −0.2527 | 0.1530 | −1.2221 | −0.8715 | 0.4357 | −0.9488 | −0.5732 | 0.0302 | −1.3058 | −0.5522 | −0.7981 | −0.6371 | −0.1376 | −0.9913 | −0.6748 | −0.6001 | −0.9203 | −0.0483 | −0.3883 | −0.1806 | 1 |
| 21 | −0.9218 | 1.0238 | 0.1857 | 0.1325 | −0.5060 | 0.6150 | −0.0497 | −0.6540 | 0.1856 | −1.4646 | −0.7467 | 0.1420 | 0.1219 | −0.2450 | −0.0476 | −0.2655 | −0.1910 | 0.7110 | −0.2819 | 0.5922 | 0 |
| 22 | 1.1915 | 0.5421 | 0.6284 | 0.2280 | 1.4242 | 0.2572 | −0.6487 | 0.1901 | 0.1229 | −0.4806 | 0.2446 | 0.2541 | 0.4371 | 0.5256 | 0.0108 | −0.8016 | 0.4805 | 1.4955 | −0.7911 | 1.9297 | 1 |
| 23 | −0.9997 | −1.2956 | −1.3319 | −0.9966 | −0.7991 | −1.0448 | −0.1708 | −0.6968 | −1.1930 | 0.0079 | −0.9208 | −0.8099 | −0.7786 | −1.0369 | −0.5300 | 0.1939 | −0.1510 | 0.2602 | 0.2545 | −0.6840 | 0 |
| 24 | −1.1251 | −1.3200 | −1.5176 | −1.5869 | −0.5999 | −1.2172 | −1.0349 | −0.8018 | −1.3932 | −1.3848 | −1.0472 | −1.1924 | −1.2572 | −1.0503 | −0.9622 | −1.0551 | −1.2676 | −1.0237 | −1.4197 | −1.2531 | 1 |
| 25 | 2.1527 | −0.3799 | 0.4128 | 0.0581 | −0.6111 | −0.2685 | −0.0154 | −0.5231 | 0.5592 | 0.7240 | −0.7720 | −0.2615 | 0.0890 | −0.5596 | −0.3297 | 0.5333 | 0.3943 | −0.8399 | 1.5212 | −0.0460 | 0 |
| 26 | −0.8154 | −0.3332 | −0.5275 | −0.2076 | −0.7800 | −0.6859 | −0.7967 | −0.6935 | 0.0172 | −0.8118 | −0.7327 | 0.0515 | −0.1746 | −0.8941 | −0.5018 | −0.6701 | 0.5713 | −1.2620 | −0.8807 | −0.9011 | 0 |
| 27 | −0.7735 | −0.6015 | −0.6106 | −0.5381 | −0.4543 | −0.2415 | −0.7473 | −0.5819 | 0.0240 | −0.7150 | −0.6884 | −0.0911 | −0.1966 | −0.7717 | −0.5844 | −0.6931 | 0.1300 | −1.3225 | −0.7485 | −0.8737 | 0 |
| 28 | −0.9109 | 0.5173 | 1.0975 | 0.5547 | −0.6970 | 0.2649 | 0.2318 | −0.5522 | 2.1660 | 1.0763 | −0.0386 | 0.3183 | 0.7718 | 0.4314 | 0.4805 | 0.5893 | 1.3612 | −0.5206 | 0.9256 | −0.2098 | 0 |
| 29 | −0.5721 | 2.7545 | 3.1274 | 1.9689 | −0.0094 | 1.1863 | −0.0478 | −0.1891 | 2.9906 | 0.4199 | 0.3571 | 2.0584 | 3.0102 | −0.1471 | 1.0052 | −0.1387 | 2.7865 | −0.6162 | −0.1087 | 0.4574 | 0 |
| 30 | −0.9161 | 1.6600 | 1.1954 | 2.3194 | −0.4940 | 1.7943 | 0.4048 | −0.3110 | 1.4713 | 0.4678 | −0.3249 | 1.0437 | 1.8333 | 0.1317 | 0.9502 | 0.3860 | 2.1646 | −0.3548 | 0.6085 | −0.0898 | 0 |
| 31 | −1.1165 | 0.8826 | 1.8629 | 0.2982 | −0.3599 | 0.2675 | 0.3571 | 0.3901 | 2.5001 | 0.9578 | 4.1938 | 0.2754 | 1.2105 | 1.0568 | −0.0928 | 0.2485 | 0.4813 | −0.8199 | 0.3256 | 0.5439 | 0 |
| 32 | −0.9831 | −0.1959 | −1.3571 | −0.4143 | 0.4154 | 0.5795 | −0.5073 | 0.1110 | −0.9439 | −0.7458 | −0.4805 | 0.1408 | −0.2808 | −1.0085 | −0.2011 | −0.2811 | −0.3807 | −1.1552 | −0.5167 | −0.8709 | 0 |
| 33 | −0.4961 | −0.0128 | −1.1792 | −0.2391 | −0.5376 | −0.3355 | −0.3795 | −0.6084 | −0.9436 | −0.1182 | −1.0148 | −0.3036 | −0.0893 | −1.0762 | −0.4375 | 0.3608 | −0.3553 | −1.2858 | 0.4535 | −0.4651 | 0 |
| 34 | 0.9835 | 0.1354 | 0.8543 | 0.1376 | −0.4418 | 0.0355 | 0.7054 | −0.2009 | 0.8813 | 1.0063 | 0.3341 | −0.1728 | 0.4361 | 0.3466 | −0.1904 | 0.1423 | 0.3706 | −0.4896 | 0.3484 | −0.3974 | 0 |
| 35 | −0.4833 | −0.3394 | −0.6218 | −0.1287 | −0.6790 | 0.1834 | −0.3002 | −0.5671 | −0.1395 | −0.4042 | −0.2245 | −0.3249 | −0.6001 | −0.8524 | −0.2944 | −0.3355 | −1.0430 | −1.0594 | −0.8860 | −0.8007 | 0 |
| 36 | −0.7281 | −0.8700 | −1.0871 | −0.8550 | −0.8165 | 0.0969 | 0.5636 | −0.5480 | −0.1181 | 0.3411 | −0.6805 | −0.2281 | −0.8138 | −0.8964 | −0.6067 | 0.6057 | −0.4189 | −0.8495 | 0.5910 | −1.1440 | 0 |
| 37 | −0.3399 | 3.0909 | 2.7314 | 0.6380 | −0.2966 | −0.2066 | −0.4223 | −0.7412 | 0.7620 | −0.6222 | −0.1975 | 0.1739 | 3.1760 | −0.7350 | 0.3958 | −0.5145 | 0.1316 | −0.5937 | −0.5462 | −0.0562 | 0 |
| 38 | −1.1911 | −0.9283 | −1.0429 | −1.0896 | −0.8135 | −0.6132 | 0.2649 | −0.7337 | −0.1926 | 0.4481 | −0.8865 | 0.0863 | −0.7292 | −1.0073 | −0.6858 | 0.5026 | 0.2004 | −1.0883 | 1.3413 | −1.1143 | 0 |
| 39 | −0.3071 | −1.2474 | 0.0860 | −1.0948 | 1.0319 | −0.8550 | −0.5113 | 0.4325 | −0.1901 | −0.4962 | 0.3330 | −0.4471 | −0.5249 | 1.0173 | −0.7199 | −0.5768 | −0.2850 | −0.7477 | −0.7928 | 0.4443 | 1 |
| 40 | 0.2579 | −0.6935 | −0.3056 | 0.8062 | −0.5388 | 0.4405 | −0.2563 | −0.1171 | 0.4400 | 0.0777 | −0.3132 | 0.0615 | −0.4472 | −0.1293 | 0.3970 | −0.2903 | 0.0332 | −0.5678 | 0.7060 | −0.1725 | 0 |
| 41 | −0.6221 | −1.2147 | −0.2310 | −0.9927 | −0.8238 | −0.7229 | −0.5687 | −0.6551 | 0.2961 | −0.7430 | −0.0772 | −0.9459 | −1.1104 | −0.4439 | −0.3033 | −0.6929 | −1.0863 | −1.1514 | −0.7252 | −0.9379 | 0 |
| 42 | 0.3246 | 0.1301 | −0.0365 | 0.7536 | 1.1815 | −0.1630 | 0.2493 | 0.8735 | −0.3192 | 0.2676 | −0.1930 | −0.4060 | −0.2470 | 0.7416 | 0.3565 | −0.0454 | −0.7271 | −0.2528 | −0.6329 | 1.2129 | 1 |
| 43 | 1.9695 | 0.9077 | 0.8436 | 2.2059 | 2.5041 | 1.1565 | 1.2523 | 5.0353 | 0.3198 | 0.6450 | 2.0593 | 1.5501 | 0.7583 | 2.0433 | 0.0396 | 1.7493 | 0.7779 | 0.7293 | 0.2850 | 2.0997 | 1 |
| 44 | −0.6286 | 1.0261 | 0.3036 | 2.5481 | −0.0714 | 3.8247 | 2.3460 | 0.2476 | 0.7710 | 1.8683 | 0.1706 | 2.3022 | 0.3095 | −0.1948 | 2.7512 | 2.0757 | 1.7530 | 0.9490 | 1.9587 | 0.6909 | 0 |
| 45 | 1.0009 | 0.7865 | 2.4011 | 1.5617 | 3.0845 | 0.8111 | 0.5197 | 1.2789 | 1.8771 | 1.1289 | 0.6236 | 2.5353 | 1.8333 | 3.4374 | 1.2130 | 0.4487 | 2.2789 | −0.2895 | 0.0593 | 3.5573 | 0 |
| 46 | 2.1343 | 0.7579 | 0.0162 | 2.6774 | 0.1163 | 2.1145 | 3.2065 | 0.5759 | 1.0534 | 3.2313 | −0.4136 | 1.3026 | 0.8483 | 0.3653 | 1.9049 | 3.1342 | 0.7206 | −0.2964 | 2.7839 | 1.5164 | 0 |
| 47 | 0.7965 | 0.3297 | 0.7166 | 0.4364 | 0.0677 | 0.8326 | 4.9333 | −0.0258 | 1.9736 | 2.0418 | −0.3203 | 2.9719 | 1.5455 | 0.5613 | 1.3445 | 4.5603 | 1.3877 | 2.1283 | 2.2865 | 2.0161 | 0 |
| 48 | −0.2596 | −0.3361 | 0.4716 | −0.1349 | 4.0534 | −0.1928 | 0.0167 | 1.3918 | 0.4278 | −0.2243 | 1.5200 | 0.8905 | 1.2019 | 1.4025 | 0.0835 | 0.0383 | 0.5237 | −0.1815 | −1.0054 | 1.6031 | 1 |
| 49 | −0.0226 | −0.4900 | −0.4330 | −0.3172 | 1.4328 | 0.0199 | −0.6444 | 0.1271 | −0.5104 | −0.9655 | −0.0533 | 0.3951 | −0.3541 | 0.9183 | 0.1715 | −0.7495 | −0.5247 | −0.9193 | −1.0624 | 0.2604 | 0 |
| 50 | 0.2691 | −0.7205 | −0.2008 | 0.2954 | 1.0927 | −1.0150 | −0.3882 | 3.2096 | −0.1876 | 0.6101 | 0.8424 | 0.0779 | −0.4090 | 1.3963 | −0.1282 | −0.0492 | 1.0513 | 1.5847 | −0.0730 | 0.8097 | 1 |
| 51 | −1.0684 | −0.6466 | −0.6541 | −0.6908 | −0.1836 | 0.3447 | −0.5880 | −0.5316 | 0.4261 | −0.2238 | −0.7169 | −0.1310 | −0.0254 | −0.4201 | −0.5104 | −0.3398 | −0.0721 | 1.8853 | −0.1081 | 1.6590 | 1 |
| 52 | 0.3214 | −0.0499 | 0.1444 | 1.4566 | −0.3449 | 2.2177 | 2.1168 | 0.0888 | 1.0617 | 3.9229 | −0.6109 | 0.4205 | −0.0079 | 0.4511 | 0.6379 | 1.6055 | 1.0541 | 3.2448 | 3.2324 | 0.6117 | 0 |
| 53 | −0.6844 | −0.8219 | −0.0865 | −0.7453 | −0.5648 | −0.0203 | −0.0723 | −0.4516 | 0.3664 | 0.3297 | 0.2201 | −0.4875 | −0.5796 | −0.2607 | −0.4161 | −0.4218 | −0.4390 | −0.3909 | 0.5963 | −0.0209 | 0 |
| 54 | −0.2355 | −0.6462 | 0.6279 | −0.3769 | 0.6364 | −0.7139 | 0.0674 | 1.7679 | 0.6581 | 0.3100 | 1.5562 | −0.2193 | 0.1656 | 2.0470 | −0.4447 | 0.0956 | −0.7435 | 0.4457 | −0.1701 | 1.7074 | 1 |
| 55 | 2.1278 | −0.4153 | −0.3230 | −0.1529 | −0.5469 | −0.2273 | −0.4385 | −0.1479 | −0.3919 | −0.0556 | 0.1997 | 0.0787 | −0.3176 | −0.1840 | −0.2261 | −0.4615 | −0.2847 | −0.8512 | −0.6685 | −0.2232 | 1 |
| 56 | 0.2032 | −0.4662 | −0.6407 | −0.5504 | 0.8009 | −0.6042 | −0.0797 | 1.3601 | −0.4682 | −0.4208 | 0.5604 | −0.3195 | −0.4353 | 0.4957 | −0.7274 | −0.1458 | −0.4557 | −0.6586 | −0.5978 | −0.1099 | 0 |
| 57 | 2.0444 | −0.8584 | 0.8658 | −0.9170 | −0.7307 | −0.6747 | −0.7436 | −0.2323 | −0.0305 | −0.4500 | 3.1704 | −0.4119 | −0.6079 | 1.3337 | −0.8944 | −0.6458 | −0.6273 | 0.9960 | −0.7052 | −0.3002 | 0 |
| 58 | −0.6652 | −0.9762 | −0.4155 | −0.7555 | −0.9611 | −0.8140 | −0.7491 | −0.8122 | 0.0495 | −1.1010 | 0.0414 | −0.8919 | −1.0581 | −0.5954 | −0.5056 | −0.7965 | −0.8875 | −1.2378 | −0.6453 | −1.1103 | 0 |
| 59 | −0.3915 | −0.6791 | −0.6367 | −0.6560 | −0.3747 | 0.3404 | 0.4901 | −0.1944 | −0.5081 | 0.3228 | −0.3809 | 0.6629 | −0.5926 | 0.1046 | −0.5600 | 0.3525 | 1.1441 | −0.1059 | 0.7094 | 0.4779 | 0 |
| 60 | −0.0889 | −0.5860 | −0.1552 | −0.2093 | 1.5965 | 0.1178 | −0.6404 | 0.4750 | 0.2935 | −1.1046 | 0.8902 | 0.6335 | −0.0928 | 1.8118 | 0.3945 | −0.3636 | −0.1894 | −0.2347 | −1.0748 | 0.5274 | 1 |
| 61 | 0.2936 | −0.5280 | −0.8700 | −0.2632 | −0.6020 | 0.0362 | −0.0931 | −0.6343 | 0.1302 | −0.6866 | −0.5733 | −0.4376 | −0.7027 | −0.3033 | −0.1950 | −0.3600 | −0.6969 | −0.7171 | −0.5882 | −0.5232 | 0 |
| 62 | 1.5230 | −0.4513 | −0.1981 | −0.1999 | 0.6194 | −0.3169 | −0.2257 | 1.2388 | −0.3746 | −0.2321 | 0.6893 | −0.2619 | −0.3217 | 1.6116 | −0.1817 | −0.0748 | −0.4894 | 0.3250 | −0.4697 | 0.0995 | 1 |
| Method | Number of Genes | Accuracy (%) |
|---|---|---|
| LogitBoot | 2000 | 85.48 |
| Random Forest | 2000 | 84.10 |
| SVM | 2000 | 82.26 |
| Two-way clustering | 2000 | 87.10 |
| Chameleon algorithm | 5 | 85.48 |
| Supervised group Lasso | 22 | 85.48 |
| Z-FS-KM-MHS (our method) | 8 | 93.46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, J.H.; Kim, M.; Lim, J.S.; Geem, Z.W. Feature Selection for Colon Cancer Detection Using K-Means Clustering and Modified Harmony Search Algorithm. Mathematics 2021, 9, 570. https://doi.org/10.3390/math9050570
Bae JH, Kim M, Lim JS, Geem ZW. Feature Selection for Colon Cancer Detection Using K-Means Clustering and Modified Harmony Search Algorithm. Mathematics. 2021; 9(5):570. https://doi.org/10.3390/math9050570
Chicago/Turabian StyleBae, Jin Hee, Minwoo Kim, J.S. Lim, and Zong Woo Geem. 2021. "Feature Selection for Colon Cancer Detection Using K-Means Clustering and Modified Harmony Search Algorithm" Mathematics 9, no. 5: 570. https://doi.org/10.3390/math9050570
APA StyleBae, J. H., Kim, M., Lim, J. S., & Geem, Z. W. (2021). Feature Selection for Colon Cancer Detection Using K-Means Clustering and Modified Harmony Search Algorithm. Mathematics, 9(5), 570. https://doi.org/10.3390/math9050570