
A Mathematical Analysis of RNA Structural Motifs in Viruses


Abstract
:1. Introduction
2. Materials and Methods
2.1. The Definitions of the Laplacian Matrix of a Tree Graph and the Algebraic Connectivity
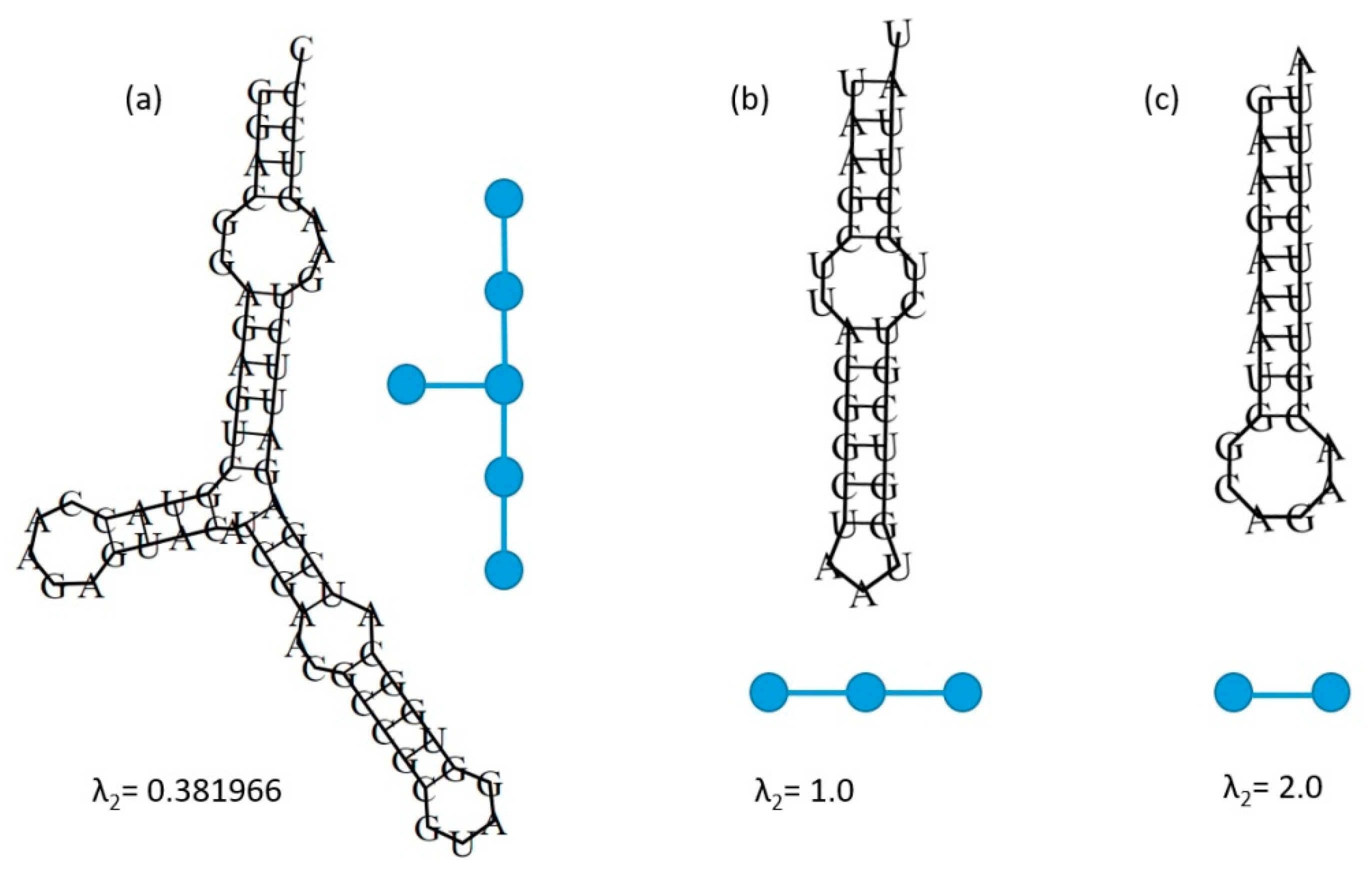
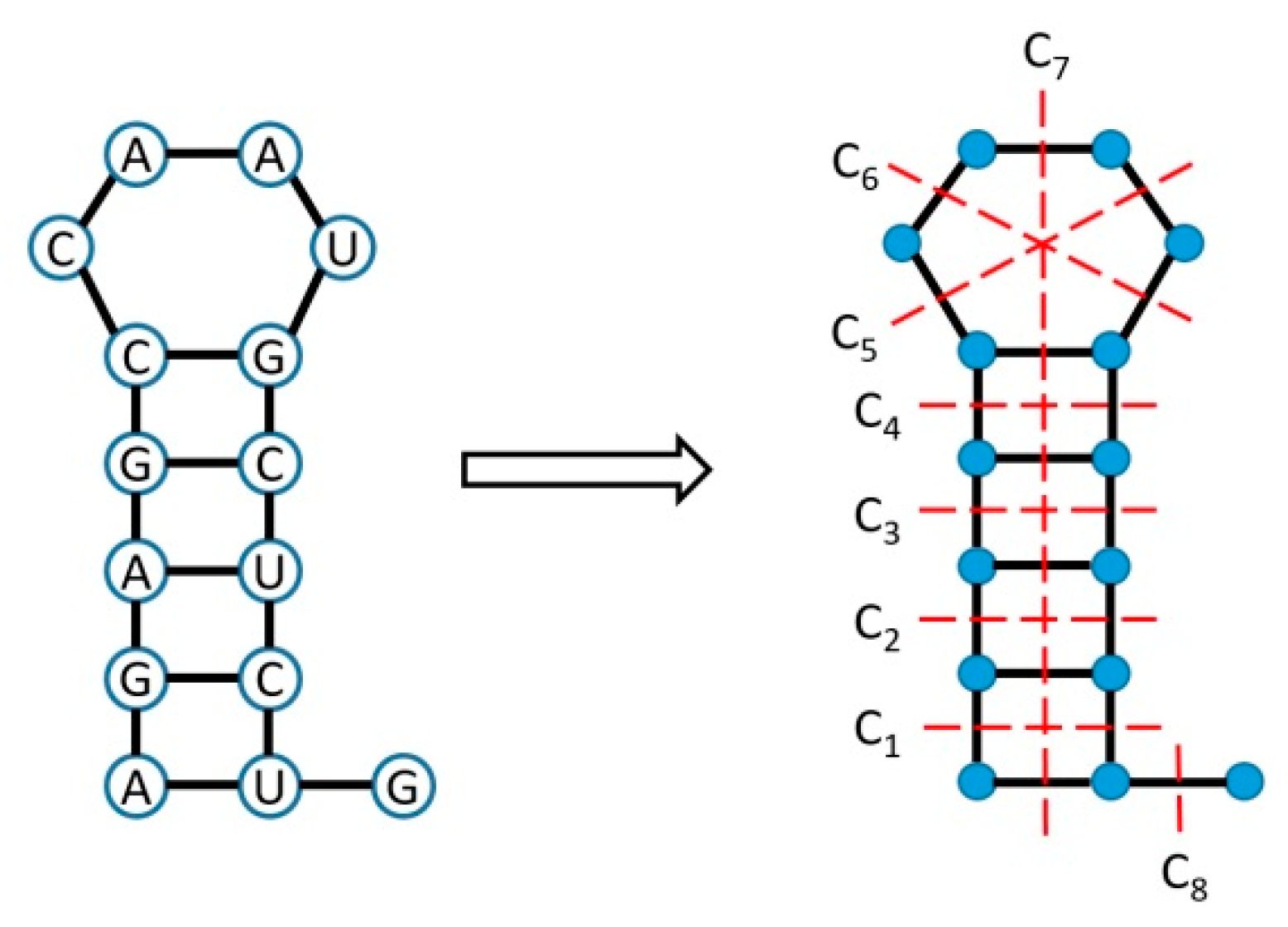
2.2. An Illustrative Example for the Algebraic Connectivity of an RNA Structure Coarse Grain Tree Graph Represented by a Laplacian Matrix
2.3. The Definitions of the Wiener and Szeged Topological Indices
2.4. Calculation of the Szeged Index of RNA Secondary Structure Graphs from Elementary Cuts
2.5. Mutational Robustness and Thermodynamic Stability
3. Results
3.1. Eigenvalue Statistics: Algebraic Connectivity Distribution
3.2. Wiener/Szeged Topological Index Distribtuion
3.3. Mutational Robustness and Thermodynamic Stability
4. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Details of the Algebraic Connectivity
- (A).
- (B).
- (C).
- (D).
References
- Hofacker, I.L.; Stadler, P.F.; Stocsits, R.R. Conserved RNA secondary structure in viral genomes: A survey. Bioinformatics 2004, 20, 1495–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marz, M.; Beerenwinkel, N.; Droste, C.; Fricke, M.; Frishman, D.; Hofacker, I.L.; Hoffmann, D.; Middendorf, M.; Rattei, T.; Stadler, P.F.; et al. Challenges in RNA virus bioinformatics. Bioinformatics 2014, 30, 1793–1799. [Google Scholar] [CrossRef] [Green Version]
- You, S.; Stump, D.D.; Branch, A.D.; Rice, C.M. A cis-acting replication element in the sequence encoding the NS5B RNA-dependent polymerase is required for hepatitis C virus RNA replication. J. Virol. 2004, 78, 1352–1356. [Google Scholar] [CrossRef] [Green Version]
- Tuplin, A.; Evans, D.J.; Simmonds, P. Detailed mapping of RNA secondary structures in core and NS5B-encoding region sequences of hepatitis C virus by RNase cleavage and novel bioinformatic prediction methods. J. Gen. Virol. 2004, 85, 3037–3047. [Google Scholar] [CrossRef]
- Vassilaki, N.; Friebe, P.; Meuleman, P.; Kallis, S.; Kaul, A.; Paranhos-Baccalà, G.; Leroux-Roels, G.; Mavromara, P.; Bartenschlager, R. Role of the hepatitis C virus core +1 open reading frame and core cis-acting RNA elements in viral RNA translation and replication. J. Virol. 2008, 82, 11503–11515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevilacqua, P.C.; Ritchey, L.E.; Su, Z.; Assmann, S.M. Genome-wide analysis of RNA secondary structures. Annu. Rev. Genet. 2016, 50, 235–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshman, D.K.; Tavantzis, S.M. Primary and secondary structure of a 360-nucleotide isolate of potato spindle tuber viroid. Arch. Virol. 1993, 128, 319–331. [Google Scholar] [CrossRef]
- Ochsenreiter, R.; Hofacker, I.L.; Wolfinger, M.T. Functional RNA structures in the 3′UTR of tick-borne, insect-specific and no-known-vector Flaviviruses. Viruses 2019, 11, 298. [Google Scholar] [CrossRef] [Green Version]
- Cuceanu, N.M.; Tuplin, A.; Simmonds, P. Evolutionary conserved RNA secondary structures in coding and non-coding sequences at the 3’ end of the hepatitis G virus/GB-virus C genome. J. Gen. Virol. 2001, 82, 713–722. [Google Scholar] [CrossRef] [Green Version]
- Waterman, M.S. Secondary structure of single stranded nucleic acids. Adv. Math. Suppl. Stud. 1978, 1, 167–212. [Google Scholar]
- Shapiro, B.A. An algorithm for comparing multiple RNA secondary structures. Comput. Appl. Biosci. 1988, 4, 387–393. [Google Scholar] [CrossRef]
- Fontana, W.; Konings, D.A.M.; Stadler, P.F.; Schuster, P. Statistics of RNA secondary structures. Biopolymers 1993, 33, 1389–1404. [Google Scholar] [CrossRef]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, S.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Mon. Chem. Chem. Mon. 1994, 124, 167–188. [Google Scholar] [CrossRef]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, R.; Bernhart, S.H.F.; Zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Computer prediction of RNA secondary structure. Methods Enzymol. 1989, 180, 262–288. [Google Scholar]
- Zuker, M. Mfold webserver for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Markham, N.R.; Zuker, M. UNAFold: Software for nucleic acid folding and hybridization. Methods Mol. Biol. 2008, 453, 3–31. [Google Scholar]
- Le, S.Y.; Nussinov, R.; Maizel, J.V. Tree graphs of RNA secondary structures and their comparison. Comput. Appl. Biosci. 1989, 22, 461–473. [Google Scholar] [CrossRef]
- Benedetti, G.; Morosetti, S. A graph-topological approach to recognition of pattern and similarity in RNA secondary structures. Biophys. Chem. 1996, 59, 179–184. [Google Scholar] [CrossRef]
- Barash, D. Deleterious mutation prediction in the secondary structure of RNAs. Nucleic Acids Res. 2003, 31, 6578–6584. [Google Scholar] [CrossRef] [Green Version]
- Barash, D. Second eigenvalue of the Laplacian matrix for predicting RNA conformational switch by mutation. Bioinformatics 2004, 20, 1861–1869. [Google Scholar] [CrossRef]
- Churkin, A.; Barash, D. RNAmute: RNA secondary structure mutation analysis tool. BMC Bioinform. 2006, 7, 221. [Google Scholar] [CrossRef] [Green Version]
- Giegerich, R.; Voss, B.; Rehmsmeier, M. Abstract shapes of RNA. Nucleic Acids Res. 2004, 32, 4843–4851. [Google Scholar] [CrossRef]
- Churkin, A.; Barash, D. An efficient method for the prediction of deleterious multiple-point mutations in the secondary structure of RNAs using suboptimal folding solutions. BMC Bioinform. 2008, 9, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barash, D.; Churkin, A. Mutational analysis in RNAs: Comparing programs for RNA deleterious mutation prediction. Brief. Bioinform. 2011, 12, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Shu, W.; Bo, X.; Liu, R.; Zhao, D.; Zheng, Z.; Wang, S. RDMAS: A webserver for RNA deleterious mutation analysis. BMC Bioinform. 2006, 7, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, W.; Bo, X.; Zheng, Z.; Wang, S. A novel representation of RNA secondary structure based on element-contact graphs. BMC Bioinform. 2008, 8, 188. [Google Scholar] [CrossRef] [Green Version]
- Fiedler, M. Algebraic connectivity of graphs. Czechoslov. Math. J. 1973, 23, 298–305. [Google Scholar] [CrossRef]
- Merris, R. Characteristic vertices of trees. Linear Multilinear Algebra 1987, 22, 115–131. [Google Scholar] [CrossRef]
- Wiener, H. Structural determination of paraffin boiling points. J. Am. Chem. Soc. 1947, 69, 17–20. [Google Scholar] [CrossRef]
- Merris, R. An edge-version of the matrix-tree theorem and the Wiener index. Linear Multilinear Algebra 1989, 25, 291–296. [Google Scholar] [CrossRef]
- Merris, R. Laplacian matrices of graphs: A survey. Linear Algebra Appl. 1994, 197, 143–176. [Google Scholar] [CrossRef] [Green Version]
- Churkin, A.; Gabdank, I.; Barash, D. On topological indices for small RNA graphs. Comput. Biol. Chem. 2012, 41, 35–40. [Google Scholar] [CrossRef]
- Hosoya, H. Topological index. A newly proposed quantity characterizing the topological nature of structural isomers of saturated hydrocarbons. Bull. Chem. Soc. Jpn. 1971, 44, 2332–2339. [Google Scholar] [CrossRef] [Green Version]
- Gutman, I. A formula for the Wiener number of trees and its extension to graphs containing cycles. Graph Theory Notes N. Y. 1994, 27, 9–15. [Google Scholar]
- Kiening, M.; Ochsenreiter, R.; Hellinger, H.J.; Rattei, T.; Hofacker, I.L.; Frishman, D. Conserved secondary structures in viral mRNAs. Viruses 2019, 11, 401. [Google Scholar] [CrossRef] [Green Version]
- Gutman, I.; Klavžar, S. An algorithm for the calculation of the Szeged index of benzenoid hydrocarbons. J. Chem. Inf. Comput. Sci. 1995, 35, 1011–1114. [Google Scholar] [CrossRef]
- Bašić, N.; Berkemer, S.J.; Fallmann, J.; Fowler, P.W.; Gatter, T.; Pisanski, T.; Retzlaff, N.; Stadler, P.F.; Zemljič, S.S. Convexity deficit of benzenoids. Croat. Chem. Acta 2020, 92, 457–466. [Google Scholar] [CrossRef]
- Strang, G.; Macnamara, S. Functions of difference matrices are Toeplitz plus Hankel. SIAM Rev. 2014, 56, 525–546. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eigenvalue | 1H Structures: Total = 594,937 | 2H Structures:Total = 82,564 |
|---|---|---|
| 2.0 | 361,043 | None |
| 1.0 | 137,616 | 41,858 |
| <1.0 | 96,278 | 40,706 |
| Eigenvalue | 1H Structures: Total = 594,937 |
|---|---|
| 2.0 | 361,043 |
| 1.0 | 137,616 |
| 0.585786 | 67,504 |
| 0.381966 | 24,744 |
| 0.267949 | 3843 |
| 0.198062 | 186 |
| 0.152241 | 1 |
| C | r | nR′(0) | nR″(0) | r × nR′(0) × nR″(0) |
|---|---|---|---|---|
| 2 | 3 | 12 | 72 | |
| 2 | 5 | 10 | 100 | |
| 2 | 7 | 8 | 112 | |
| 2 | 9 | 6 | 108 | |
| 2 | 12 | 3 | 72 | |
| 2 | 12 | 3 | 72 | |
| 6 | 7 | 8 | 336 | |
| 1 | 14 | 1 | 14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Churkin, A.; Totzeck, F.; Zakh, R.; Parr, M.; Tuller, T.; Frishman, D.; Barash, D. A Mathematical Analysis of RNA Structural Motifs in Viruses. Mathematics 2021, 9, 585. https://doi.org/10.3390/math9060585
Churkin A, Totzeck F, Zakh R, Parr M, Tuller T, Frishman D, Barash D. A Mathematical Analysis of RNA Structural Motifs in Viruses. Mathematics. 2021; 9(6):585. https://doi.org/10.3390/math9060585
Chicago/Turabian StyleChurkin, Alexander, Franziska Totzeck, Rami Zakh, Marina Parr, Tamir Tuller, Dmitrij Frishman, and Danny Barash. 2021. "A Mathematical Analysis of RNA Structural Motifs in Viruses" Mathematics 9, no. 6: 585. https://doi.org/10.3390/math9060585