
The Role of Methyl Donors of the Methionine Cycle in Gastrointestinal Infection and Inflammation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
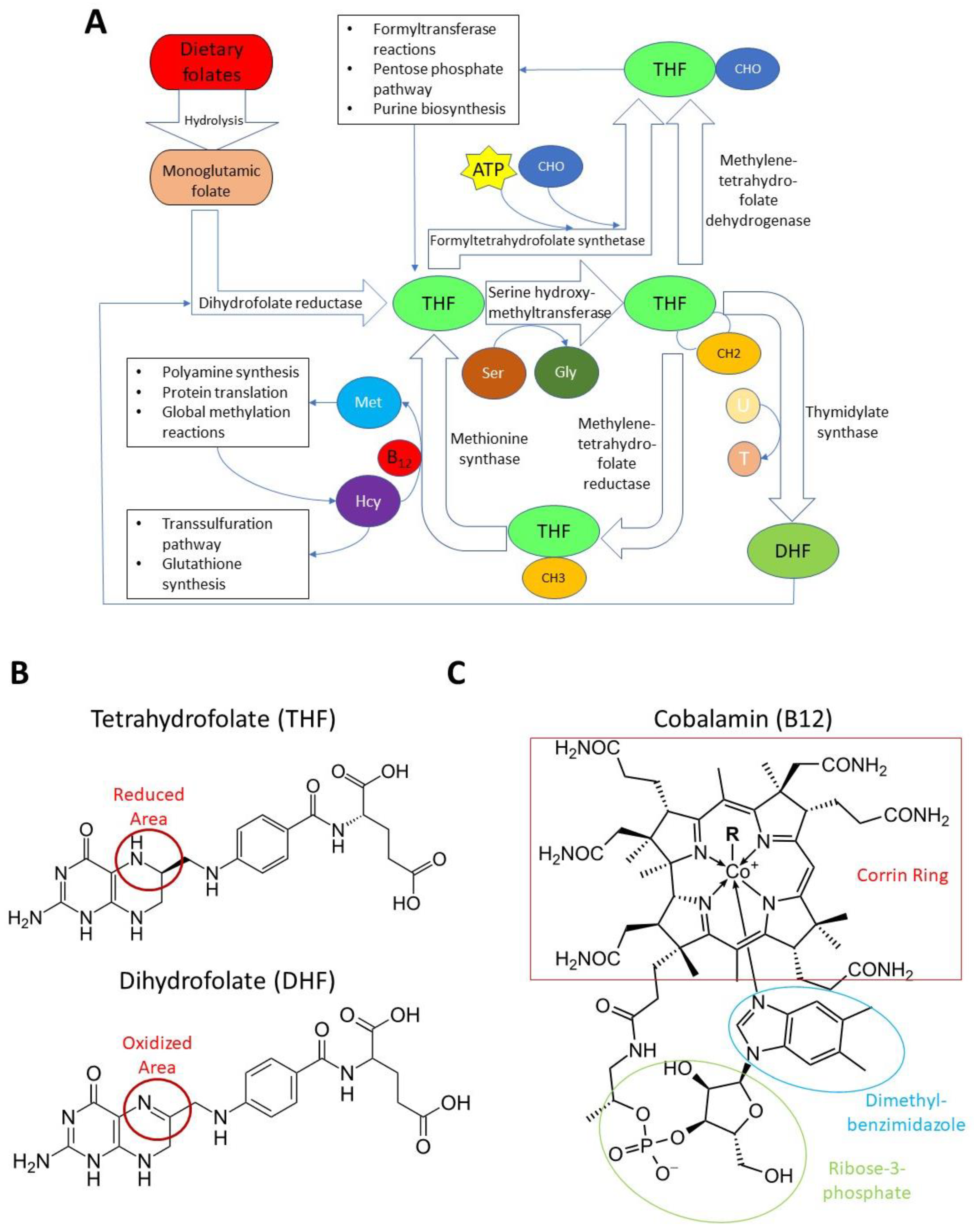
2. Folate, B12, and the Methionine Cycle
2.1. Folate and B12
2.2. S-Adenosyl Methionine
3. Folate, B12, and SAM: Links to Tissue-Specific Inflammation
3.1. The Gastrointestinal Tract
3.2. Systemic Inflammation
3.3. Immune Cells
3.4. The Nervous System
3.5. The Liver
3.6. Other Tissues
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BHMT | Betaine homocysteine methyltransferase |
| Ccl3 | Chemokine (C-C motif) ligand 3 |
| CD | Crohn’s disease |
| CD4+ | Cluster of differentiation 4-positive |
| CD8+ | Cluster of differentiation 8-positive |
| DHF | Dihydrofolate |
| DHFR | Dihydrofolate reductase |
| ER | Endoplasmic reticulum |
| ERK1/2 | Extracellular signal-regulated protein kinases 1 and 2 |
| FTHFS | Formyltetrahydrofolate synthase |
| Gly | Glycine |
| Hcy | Homocysteine |
| IBD | Inflammatory bowel disease |
| IF | Intrinsic factor |
| IgA | Immunoglobulin A |
| IL-6 | Interleukin 6 |
| IRES | Internal ribosome entry site |
| JNK | C-Jun N-terminal kinase |
| LPS | Lipopolysaccharide |
| Lyz1/2 | Lysozyme 1 and 2 |
| MAT | Methionine adenosyltransferase |
| Met | Methionine |
| MS | Methionine synthase |
| MTases | Methyltransferases |
| MTHFD | Methylenetetrahydrofolate dehydrogenase |
| MTHFR | Methylenetetrahydrofolate reductase |
| NK cell | Natural killer cells |
| NLRP3 | NLR family pyrin domain containing 3 |
| NOS | Nitric oxide synthase |
| SAM | S-adenosyl-L-methionine |
| SAH | S-adenosyl homocysteine |
| Ser | Serine |
| SHMT | Serine hydroxymethyltransferase |
| STAT1/3 | Signal transducer and activator of transcription 1 and 3 |
| THF | Tetrahydrofolate |
| TNF | Tumor necrosis factor |
| TNXIP | Thioredoxin interacting protein |
| TS | Thymidylate synthase |
| UC | Ulcerative colitis |
References
- Mitchell, H.K.; Snell, E.E.; Williams, R.J. The concentration of “folic acid”. J. Am. Chem. Soc. 1941, 63, 2284. [Google Scholar] [CrossRef]
- Davidson, L.; Girdwood, R.; Innes, E. Folic Acid in the Treatment of the Sprue Syndrome. Lancet 1947, 249, 511–515. [Google Scholar] [CrossRef]
- Mosley, B.S.; Cleves, M.A.; Siega-Riz, A.M.; Shaw, G.M.; Canfield, M.A.; Waller, D.K.; Werler, M.M.; Hobbs, C.A. National Birth Defects Prevention Study Neural Tube Defects and Maternal Folate Intake Among Pregnancies Conceived After Folic Acid Fortification in the United States. Am. J. Epidemiol. 2008, 169, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konings, E.J.; Roomans, H.H.; Dorant, E.; Goldbohm, R.A.; Saris, W.H.; Brandt, P.V.D. Folate intake of the Dutch population according to newly established liquid chromatography data for foods. Am. J. Clin. Nutr. 2001, 73, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engevik, M.; Morra, C.N.; Röth, D.; Engevik, K.; Spinler, J.K.; Devaraj, S.; Crawford, S.E.; Estes, M.K.; Kalkum, M.; Versalovic, J. Microbial Metabolic Capacity for Intestinal Folate Production and Modulation of Host Folate Receptors. Front. Microbiol. 2019, 10, 2305. [Google Scholar] [CrossRef] [PubMed]
- Levit, R.; De Giori, G.S.; Leblanc, A.D.M.D.; Leblanc, J.G. Folate-producing lactic acid bacteria reduce inflammation in mice with induced intestinal mucositis. J. Appl. Microbiol. 2018, 125, 1494–1501. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.W. Partial villous atrophy in nutritional megaloblastic anaemia corrected by folic acid therapy. J. Clin. Pathol. 1971, 24, 131–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurence, K.M.; James, N.; Miller, M.H.; Tennant, G.B.; Campbell, H. Double-blind randomised controlled trial of folate treatment before conception to prevent recurrence of neural-tube defects. BMJ 1981, 282, 1509–1511. [Google Scholar] [CrossRef] [Green Version]
- Pereza, N.; Ostojić, S.; Kapović, M.; Peterlin, B. Systematic review and meta-analysis of genetic association studies in idiopathic recurrent spontaneous abortion. Fertil. Steril. 2017, 107, 150–159.e2. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Liu, M.; Wang, Y.; Dai, J.; Tao, J.; Wang, S.; Zhong, N.; Chen, Y. Association between SNPs in genes involved in folate metabolism and preterm birth risk. Genet. Mol. Res. 2015, 14, 850–859. [Google Scholar] [CrossRef]
- Wu, A.; Chanarin, I.; Slavin, G.; Levi, A.J. Folate Deficiency in the Alcoholic—its Relationship to Clinical and Haematological Abnormalities, Liver Disease and Folate Stores. Br. J. Haematol. 1975, 29, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, S.; Hirayama, C.; Yamamoto, S.; Koda, M.; Udagawa, A.; Kadowaki, Y.; Inoue, M.; Sagayama, A.; Umeki, K. Red blood cell status in alcoholic and non-alcoholic liver disease. J. Lab. Clin. Med. 2001, 138, 332–337. [Google Scholar] [CrossRef]
- Pan, Y.; Liu, Y.; Guo, H.; Jabir, M.S.; Liu, X.; Cui, W.; Li, D. Associations between Folate and Vitamin B12 Levels and Inflammatory Bowel Disease: A Meta-Analysis. Nutrients 2017, 9, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballestero-Fernández, C.; Varela-Moreiras, G.; Úbeda, N.; Alonso-Aperte, E. Nutritional Status in Spanish Adults with Celiac Disease Following a Long-Term Gluten-Free Diet Is Similar to Non-Celiac. Nutrients 2021, 13, 1626. [Google Scholar] [CrossRef]
- O’Brien, W.; England, N.W. Folate Deficiency in Acute Tropical Sprue. Br. Med. J. 1964, 2, 1573–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angier, R.B.; Boothe, J.H.; Hutchings, B.L.; Mowat, J.H.; Semb, J.; Stokstad, E.L.R.; Subbarow, Y.; Waller, C.W.; Cosulich, D.B.; Fahrenbach, M.J.; et al. Synthesis of a Compound Identical with the L. casei Factor Isolated from Liver. Science 1945, 102, 227. [Google Scholar] [CrossRef] [PubMed]
- Angier, R.B.; Boothe, J.H.; Hutchings, B.L.; Mowat, J.H.; Semb, J.; Stokstad, E.L.R.; SubbaRow, Y.; Waller, C.W.; Cosulich, D.B.; Fahrenbach, M.J.; et al. The Structure and Synthesis of the Liver L. casei Factor. Science 1946, 103, 667–669. [Google Scholar] [CrossRef]
- Pfiffner, J.J.; Calkins, D.G.; Bloom, E.S.; O’Dell, B.L. On the Peptide Nature of Vitamin Bc Conjugate from Yeast. J. Am. Chem. Soc. 1946, 68, 1392. [Google Scholar] [CrossRef]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an Intestinal Folate Transporter and the Molecular Basis for Hereditary Folate Malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selhub, J.; Rosenberg, I.H. Folate transport in isolated brush border membrane vesicles from rat intestine. J. Biol. Chem. 1981, 256, 4489–4493. [Google Scholar] [CrossRef]
- Garrow, T.A.; Brenner, A.A.; Whitehead, V.M.; Chen, X.N.; Duncan, R.G.; Korenberg, J.R.; Shane, B. Cloning of human cDNAs encoding mitochondrial and cytosolic serine hydroxymethyltransferases and chromosomal localization. J. Biol. Chem. 1993, 268, 11910–11916. [Google Scholar] [CrossRef]
- Katzen, H.M.; Buchanan, J.M. Enzymatic synthesis of the methyl group of methionine. 8. repression-derepression, purification, and properties of 5,10-methylenetetrahydrofolate reductase from Escherichia coli. J. Biol. Chem. 1965, 240, 825–835. [Google Scholar] [CrossRef]
- Wilson, R.S.; Mertes, M.P. Chemical model for thymidylate synthetase catalysis. J. Am. Chem. Soc. 1972, 94, 7182–7183. [Google Scholar] [CrossRef]
- Himes, R.H.; Rabinowitz, J.C. Formyltetrahydrofolate synthetase. II. Characteristics of the enzyme and the enzymic reaction. J. Biol. Chem. 1962, 237, 2903–2914. [Google Scholar] [CrossRef]
- Wasserman, G.F.; Mueller, W.T.; Benkovic, S.J.; Liao, W.S.L.; Taylor, J. Evidence that the folate-requiring enzymes of de novo purine biosynthesis are encoded by individual mRNAs. Biochemistry 1984, 23, 6704–6710. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.U.; Drury, E.J.; MacKenzie, R.E. Methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase-formyltetrahydrofolate synthetase. A multifunctional protein from porcine liver. J. Biol. Chem. 1977, 252, 1117–1122. [Google Scholar] [CrossRef]
- Goldblatt, M.W. Occupational diseases. Br. Med. J. 1955, 2, 611. [Google Scholar] [CrossRef]
- Doddrell, D.; Allerhand, A. Assignments in the carbon-13 nuclear magnetic resonance spectra of vitamin B12’ coenzyme B12’ and other corrinoids: Application of partially-relaxed fourier transform spectroscopy. Proc. Natl. Acad. Sci. USA 1971, 68, 1083–1088. [Google Scholar] [CrossRef] [Green Version]
- Hodgkin, D.C.; Kamper, J.; Mackay, M.; Pickworth, J.; Trueblood, K.N.; White, J.G. Structure of vitamin B12. Nature 1956, 178, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Ruiz, A.; Lanza, F.; Haange, S.-B.; Oberbach, A.; Till, H.; Bargiela, R.; Campoy, C.; Segura, M.T.; Richter, M.; et al. Microbiota from the distal guts of lean and obese adolescents exhibit partial functional redundancy besides clear differences in community structure. Environ. Microbiol. 2013, 15, 211–226. [Google Scholar] [CrossRef]
- Allen, L.H.; Miller, J.W.; de Groot, L.; Rosenberg, I.H.; Smith, A.D.; Refsum, H.; Raiten, D.J. Biomarkers of Nutrition for Development (BOND): Vitamin B-12 Review. J. Nutr. 2018, 148, 1995s–2027s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickes, E.L.; Brink, N.G.; Koniuszy, F.R.; Wood, T.R.; Folkers, K. Crystalline Vitamin B12. Science 1948, 107, 396–397. [Google Scholar] [CrossRef] [PubMed]
- Conley, C.L.; Krevans, J.R. Development of Neurologic Manifestations of Pernicious Anemia during Multivitamin Therapy. N. Engl. J. Med. 1951, 245, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Karnaze, D.S.; Carmel, R. Neurologic and evoked potential abnormalities in subtle cobalamin deficiency states, including deficiency without anemia and with normal absorption of free cobalamin. Arch. Neurol. 1990, 47, 1008–1012. [Google Scholar] [CrossRef]
- Lindenbaum, J.; Healton, E.B.; Savage, D.G.; Brust, J.C.M.; Garrett, T.J.; Podell, E.R.; Margell, P.D.; Stabler, S.P.; Allen, R.H. Neuropsychiatric Disorders Caused by Cobalamin Deficiency in the Absence of Anemia or Macrocytosis. N. Engl. J. Med. 1988, 318, 1720–1728. [Google Scholar] [CrossRef]
- Bousselamti, A.; El Hasbaoui, B.; Echahdi, H.; Krouile, Y. Psychomotor regression due to vitamin B12 deficiency. Pan Afr. Med. J. 2018, 30, 152. [Google Scholar] [CrossRef]
- Hasbaoui, B.E.; Mebrouk, N.; Saghir, S.; Yajouri, A.E.; Abilkassem, R.; Agadr, A. Vitamin B12 deficiency: Case report and review of literature. Pan Afr. Med. J. 2021, 38, 237. [Google Scholar] [CrossRef]
- Kocaoglu, C.; Akin, F.; Caksen, H.; Böke, S.B.; Arslan, S.; Aygün, S. Cerebral atrophy in a vitamin B12-deficient infant of a vegetarian mother. J. Health Popul. Nutr. 2014, 32, 367–371. [Google Scholar]
- Allen, R.H.; Seetharam, B.; Podell, E.; Alpers, D.H. Effect of proteolytic enzymes on the binding of cobalamin to R protein and intrinsic factor. In vitro evidence that a failure to partially degrade R protein is responsible for cobalamin malabsorption in pancreatic insufficiency. J. Clin. Investig. 1978, 61, 47–54. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Madsen, M.; Kilkenney, A.; Gregory, B.; Christensen, E.I.; Vorum, H.; Højrup, P.; Schäffer, A.A.; Kirkness, E.F.; Tanner, S.M.; et al. Amnionless function is required for cubilin brush-border expression and intrinsic factor-cobalamin (vitamin B12) absorption in vivo. Blood 2005, 106, 1447–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappazzo, M.E.; Hall, C.A. Transport function of transcobalamin II. J. Clin. Investig. 1972, 51, 1915–1918. [Google Scholar] [CrossRef]
- Beedholm-Ebsen, R.; van de Wetering, K.; Hardlei, T.; Nexø, E.; Borst, P.; Moestrup, S.K. Identification of multidrug resistance protein 1 (MRP1/ABCC1) as a molecular gate for cellular export of cobalamin. Blood 2010, 115, 1632–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mørkbak, A.L.; Hvas, A.-M.; Lloyd-Wright, Z.; Sanders, T.A.B.; Bleie, Ø.; Refsum, H.; Nygaard, O.K.; Nexø, E. Effect of Vitamin B12 Treatment on Haptocorrin. Clin. Chem. 2006, 52, 1104–1111. [Google Scholar] [CrossRef] [Green Version]
- Quadros, E.V.; Nakayama, Y.; Sequeira, J.M. The protein and the gene encoding the receptor for the cellular uptake of transcobalamin-bound cobalamin. Blood 2009, 113, 186–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youngdahl-Turner, P.; Mellman, I.S.; Allen, R.H.; Rosenberg, L.E. Protein mediated vitamin uptake: Adsorptive endocytosis of the transcobalamin II—Cobalamin complex by cultured human fibroblasts. Exp. Cell Res. 1979, 118, 127–134. [Google Scholar] [CrossRef]
- Coelho, D.; Kim, J.C.; Miousse, I.R.; Fung, S.; du Moulin, M.; Buers, I.; Suormala, T.; Burda, P.; Frapolli, M.; Stucki, M.; et al. Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat. Genet. 2012, 44, 1152–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutsch, F.; Gailus, S.; Miousse, I.R.; Suormala, T.; Sagné, C.; Toliat, M.R.; Nürnberg, G.; Wittkampf, T.; Buers, I.; Sharifi, A.; et al. Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Nat. Genet. 2009, 41, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, L.; Kim, J.; Brasch, N.E.; Wang, S.; Rosenblatt, D.S.; Banerjee, R.; Jacobsen, D.W. Processing of alkylcobalamins in mammalian cells: A role for the MMACHC (cblC) gene product. Mol. Genet. Metab. 2009, 97, 260–266. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Gherasim, C.; Banerjee, R. Decyanation of vitamin B12 by a trafficking chaperone. Proc. Natl. Acad. Sci. USA 2008, 105, 14551–14554. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Gherasim, C.; Banerjee, R.; Koutmos, M. Structure of Human B12 Trafficking Protein CblD Reveals Molecular Mimicry and Identifies a New Subfamily of Nitro-FMN Reductases. J. Biol. Chem. 2015, 290, 29155–29166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassila, C.; Ghemrawi, R.; Flayac, J.; Froese, D.S.; Baumgartner, M.R.; Guéant, J.L.; Coelho, D. Methionine synthase and methionine synthase reductase interact with MMACHC and with MMADHC. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 103–112. [Google Scholar] [CrossRef]
- McDonald, M.K.; Fritz, J.-A.; Jia, D.; Scheuchner, D.; Snyder, F.F.; Stanislaus, A.; Curle, J.; Li, L.; Stabler, S.P.; Allen, R.H.; et al. Identification of ABC transporters acting in vitamin B12 metabolism in Caenorhabditis elegans. Mol. Genet. Metab. 2017, 122, 160–171. [Google Scholar] [CrossRef]
- Karlsson, F.A.; Burman, P.; Lööf, L.; Mårdh, S. Major parietal cell antigen in autoimmune gastritis with pernicious anemia is the acid-producing H+,K+-adenosine triphosphatase of the stomach. J. Clin. Investig. 1988, 81, 475–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, S.M.; Li, Z.; Perko, J.D.; Oner, C.; Cetin, M.; Altay, C.; Yurtsever, Z.; David, K.L.; Faivre, L.; Ismail, E.A.; et al. Hereditary juvenile cobalamin deficiency caused by mutations in the intrinsic factor gene. Proc. Natl. Acad. Sci. USA 2005, 102, 4130–4133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeken, W.L. Remediable defects in Crohn disease: A prospective study of 63 patients. Arch. Intern. Med. 1975, 135, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Halsted, J.A. Megaloblastic anemia, associated with surgically produced gastrointestinal abnormalities. Calif. Med. 1955, 83, 212–217. [Google Scholar] [PubMed]
- Aroda, V.R.; Edelstein, S.L.; Goldberg, R.B.; Knowler, W.C.; Marcovina, S.M.; Orchard, T.J.; Bray, G.A.; Schade, D.S.; Temprosa, M.G.; White, N.H.; et al. Long-term Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J. Clin. Endocrinol. Metab. 2016, 101, 1754–1761. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Kumar, A.; Karmakar, D.; Jha, R.K. Association of B12 deficiency and clinical neuropathy with metformin use in type 2 diabetes patients. J. Postgrad. Med. 2013, 59, 253–257. [Google Scholar] [CrossRef]
- Roze, E.; Gervais, D.; Demeret, S.; Ogier de Baulny, H.; Zittoun, J.; Benoist, J.F.; Said, G.; Pierrot-Deseilligny, C.; Bolgert, F. Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch. Neurol. 2003, 60, 1457–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannella, R.A.; Broitman, S.A.; Zamcheck, N. Vitamin B12 uptake by intestinal microorganisms: Mechanism and relevance to syndromes of intestinal bacterial overgrowth. J. Clin. Investig. 1971, 50, 1100–1107. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, D.K.; Chatterjea, J.B. Serum vitamin B12 in vegetarians. Br. Med. J. 1960, 2, 992–994. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, W.; Obeid, R.; Schorr, H.; Hübner, U.; Geisel, J.; Sand-Hill, M.; Ali, N.; Herrmann, M. Enhanced bone metabolism in vegetarians–the role of vitamin B12 deficiency. Clin. Chem. Lab. Med. 2009, 47, 1381–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, W.; Schorr, H.; Obeid, R.; Geisel, J. Vitamin B-12 status, particularly holotranscobalamin II and methylmalonic acid concentrations, and hyperhomocysteinemia in vegetarians. Am. J. Clin. Nutr. 2003, 78, 131–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlak, R.; Parrott, S.J.; Raj, S.; Cullum-Dugan, D.; Lucus, D. How prevalent is vitamin B12 deficiency among vegetarians? Nutr. Rev. 2013, 71, 110–117. [Google Scholar] [CrossRef]
- Zeisel, S. Choline, Other Methyl-Donors and Epigenetics. Nutrients 2017, 9, 445. [Google Scholar] [CrossRef]
- Lyon, P.; Strippoli, V.; Fang, B.; Cimmino, L. B Vitamins and One-Carbon Metabolism: Implications in Human Health and Disease. Nutrients 2020, 12, 2867. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.L. The methionine-homocysteine cycle and its effects on cognitive diseases. Altern. Med. Rev. 2003, 8, 7–19. [Google Scholar]
- Krebs, H.A.; Hems, R.; Tyler, B. The regulation of folate and methionine metabolism. Biochem. J. 1976, 158, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, D.; Wilson, A.; Dumas, R.; Gafuik, C.; Song, D.; Watkins, D.; Heng, H.H.; Rommens, J.M.; Scherer, S.W.; Rosenblatt, D.S.; et al. Cloning and mapping of a cDNA for methionine synthase reductase, a flavoprotein defective in patients with homocystinuria. Proc. Natl. Acad Sci. USA 1998, 95, 3059–3064. [Google Scholar] [CrossRef] [Green Version]
- Bressenot, A.; Pooya, S.; Bossenmeyer-Pourie, C.; Gauchotte, G.; Germain, A.; Chevaux, J.-B.; Coste, F.; Vignaud, J.-M.; Guéant, J.-L.; Peyrin-Biroulet, L. Methyl donor deficiency affects small-intestinal differentiation and barrier function in rats. Br. J. Nutr. 2013, 109, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Muehlmann, A.M.; Bliznyuk, N.; Duerr, I.; Yang, T.P.; Lewis, M.H. Early exposure to a methyl donor supplemented diet and the development of repetitive motor behavior in a mouse model. Dev. Psychobiol. 2020, 62, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Sahara, Y.; Matsuzawa, D.; Ishii, D.; Fuchida, T.; Goto, T.; Sutoh, C.; Shimizu, E. Paternal methyl donor deficient diets during development affect male offspring behavior and memory-related gene expression in mice. Dev. Psychobiol. 2019, 61, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Kurogi, T.; Inoue, H.; Guo, Y.; Nobukiyo, A.; Nohara, K.; Kanno, M. A methyl-deficient diet modifies early B cell development. Pathobiology 2012, 79, 209–218. [Google Scholar] [CrossRef]
- Shou, L.; Pan, F.; Chin, S.-f. Pancreatic Hormones and Hepatic Methionine Adenosyltransferase in the Rat. Proc. Soc. Exp. Biol. Med. 1969, 131, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Soda, K. Polyamine Metabolism and Gene Methylation in Conjunction with One-Carbon Metabolism. Int. J. Mol. Sci. 2018, 19, 3106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.H.; Wu, K.T.; Hung, C.J.; Hsieh, M.; Li, C. Effects of adenosine dialdehyde treatment on in vitro and in vivo stable protein methylation in HeLa cells. J. Biochem. 2004, 136, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Hop, C.E.; Bakhtiar, R. Homocysteine thiolactone and protein homocysteinylation: Mechanistic studies with model peptides and proteins. Rapid Commun. Mass Spectrom. 2002, 16, 1049–1053. [Google Scholar] [CrossRef]
- Lu, S.C. S-Adenosylmethionine. Int. J. Biochem. Cell Biol. 2000, 32, 391–395. [Google Scholar] [CrossRef]
- Ericson, L.E.; Williams, J.N., Jr.; Elvehjem, C.A. Studies on partially purified betaine-homocysteine transmethylase of liver. J. Biol. Chem. 1955, 212, 537–544. [Google Scholar] [CrossRef]
- Kieliszek, M. Selenium–Fascinating Microelement, Properties and Sources in Food. Molecules 2019, 24, 1298. [Google Scholar] [CrossRef] [Green Version]
- Roman, M.; Jitaru, P.; Barbante, C. Selenium biochemistry and its role for human health. Metallomics 2014, 6, 25–54. [Google Scholar] [CrossRef]
- Chen, S.; Dong, Z.; Cheng, M.; Zhao, Y.; Wang, M.; Sai, N.; Wang, X.; Liu, H.; Huang, G.; Zhang, X. Homocysteine exaggerates microglia activation and neuroinflammation through microglia localized STAT3 overactivation following ischemic stroke. J. Neuroinflamm. 2017, 14, 187. [Google Scholar] [CrossRef] [Green Version]
- Leng, Y.P.; Ma, Y.S.; Li, X.G.; Chen, R.F.; Zeng, P.Y.; Li, X.H.; Qiu, C.F.; Li, Y.P.; Zhang, Z.; Chen, A.F. l-Homocysteine-induced cathepsin V mediates the vascular endothelial inflammation in hyperhomocysteinaemia. Br. J. Pharmacol. 2018, 175, 1157–1172. [Google Scholar] [CrossRef]
- Nilsson, K.; Gustafson, L.; Hultberg, B.r. The Plasma Homocysteine Concentration Is Better Than That of Serum Methylmalonic Acid as a Marker for Sociopsychological Performance in a Psychogeriatric Population. Clin. Chem. 2000, 46, 691–696. [Google Scholar] [CrossRef]
- Perła-Kaján, J.; Jakubowski, H. Dysregulation of Epigenetic Mechanisms of Gene Expression in the Pathologies of Hyperhomocysteinemia. Int. J. Mol. Sci. 2019, 20, 3140. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, D.; Das, C.R.; Sultana, R.; Kashyap, N.; Islam, M.; Bose, P.D.; Saikia, A.K.; Bose, S. Increased homocysteine mediated oxidative stress as key determinant of hepatitis E virus (HEV) infected pregnancy complication and outcome: A study from Northeast India. Infect. Genet. Evol. 2021, 92, 104882. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Tami, A.; Wildemann, B.; Wolny, M.; Wagner, A.; Schorr, H.; Taban-Shomal, O.; Umanskaya, N.; Ross, S.; Garcia, P.; et al. Hyperhomocysteinemia induces a tissue specific accumulation of homocysteine in bone by collagen binding and adversely affects bone. Bone 2009, 44, 467–475. [Google Scholar] [CrossRef]
- Roy, D.G.; Chen, J.; Mamane, V.; Ma, E.H.; Muhire, B.M.; Sheldon, R.D.; Shorstova, T.; Koning, R.; Johnson, R.M.; Esaulova, E.; et al. Methionine Metabolism Shapes T Helper Cell Responses through Regulation of Epigenetic Reprogramming. Cell Metab. 2020, 31, 250–266.e259. [Google Scholar] [CrossRef] [PubMed]
- Lenherr, N.; Christodoulou, J.; Duley, J.; Dobritzsch, D.; Fairbanks, L.; Datta, A.N.; Filges, I.; Gürtler, N.; Roelofsen, J.; van Kuilenburg, A.B.P.; et al. Co-therapy with S-adenosylmethionine and nicotinamide riboside improves t-cell survival and function in Arts Syndrome (PRPS1 deficiency). Mol. Genet. Metab. Rep. 2021, 26, 100709. [Google Scholar] [CrossRef] [PubMed]
- Hoffbrand, A.V.; Stewart, J.S.; Booth, C.C.; Mollin, D.L. Folate deficiency in Crohn’s disease: Incidence, pathogenesis, and treatment. Br. Med. J. 1968, 2, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolho, K.-L.; Pessia, A.; Jaakkola, T.; de Vos, W.M.; Velagapudi, V. Faecal and Serum Metabolomics in Paediatric Inflammatory Bowel Disease. J. Crohn Colitis 2017, 11, 321–334. [Google Scholar] [CrossRef]
- Holland, N.; Harmatz, P.; Golden, D.; Hubbard, A.; Wu, Y.-Y.; Bae, J.; Chen, C.; Huen, K.; Heyman, M.B. Cytogenetic Damage in Blood Lymphocytes and Exfoliated Epithelial Cells of Children With Inflammatory Bowel Disease. Pediatr. Res. 2007, 61, 209–214. [Google Scholar] [CrossRef]
- Fischer, E.A.; Hetland, M.L.; Krabbe, S. Folinic acid alleviates side effects of methotrexate in arthritis patients with side effects despite folic acid supplementation: An observational cohort study. Rheumatology 2020, 59, 3566–3568. [Google Scholar] [CrossRef]
- Saibeni, S.; Bollani, S.; Losco, A.; Michielan, A.; Sostegni, R.; Devani, M.; Lupinacci, G.; Pirola, L.; Cucino, C.; Meucci, G.; et al. The use of methotrexate for treatment of inflammatory bowel disease in clinical practice. Dig. Liver Dis. 2012, 44, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Piovani, D.; Danese, S.; Peyrin-Biroulet, L.; Nikolopoulos, G.K.; Lytras, T.; Bonovas, S. Environmental Risk Factors for Inflammatory Bowel Diseases: An Umbrella Review of Meta-analyses. Gastroenterology 2019, 157, 647–659.e644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zintzaras, E. Genetic variants of homocysteine/folate metabolism pathway and risk of inflammatory bowel disease: A synopsis and meta-analysis of genetic association studies. Biomarkers 2010, 15, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, M.A.Y.; Imhann, F.; Collij, V.; Fu, J.; Wijmenga, C.; Zhernakova, A.; Dijkstra, G.; Festen, E.A.M.; Gacesa, R.; Vich Vila, A.; et al. Anti-inflammatory Gut Microbial Pathways Are Decreased During Crohn’s Disease Exacerbations. J. Crohns Colitis 2019, 13, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Gimier, E.; Chervy, M.; Agus, A.; Sivignon, A.; Billard, E.; Privat, M.; Viala, S.; Minet-Quinard, R.; Buisson, A.; Vazeille, E.; et al. Methyl-donor supplementation prevents intestinal colonization by Adherent-Invasive E. coli in a mouse model of Crohn’s disease. Sci. Rep. 2020, 10, 12922. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.D.; Haltalin, K.C. Effect of neonatal folic acid deprivation on later growth and susceptibility to Shigella infection in the guinea pig. Am. J. Clin. Nutr. 1972, 25, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Park, A.M.; Omura, S.; Fujita, M.; Sato, F.; Tsunoda, I. Helicobacter pylori and gut microbiota in multiple sclerosis versus Alzheimer’s disease: 10 pitfalls of microbiome studies. Clin. Exp. Neuroimmunol. 2017, 8, 215–232. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Yang, W.; Wu, C.; Lin, D.; Ding, R.; Sun, L.; Jiang, L. Association of ulcerative colitis with transcobalamin II gene polymorphisms and serum homocysteine, vitamin B12, and folate levels in Chinese patients. Immunogenetics 2017, 69, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Madanchi, M.; Fagagnini, S.; Fournier, N.; Biedermann, L.; Zeitz, J.; Battegay, E.; Zimmerli, L.; Vavricka, S.R.; Rogler, G.; Scharl, M.; et al. The Relevance of Vitamin and Iron Deficiency in Patients with Inflammatory Bowel Diseases in Patients of the Swiss IBD Cohort. Inflamm. Bowel Dis. 2018, 24, 1768–1779. [Google Scholar] [CrossRef]
- Lurz, E.; Horne, R.G.; Määttänen, P.; Wu, R.Y.; Botts, S.R.; Li, B.; Rossi, L.; Johnson-Henry, K.C.; Pierro, A.; Surette, M.G.; et al. Vitamin B12 Deficiency Alters the Gut Microbiota in a Murine Model of Colitis. Front. Nutr. 2020, 7, 83. [Google Scholar] [CrossRef]
- Tamura, J.; Kubota, K.; Murakami, H.; Sawamura, M.; Matsushima, T.; Tamura, T.; Saitoh, T.; Kurabayshi, H.; Naruse, T. Immunomodulation by vitamin B12: Augmentation of CD8+ T lymphocytes and natural killer (NK) cell activity in vitamin B12-deficient patients by methyl-B12 treatment. Clin. Exp. Immunol. 1999, 116, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Xiang, S.; Feng, X.; Wang, H.; Tian, S.; Xu, Y.; Shi, L.; Yang, L.; Li, M.; Shen, Y.; et al. Impact of Cyanocobalamin and Methylcobalamin on Inflammatory Bowel Disease and the Intestinal Microbiota Composition. J. Agric. Food Chem. 2018, 67, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Melhem, H.; Hansmannel, F.; Bressenot, A.; Battaglia-Hsu, S.-F.; Billioud, V.; Alberto, J.M.; Gueant, J.L.; Peyrin-Biroulet, L. Methyl-deficient diet promotes colitis and SIRT1-mediated endoplasmic reticulum stress. Gut 2016, 65, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Roblin, X.; Phelip, J.M.; Genevois, M.; Ducros, V.; Bonaz, B. Hyperhomocysteinaemia is associated with osteoporosis in patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2007, 25, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Harb, Z.; Deckert, V.; Bressenot, A.M.; Christov, C.; Guéant-Rodriguez, R.-M.; Raso, J.; Alberto, J.M.; de Barros, J.-P.P.; Umoret, R.; Peyrin-Biroulet, L.; et al. The deficit in folate and vitamin B12 triggers liver macrovesicular steatosis and inflammation in rats with dextran sodium sulfate-induced colitis. J. Nutr. Biochem. 2020, 84, 108415. [Google Scholar] [CrossRef] [PubMed]
- Oz, H.S.; Chen, T.S.; McClain, C.J.; de Villiers, W.J. Antioxidants as novel therapy in a murine model of colitis. J. Nutr. Biochem. 2005, 16, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Schmedes, A.; Nielsen, J.N.; Hey, H.; Brandslund, I. Low S-adenosylmethionine concentrations found in patients with severe inflammatory bowel disease. Clin. Chem. Lab. Med. 2004, 42, 648–653. [Google Scholar] [CrossRef]
- Bermejo, F.; Algaba, A.; Guerra, I.; Chaparro, M.; De-La-Poza, G.; Valer, P.; Piqueras, B.; Bermejo, A.; García-Alonso, J.; Pérez, M.-J.; et al. Should we monitor vitamin B12and folate levels in Crohn’s disease patients? Scand. J. Gastroenterol. 2013, 48, 1272–1277. [Google Scholar] [CrossRef]
- Ao, M.; Tsuji, H.; Shide, K.; Kosaka, Y.; Noda, A.; Inagaki, N.; Nakase, H.; Tanaka, K. High prevalence of vitamin B-12 insufficiency in patients with Crohn’s disease. Asia Pac. J. Clin. Nutr. 2017, 26, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Casella, G.; Antonelli, E.; Di Bella, C.; Di Marco, E.; Piatti, M.; Villanacci, V.; Bologna, S.; Baldini, V.; Bassotti, G. Hyperhomocysteinemia in patients with Crohn’s disease. Tech. Coloproctol. 2013, 17, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.; Benhayoun, S.; Adjalla, C.; Gélot, M.-A.; Renkes, P.; Felden, F.; Gérard, P.; Belleville, F.; Gaucher, P.; Guéant, J.-L.; et al. Crohn’s disease and vitamin B12 metabolism. Dig. Dis. Sci. 1996, 41, 1417–1422. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, A.L.F.; Dalli, J.; Brancaleone, V.; D’Acquisto, F.; Perretti, M.; Wheatley, C. Biphasic Modulation of NOS Expression, Protein and Nitrite Products by Hydroxocobalamin Underlies Its Protective Effect in Endotoxemic Shock: Downstream Regulation of COX-2, IL-1β, TNF-α, IL-6, and HMGB1 Expression. Mediat. Inflamm. 2013, 2013, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-J.; Wang, M.-Y.; Lin, M.-C.; Lin, P.-T. Associations between Vitamin B-12 Status and Oxidative Stress and Inflammation in Diabetic Vegetarians and Omnivores. Nutrients 2016, 8, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, H.; Mu, J.; Ma, J.; Gong, J.; Li, J.; Wang, J.; Gao, T.; Zhu, P.; Zheng, S.; Xie, J.; et al. Selenium Inhibits Homocysteine-Induced Endothelial Dysfunction and Apoptosis via Activation of AKT. Cell. Physiol. Biochem. 2016, 38, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Yıldızhan, K.; Nazıroğlu, M. Protective role of selenium on MPP(+) and homocysteine-induced TRPM2 channel activation in SH-SY5Y cells. J. Recept. Signal Transduct. Res. 2021, 1–10. [Google Scholar] [CrossRef]
- Kieliszek, M.; Lipinski, B. Pathophysiological significance of protein hydrophobic interactions: An emerging hypothesis. Med. Hypotheses 2018, 110, 15–22. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Li, G.; Chen, Y.; Conley, S.M.; Gehr, T.W.B.; Boini, K.M.; Li, P.-L. Nod-like Receptor Protein 3 (NLRP3) Inflammasome Activation and Podocyte Injury via Thioredoxin-Interacting Protein (TXNIP) during Hyperhomocysteinemia. J. Biol. Chem. 2014, 289, 27159–27168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, A.F.; Petrie, L. Folate deficiency enhances the inflammatory response of macrophages. Mol. Immunol. 2013, 54, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Samblas, M.; Martínez, J.A.; Milagro, F. Folic Acid Improves the Inflammatory Response in LPS-Activated THP-1 Macrophages. Mediat. Inflamm. 2018, 2018, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, S.S.; Basford, R.E. Effect of vitamin B12 and folic acid deficiencies on neutrophil function. Blood 1976, 47, 801–805. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Ding, N.; Zhang, H.; Liu, K.; Xiong, J.; Ma, S.; Yang, A.; Zhang, H.; Jiang, Y. SNF5 promotes IL-1β expression via H3K4me1 in atherosclerosis induced by homocysteine. Int. J. Biochem. Cell Biol. 2021, 135, 105974. [Google Scholar] [CrossRef]
- Bian, Y.; Li, W.; Kremer, D.M.; Sajjakulnukit, P.; Li, S.; Crespo, J.; Nwosu, Z.C.; Zhang, L.; Czerwonka, A.; Pawłowska, A.; et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 2020, 585, 277–282. [Google Scholar] [CrossRef]
- Hote, P.T.; Sahoo, R.; Jani, T.S.; Ghare, S.S.; Chen, T.; Joshi-Barve, S.; McClain, C.J.; Barve, S.S. Ethanol inhibits methionine adenosyltransferase II activity and S-adenosylmethionine biosynthesis and enhances caspase-3-dependent cell death in T lymphocytes: Relevance to alcohol-induced immunosuppression. J. Nutr. Biochem. 2008, 19, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, T.S.; Soares, M.S.P.; Pedra, N.S.; Bona, N.P.; Spohr, L.; Teixeira, F.C.; Couto, C.A.T.D.; Spanevello, R.M.; Deon, M.; Vargas, C.R.; et al. Characterization of macrophage phenotype, redox, and purinergic response upon chronic treatment with methionine and methionine sulfoxide in mice. Amino Acids 2020, 52, 629–638. [Google Scholar] [CrossRef]
- dos Santos, L.M.; da Silva, T.M.; Azambuja, J.H.; Ramos, P.T.; Oliveira, P.S.; da Silveira, E.F.; Pedra, N.S.; Galdino, K.; Couto, C.A.T.D.; Soares, M.S.P.; et al. Methionine and methionine sulfoxide treatment induces M1/classical macrophage polarization and modulates oxidative stress and purinergic signaling parameters. Mol. Cell. Biochem. 2017, 424, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.S.P.; Costa, M.Z.; Da Silva, T.M.; Gazal, M.; Couto, C.A.T.D.; Debom, G.N.; Rodrigues, R.; Azambuja, J.H.; Casali, E.A.; Moritz, C.E.J.; et al. Methionine and/or Methionine Sulfoxide Alter Ectoenzymes Activities in Lymphocytes and Inflammatory Parameters in Serum from Young Rats: Acute and Chronic Effects. Cell Biophys. 2017, 76, 243–253. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Li, X.; Geng, J. Preliminary analysis of immunoregulatory mechanism of hyperhomocysteinemia-induced brain injury in Wistar-Kyoto rats. Exp. Ther. Med. 2021, 21, 1–10. [Google Scholar] [CrossRef]
- Chen, H.; Liu, S.; Ji, L.; Wu, T.; Ji, Y.; Zhou, Y.; Zheng, M.; Zhang, M.; Xu, W.; Huang, G. Folic Acid Supplementation Mitigates Alzheimer’s Disease by Reducing Inflammation: A Randomized Controlled Trial. Mediat. Inflamm. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Di Rosa, G.; Attinà, S.; Spano, M.; Ingegneri, G.; Sgrò, D.L.; Pustorino, G.; Bonsignore, M.; Tortorella, G.; Trapani-Lombardo, V. Efficacy of Folic Acid in Children with Migraine, Hyperhomocysteinemia and MTHFR Polymorphisms. Headache: J. Head Face Pain 2007, 47, 1342–1344. [Google Scholar] [CrossRef]
- Dorszewska, J.; Florczak, J.; Rozycka, A.; Kempisty, B.; Jaroszewska-Kolecka, J.; Chojnacka, K.; Trzeciak, W.H.; Kozubski, W. Oxidative DNA damage and level of thiols as related to polymorphisms of MTHFR, MTR, MTHFD1 in Alzheimer’s and Parkinson’s diseases. Acta Neurobiol. Exp. 2007, 67, 113–129. [Google Scholar]
- Zhang, Y.; Hodgson, N.W.; Trivedi, M.S.; Abdolmaleky, H.M.; Fournier, M.; Cuenod, M.; Do, K.Q.; Deth, R.C. Decreased Brain Levels of Vitamin B12 in Aging, Autism and Schizophrenia. PLoS ONE 2016, 11, e0146797. [Google Scholar] [CrossRef] [Green Version]
- Didangelos, T.; Karlafti, E.; Kotzakioulafi, E.; Margariti, E.; Giannoulaki, P.; Batanis, G.; Tesfaye, S.; Kantartzis, K. Vitamin B12 Supplementation in Diabetic Neuropathy: A 1-Year, Randomized, Double-Blind, Placebo-Controlled Trial. Nutrients 2021, 13, 395. [Google Scholar] [CrossRef] [PubMed]
- de Queiroz, K.B.; Cavalcante-Silva, V.; Lopes, F.L.; Rocha, G.A.; D’Almeida, V.; Coimbra, R.S. Vitamin B12 is neuroprotective in experimental pneumococcal meningitis through modulation of hippocampal DNA methylation. J. Neuroinflamm. 2020, 17, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filiz, A.K.; Gumus, E.; Karabulut, S.; Tastemur, Y.; Taskiran, A.S. Protective effects of lamotrigine and vitamin B12 on pentylenetetrazole-induced epileptogenesis in rats. Epilepsy Behav. 2021, 118, 107915. [Google Scholar] [CrossRef]
- Sudduth, T.L.; Powell, D.K.; Smith, C.D.; Greenstein, A.; Wilcock, D.M. Induction of Hyperhomocysteinemia Models Vascular Dementia by Induction of Cerebral Microhemorrhages and Neuroinflammation. J. Cereb. Blood Flow Metab. 2013, 33, 708–715. [Google Scholar] [CrossRef] [Green Version]
- Weekman, E.M.; Sudduth, T.L.; Price, B.R.; Woolums, A.E.; Hawthorne, D.; Seaks, C.E.; Wilcock, D.M. Time course of neuropathological events in hyperhomocysteinemic amyloid depositing mice reveals early neuroinflammatory changes that precede amyloid changes and cerebrovascular events. J. Neuroinflamm. 2019, 16, 284. [Google Scholar] [CrossRef]
- Ehmedah, A.; Nedeljkovic, P.; Dacic, S.; Repac, J.; Draskovic-Pavlovic, B.; Vučević, D.; Pekovic, S.; Nedeljkovic, B.B. Effect of Vitamin B Complex Treatment on Macrophages to Schwann Cells Association during Neuroinflammation after Peripheral Nerve Injury. Molecules 2020, 25, 5426. [Google Scholar] [CrossRef]
- Bakulski, K.M.; Dolinoy, D.C.; Sartor, M.A.; Paulson, H.L.; Konen, J.R.; Lieberman, A.P.; Albin, R.L.; Hu, H.; Rozek, L.S. Genome-Wide DNA Methylation Differences Between Late-Onset Alzheimer’s Disease and Cognitively Normal Controls in Human Frontal Cortex. J. Alzheimer Dis. 2012, 29, 571–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, R.L.; Lee, J.M.; Maroun, L.E. Hypomethylation of the amyloid precursor protein gene in the brain of an alzheimer’s disease patient. J. Mol. Neurosci. 1995, 6, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Carmo, S.D.; Hanzel, C.E.; Jacobs, M.L.; Machnes, Z.; Iulita, M.F.; Yang, J.; Yu, L.; Ducatenzeiler, A.; Danik, M.; Breuillaud, L.S.; et al. Rescue of Early bace-1 and Global DNA Demethylation by S-Adenosylmethionine Reduces Amyloid Pathology and Improves Cognition in an Alzheimer’s Model. Sci. Rep. 2016, 6, 34051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jindal, A.; Rajagopal, S.; Winter, L.; Miller, J.W.; Jacobsen, D.W.; Brigman, J.; Allan, A.M.; Paul, S.; Poddar, R. Hyperhomocysteinemia leads to exacerbation of ischemic brain damage: Role of GluN2A NMDA receptors. Neurobiol. Dis. 2019, 127, 287–302. [Google Scholar] [CrossRef]
- Eissa, S.; Hendy, O.; Younis, F.; Samy, A.; Abdallah, A.; Ahmed, L. Correlations of folic acid, vitamin B12, homocysteine, and thrombopoietin to platelet count in HCV infection. Egypt. J. Intern. Med. 2012, 24, 83–92. [Google Scholar] [CrossRef]
- Feld, J.J.; Modi, A.A.; El-Diwany, R.; Rotman, Y.; Thomas, E.; Ahlenstiel, G.; Titerence, R.; Koh, C.; Cherepanov, V.; Heller, T.; et al. S-Adenosyl Methionine Improves Early Viral Responses and Interferon-Stimulated Gene Induction in Hepatitis C Nonresponders. Gastroenterology 2011, 140, 830–839.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocco, A.; Compare, D.; Coccoli, P.; Esposito, C.; Di Spirito, A.; Barbato, A.; Strazzullo, P.; Nardone, G. Vitamin B12supplementation improves rates of sustained viral response in patients chronically infected with hepatitis C virus. Gut 2012, 62, 766–773. [Google Scholar] [CrossRef]
- Cabrales-Romero Mdel, P.; Márquez-Rosado, L.; Fattel-Fazenda, S.; Trejo-Solís, C.; Arce-Popoca, E.; Alemán-Lazarini, L.; Villa-Treviño, S. S-adenosyl-methionine decreases ethanol-induced apoptosis in primary hepatocyte cultures by a c-Jun N-terminal kinase activity-independent mechanism. World J. Gastroenterol. 2006, 12, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Colell, A.; Morales, A.; Pañeda, C.; Varela-Nieto, I.; García-Ruiz, C.; Fernández-Checa, J.C. Acidic sphingomyelinase downregulates the liver-specific methionine adenosyltransferase 1A, contributing to tumor necrosis factor–induced lethal hepatitis. J. Clin. Investig. 2004, 113, 895–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlesimo, M.; Mari, E.; Arcese, A.; De Angelis, F.; Palese, E.; Abruzzese, C.; De Marco, G.; Cattaruzza, M.S.; Camplone, G. Safety and Efficacy of Calcium Folinate in Psoriasis: An Observational Study. Int. J. Immunopathol. Pharmacol. 2010, 23, 649–653. [Google Scholar] [CrossRef]
- Del Duca, E.; Farnetani, F.; De Carvalho, N.; Bottoni, U.; Pellacani, G.; Nisticò, S.P. Superiority of a vitamin B12-containing emollient compared to a standard emollient in the maintenance treatment of mild-to-moderate plaque psoriasis. Int. J. Immunopathol. Pharmacol. 2017, 30, 439–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobin, A.-M.; Hughes, R.; Hand, E.B.; Leong, T.; Graham, I.M.; Kirby, B. Homocysteine status and cardiovascular risk factors in patients with psoriasis: A case-control study. Clin. Exp. Dermatol. 2010, 36, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Nistico, S.P.; Del Duca, E.; Tamburi, F.; Pignataro, E.; De Carvalho, N.; Farnetani, F.; Pellacani, G. Superiority of a vitamin B12-barrier cream compared with standard glycerol-petrolatum-based emollient cream in the treatment of atopic dermatitis: A randomized, left-to-right comparative trial. Dermatol. Ther. 2017, 30, e12523. [Google Scholar] [CrossRef] [PubMed]
- Stucker, M.; Pieck, C.; Stoerb, C.; Niedner, R.; Hartung, J.; Altmeyer, P. Topical vitamin B12-a new therapeutic approach in atopic dermatitis-evaluation of efficacy and tolerability in a randomized placebo-controlled multicentre clinical trial. Br. J. Dermatol. 2004, 150, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Strober, B.E.; Menon, K. Folate supplementation during methotrexate therapy for patients with psoriasis. J. Am. Acad. Dermatol. 2005, 53, 652–659. [Google Scholar] [CrossRef]
- Juhlin, L.; Olsson, M.J. Improvement of vitiligo after oral treatment with vitamin B12 and folic acid and the importance of sun exposure. Acta Derm. Venereol. 1997, 77, 460–462. [Google Scholar]
- Scambi, C.; De Franceschi, L.; Guarini, P.; Poli, F.; Siciliano, A.; Pattini, P.; Biondani, A.; La Verde, V.; Bortolami, O.; Turrini, F.; et al. Preliminary Evidence for Cell Membrane Amelioration in Children with Cystic Fibrosis by 5-MTHF and Vitamin B12 Supplementation: A Single Arm Trial. PLoS ONE 2009, 4, e4782. [Google Scholar] [CrossRef]
- Yoon, S.-Y.; Hong, G.H.; Kwon, H.-S.; Park, S.; Park, S.Y.; Shin, B.; Kim, T.-B.; Moon, H.-B.; Cho, Y.S. S-adenosylmethionine reduces airway inflammation and fibrosis in a murine model of chronic severe asthma via suppression of oxidative stress. Exp. Mol. Med. 2016, 48, e236. [Google Scholar] [CrossRef]
- Papamichael, M.M.; Katsardis, C.; Tsoukalas, D.; Lambert, K.; Erbas, B.; Itsiopoulos, C. Potential role of folate status on pulmonary function in pediatric asthma. Nutrients 2021, 90, 111267. [Google Scholar] [CrossRef]
- Na, J.D.; Choi, Y.J.; Jun, D.S.; Kim, Y.C. Alleviation of paraquat-induced oxidative lung injury by betaineviaregulation of sulfur-containing amino acid metabolism despite the lack of betaine-homocysteine methyltransferase (BHMT) in the lung. Food Funct. 2019, 10, 1225–1234. [Google Scholar] [CrossRef]
- Moncada, C.A.; Clarkson, A.; Perez-Leal, O.; Merali, S. Mechanism and tissue specificity of nicotine-mediated lung S-adenosylmethionine reduction. J. Biol. Chem. 2008, 283, 7690–7696. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaccaro, J.A.; Naser, S.A. The Role of Methyl Donors of the Methionine Cycle in Gastrointestinal Infection and Inflammation. Healthcare 2022, 10, 61. https://doi.org/10.3390/healthcare10010061
Vaccaro JA, Naser SA. The Role of Methyl Donors of the Methionine Cycle in Gastrointestinal Infection and Inflammation. Healthcare. 2022; 10(1):61. https://doi.org/10.3390/healthcare10010061
Chicago/Turabian StyleVaccaro, Joseph A., and Saleh A. Naser. 2022. "The Role of Methyl Donors of the Methionine Cycle in Gastrointestinal Infection and Inflammation" Healthcare 10, no. 1: 61. https://doi.org/10.3390/healthcare10010061