
Adsorption Characteristics of Carbon Monoxide on Ag- and Au-Doped HfS2 Monolayers Based on Density Functional Theory †



Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
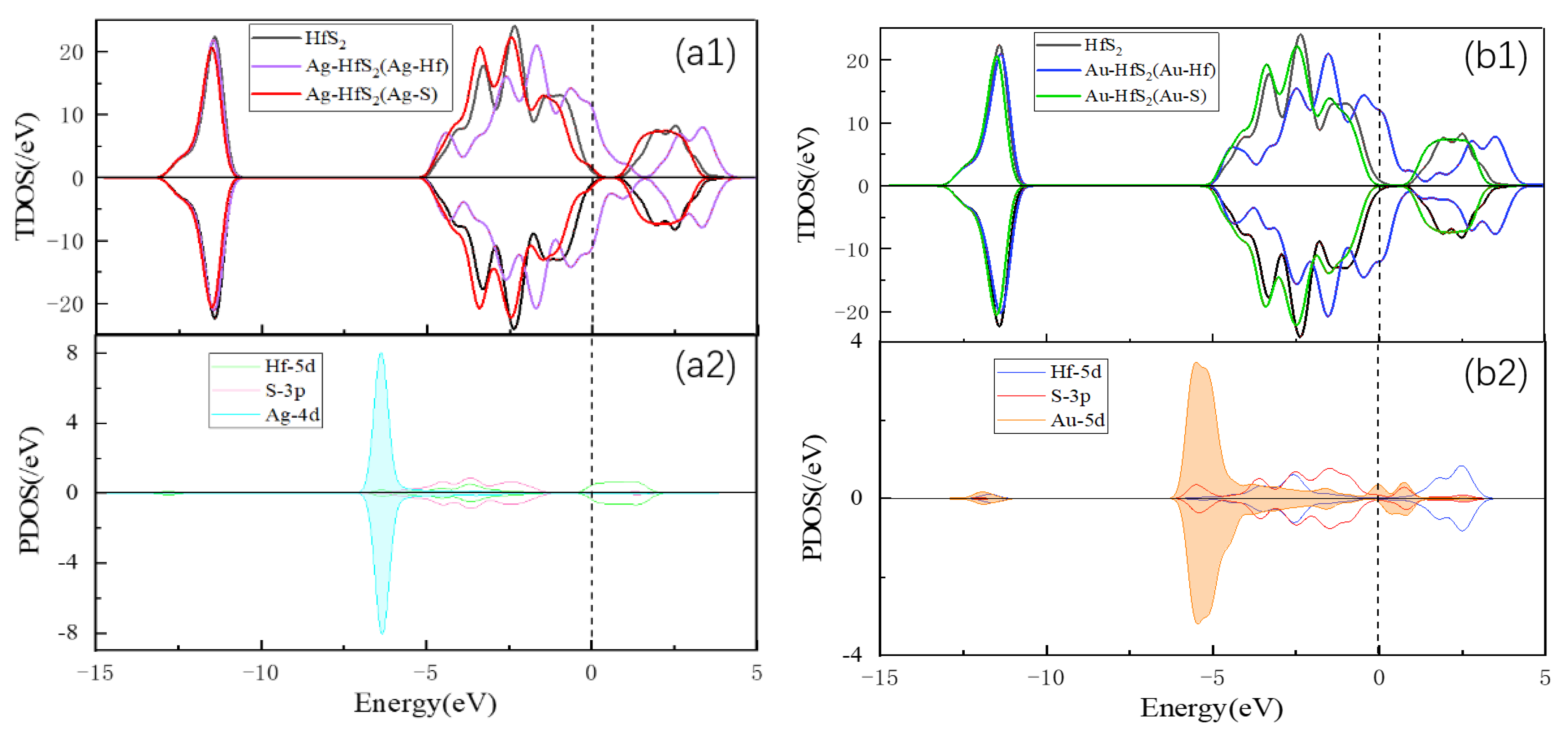
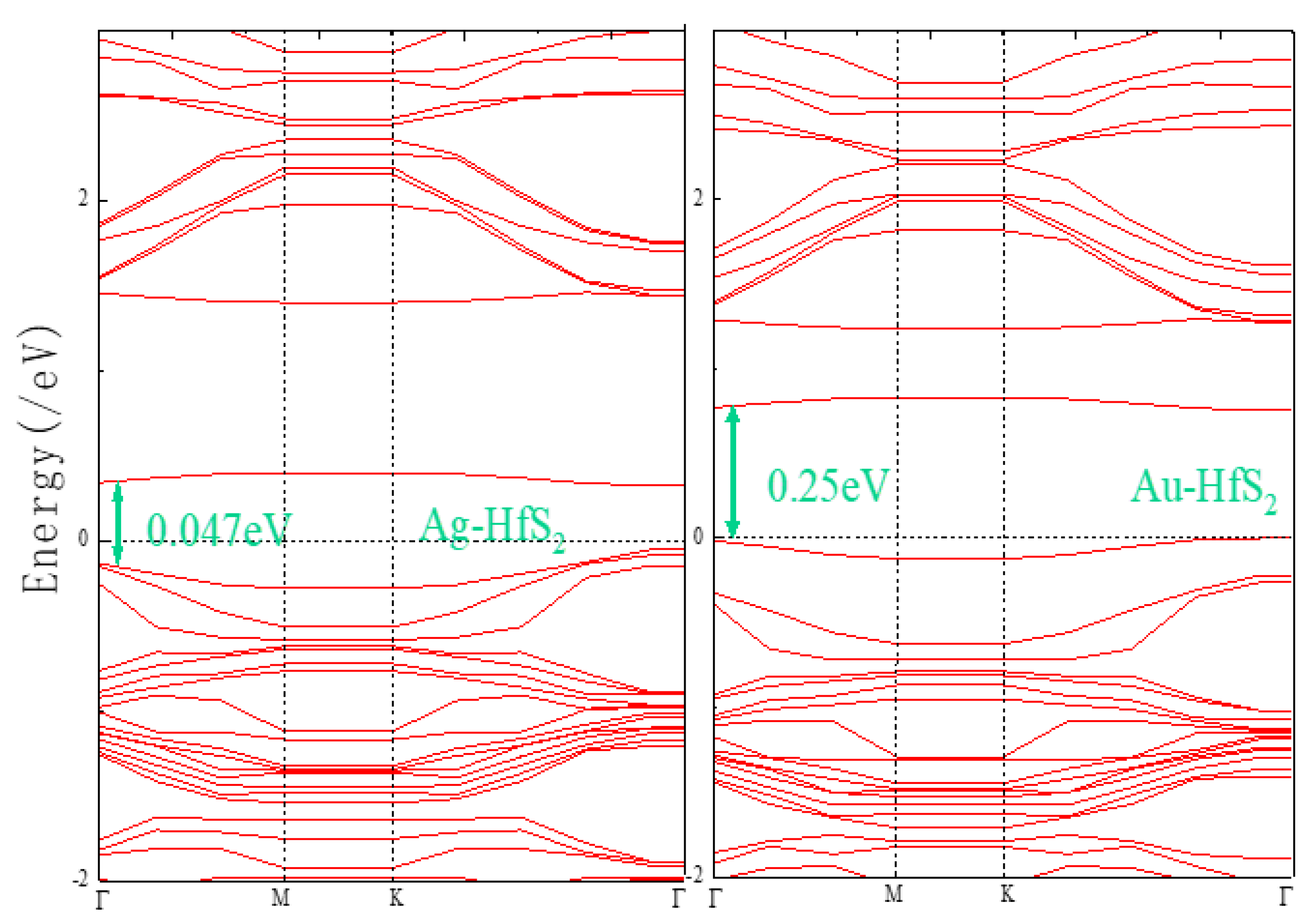
3.1. Metal Doping Method Selection
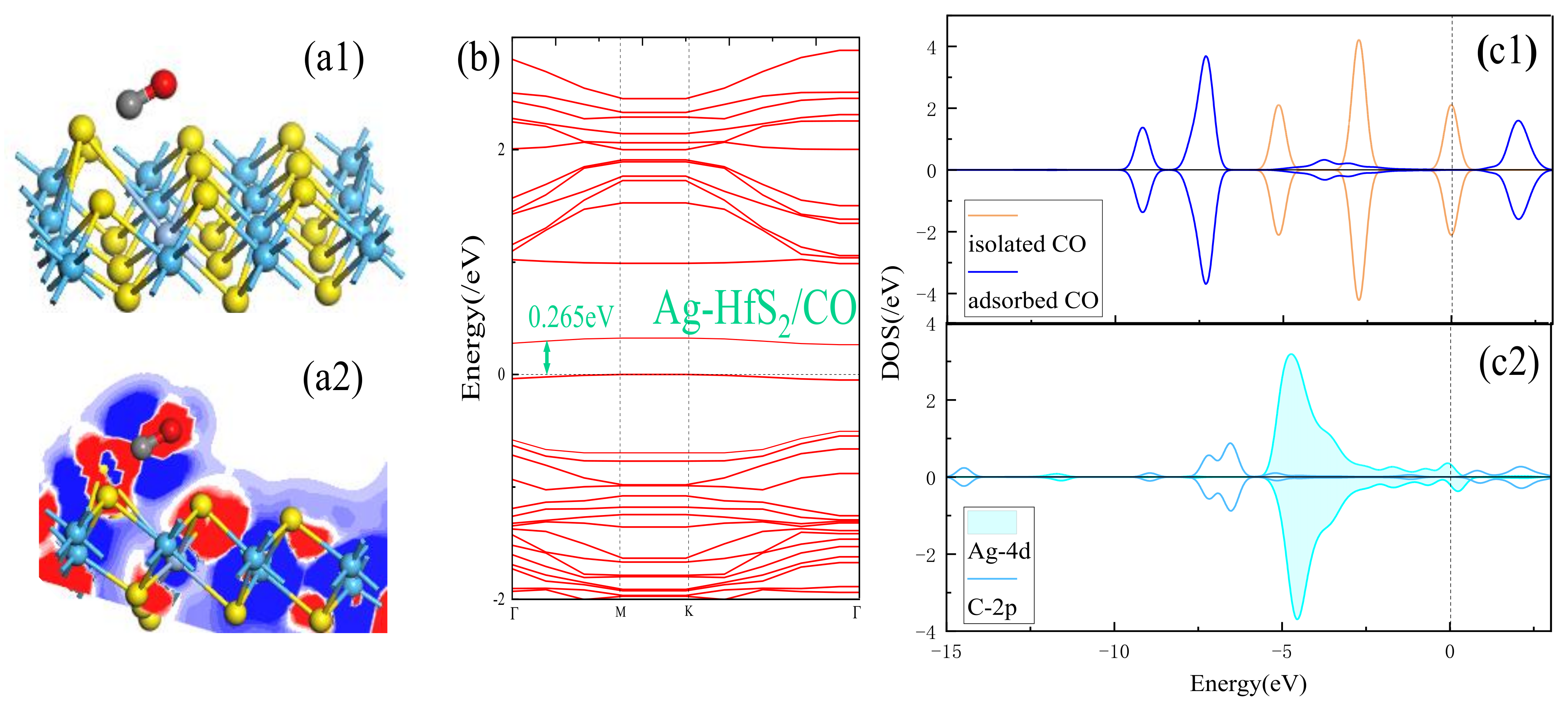
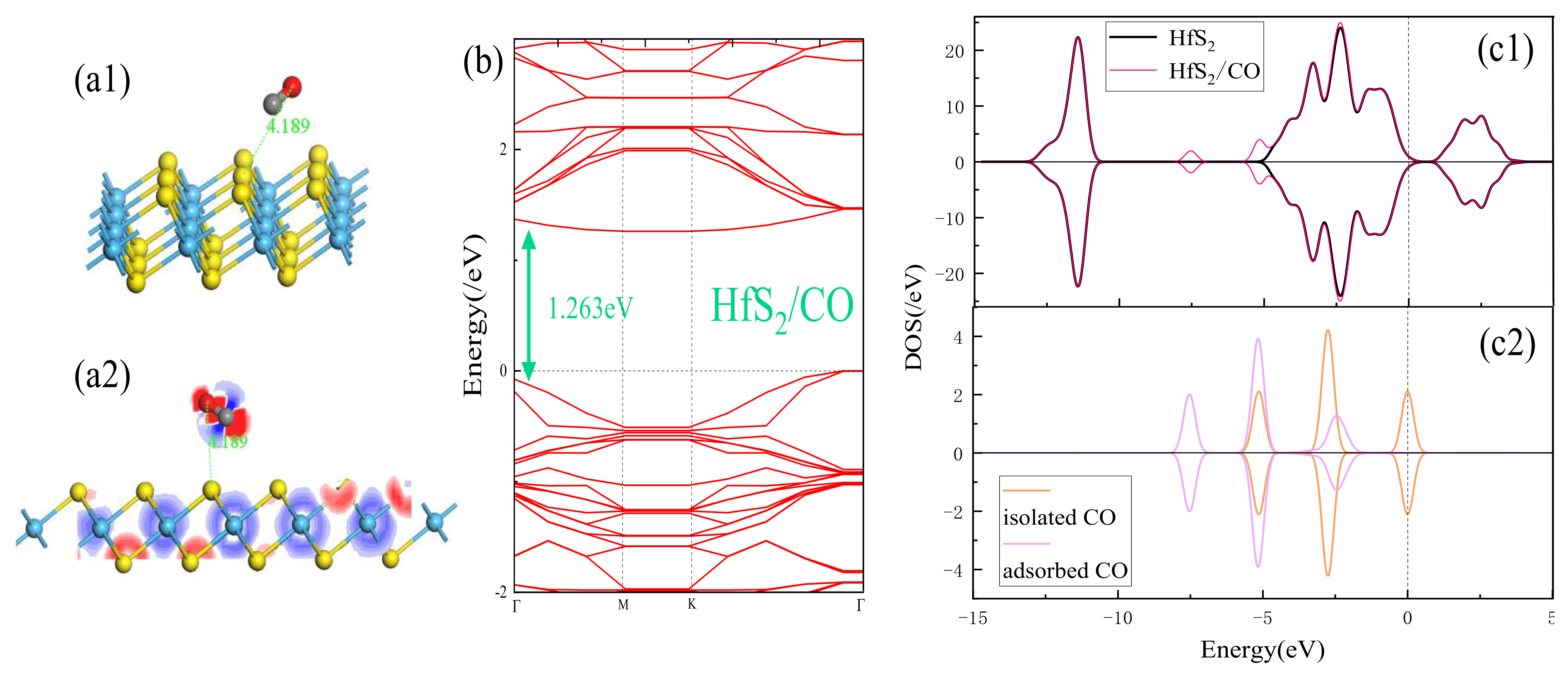
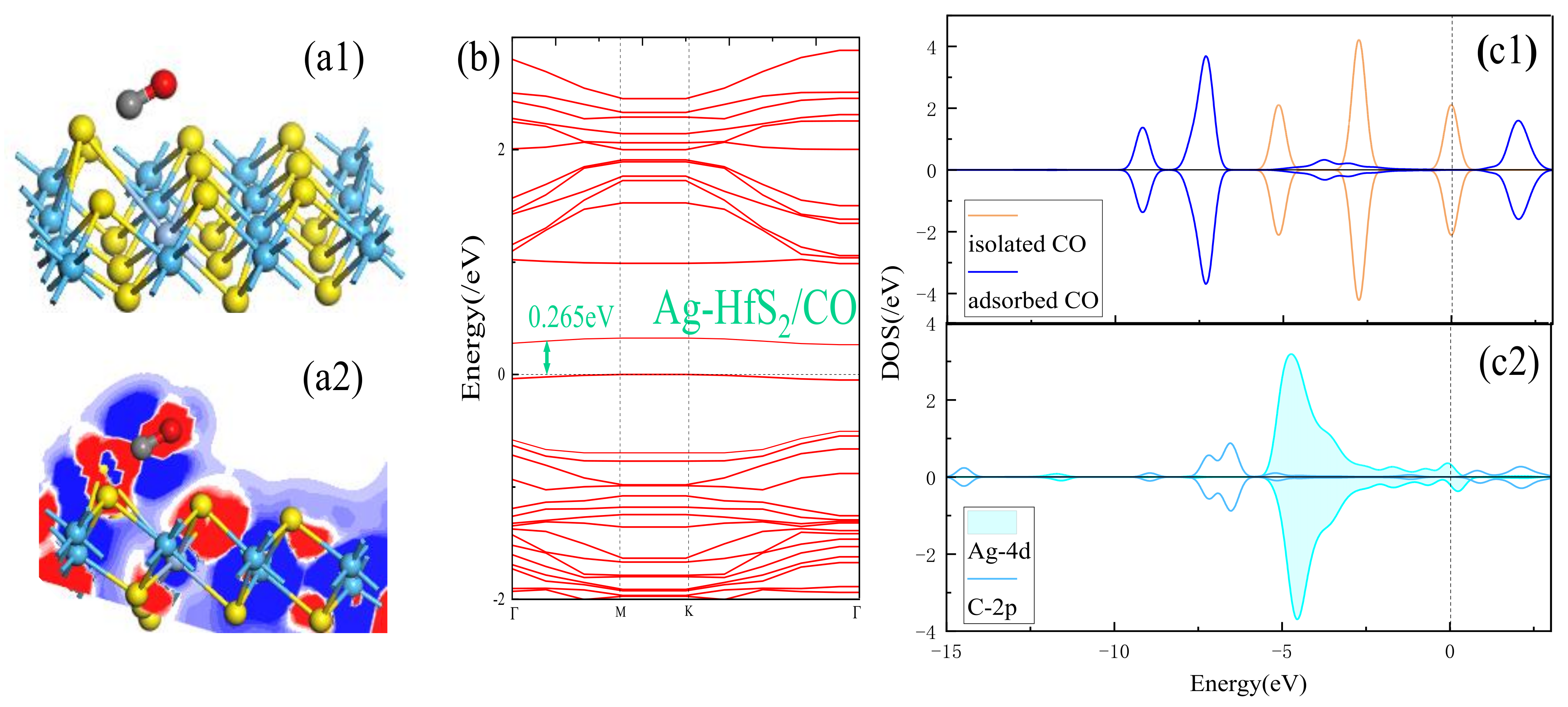
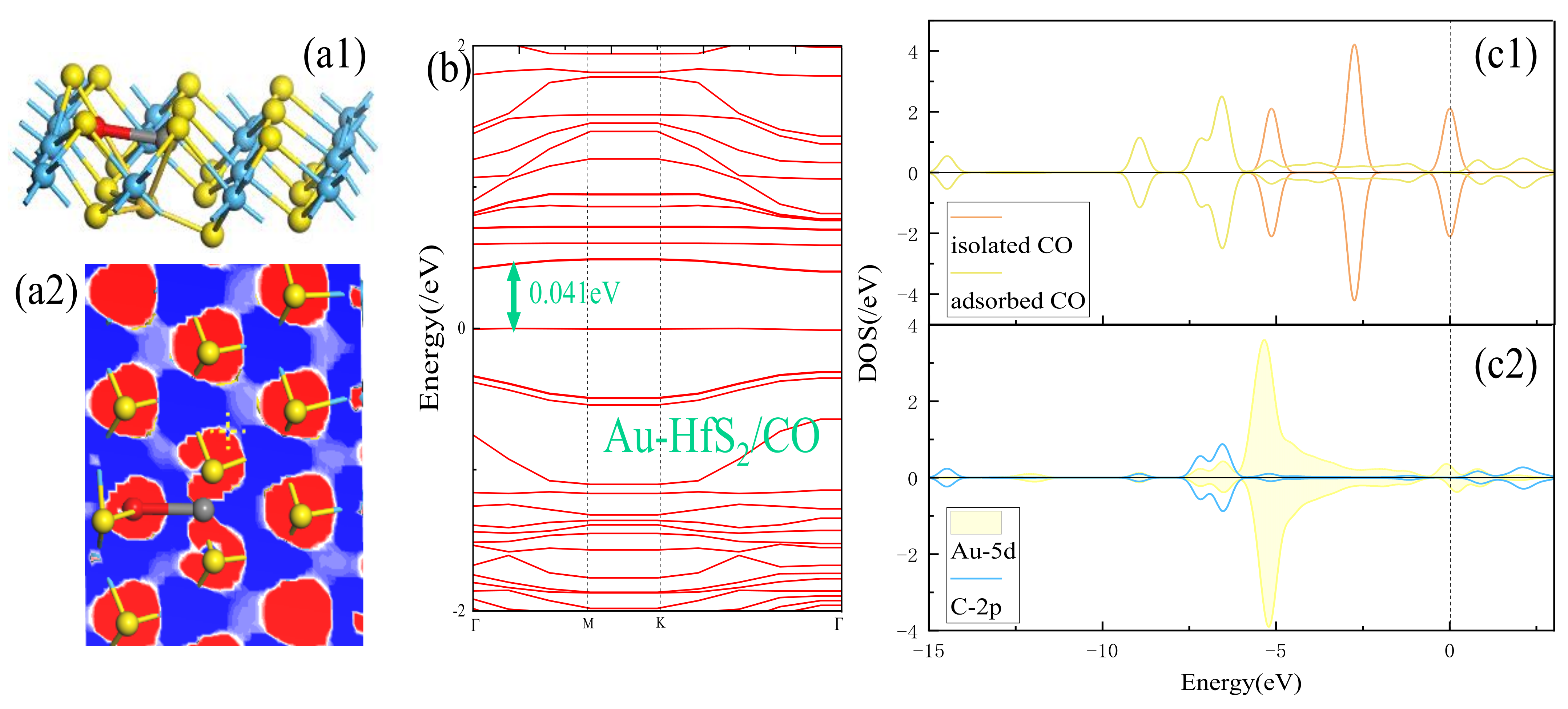
3.2. Adsorption Performance Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Liu, B.S.; Wang, D.; Ma, A.J.; Gui, Y.; Zhang, Q.S.; Zhang, J.; Gong, L.; Tian, M.; Wang, X.P. Study on the components of air under partial discharge condition in closed space. Power Syst. Prot. Control. 2021, 49, 21–28. (In Chinese) [Google Scholar]
- Tao, S.Y.; Feng, Y.; Zhang, T.C.; Cai, H.W. High voltage switch cabinet partial discharge on-line monitoring device based on pulse current method. Power Syst. Prot. Control. 2019, 47, 145–149. (In Chinese) [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.F.; Liu, S.B.; Meng, F.L.; Liu, J.Y.; Jin, Z.; Kong, L.T.; Liu, J.H. Metal oxide nanostructures and their gas sensing properties: A review. Sensors 2012, 12, 2610–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.H.; Han, L.F.; Xiao, Y.H.; Jia, D.Z.; Guo, Z.H.; Li, F. Understanding dopant and defect effect on H2S sensing performances of graphene: A first-principles study. Comput. Mater. Sci. 2013, 69, 222–228. [Google Scholar] [CrossRef]
- Feng, Z.H.; Yuan, X.; Chen, J.C.; Yu, Y.Y.; Zheng, S.J.; Zhang, R.; Li, Q.N.; Chen, X.J.; Sun, C.L.; Zhang, H.; et al. Highly sensitive MoTe2 chemical sensor with fast recovery rate through gate biasing. 2D Materials 2017, 4, 025018. [Google Scholar] [CrossRef]
- Zhang, D.Z.; Wu, J.F.; Li, P.; Cao, Y.H. Room-temperature SO2 gas-sensing properties based on a metal-doped MoS2 nanoflower: An experimental and density functional theory investigation. J. Mater. Chem. 2017, 5, 20666–20677. [Google Scholar] [CrossRef]
- Baek, J.; Yin, D.M.; Liu, N.; Omkaram, I.; Jung, C.; Im, H.; Hong, S.; Kim, S.M.; Hong, Y.K.; Hur, J.; et al. A highly sensitive chemical gas detecting transistor based on highly crystalline CVD-grown MoSe2 films. Nano Res. 2017, 10, 2904. [Google Scholar] [CrossRef] [Green Version]
- Abbasi, A.; Sardroodi, J. Adsorption of O3, SO2 and SO3 gas molecules on MoS2 monolayers: A computational investigation. Apply Surf. Sci. 2019, 469, 781–791. [Google Scholar] [CrossRef]
- Wang, J.X.; Zhou, Q.; Lu, Z.R.; Gui, Y.G.; Zeng, W. Adsorption of H2O molecule on TM (Au, Ag) doped-MoS2 monolayer: A first-principles study. Phys. E Low-Dimens. Syst. Nanostruct. 2019, 113, 72–78. [Google Scholar] [CrossRef]
- Chen, D.C.; Tang, J.; Zhang, X.X.; Li, Y.; Liu, H.J. Detecting decompositions of sulfur hexafluoride using MoS2 monolayer as gas sensor. IEEE Sens. J. 2019, 19, 9–46. [Google Scholar] [CrossRef]
- Zhang, D.Z.; Jiang, C.X.; Wu, J.F. Layer-by-layer assembled In2O3 nanocubes/flower-like MoS2 nanofilm for room temperature for maldehyde sensing. Sens. Actuators B Chem. 2018, 273, 176–184. [Google Scholar] [CrossRef]
- Wang, J.X.; Zhou, Q.; Lu, Z.R.; Wei, Z.J.; Zeng, W. Gas sensing performances and mechanism at atomic level of Au-MoS2 microspheres. Apply Surf. Sci. 2019, 490, 124–136. [Google Scholar] [CrossRef]
- Zhang, W.X.; Huang, Z.S.; Zhang, W.L.; Li, Y.R. Two-dimensional semiconductors with possible high room temperature mobility. Nano Res. 2014, 7, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Aretouli, K.; Tsipas, P.; Tsoutsou, D.; Marquez-Velasco, J.; Xenogiannopoulou, E.; Giamini, S.A.; Vassalou, E.; Kelaidis, N.; Dimoulas, A. Two-dimensional semiconductor HfSe2 and MoSe2/HfSe2 van der Waals heterostructures by molecular beam epitaxy. Appl. Phys. Lett. 2015, 106, 699. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Zhang, H.J.; Wang, W.; Colombo, L.; Wallace, R.M.; Cho, K. Band alignment of two-dimensional transition metal dichalcogenides: Application in tunnel field effect transistors. Appl. Phys. Lett. 2013, 103, 329. [Google Scholar]
- Wu, P.; Yin, N.Q.; Li, P.; Cheng, W.J.; Min, H. The adsorption and diffusion behavior of noble metal adatoms (Pd, Pt, Cu, Ag and Au) on a MoS2 monolayer: A first-principles study. Phys. Chem. Chem. Phys. 2017, 19, 20713–20722. [Google Scholar] [CrossRef] [PubMed]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Tkatchenko, A.; DiStasio, R.A., Jr.; Head-Gordon, M.; Scheffler, M. Dispersion-corrected Møller-Plesset second-order perturbation theory. J. Mater. Phys. 2009, 131, 171. [Google Scholar]
- Fonseca, G.; Handgraaf, J.; Baerends, E.; Bickelhaupt, F.M. Voronoi deformation density (VDD) charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD methods for charge analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar] [CrossRef]
- Lugo-Solis, A.; Vasiliev, I. Ab initio study of K adsorption on graphene and carbon nanotubes: Role of long-range ionic forces. Phys. Rev. B 2007, 76, 235431. [Google Scholar] [CrossRef]
- Casida, M.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diego, C.A.; Nery, V.E.; Daniela, E.O. Fe–doped graphene nanosheet as an adsorption platform of harmful gas molecules (CO, CO2, SO2 and H2S), and the co–adsorption in O2 environments. Appl. Surf. Sci. 2018, 427, 227–236. [Google Scholar]
- Yao, W.; Zhou, S.; Wang, Z.; Lu, Z.; Hou, C. Antioxidant behaviors of graphene in marine environment: A first–principles simulation. Appl. Surf. Sci. 2020, 499, 143962. [Google Scholar] [CrossRef]
- Sun, S.S.; Meng, F.C.; Wang, H.Y.; Wang, H. Novel two-dimensional semiconductor SnP3: Highest ability, tunable bandgaps and high carrier mobility explored using first-principles calculations. Mater. Chem. 2018, 6, 11890. [Google Scholar] [CrossRef]
- Yuwen, L.H.; Xu, F.; Xue, B.; Luo, Z.M.; Zhang, Q.; Bao, B.Q.; Su, S.; Weng, L.X.; Huang, W.; Wang, L.H. General synthesis of noble metal (Au, Ag, Pd, Pt) nanocrystal modified MoS2 nanosheets and the enhanced catalytic activity of Pd-MoS2 for methanol oxidation. Nanoscale 2014, 6, 5762–5769. [Google Scholar] [CrossRef] [PubMed]
- Michael, S. Air sensitivity of MoS2, MoSe2, MoTe2, HfS2 and HfSe2. J. Appl. Phys. 2016, 120, 125102. [Google Scholar]
- Chen, D.C.; Zhang, X.X.; Tang, J.; Li, Y.; Cui, Z.L.; Zhou, Q. Using Single-Layer HfS2 as Prospective Sensing Device Toward Typical Partial Discharge Gas in SF6-Based Gas-Insulated Switchgear. IEEE Trans. Electron Devices 2019, 66, 1557–9646. [Google Scholar] [CrossRef]
- Chen, D.C.; Zhang, X.X.; Xiong, H.; Tang, J.; Xiao, S.; Zhang, D.Z. A First-Principles Study of the SF6 Decomposed Products Adsorbed Over Defective WS2 Monolayer as Promising Gas Sensing Device. IEEE Trans. Device Mater. Reliab. 2019, 19, 473–483. [Google Scholar] [CrossRef]
- Wang, J.X.; Zhou, Q.; Xu, L.N.; Gao, X.; Zeng, W. Gas sensing mechanism of transformer oil decomposed gases on Ag-MoS2 monolayer: A DFT study. Phys. E Low-Dimens. Syst. Nanostruct. 2020, 118, 113947. [Google Scholar] [CrossRef]
- Liu, Y.P.; Zhou, Q.; Hou, W.J.; Li, J.; Zeng, W. Adsorption properties of Cr modified GaN monolayer for H2, CO, C2H2 and C2H4. Chem. Phys. 2021, 550, 111304. [Google Scholar] [CrossRef]
- Zeng, F.P.; Li, H.T.; Cheng, H.T.; Tang, J.; Liu, Y.L. SF6 Decomposition and Insulation Condition Monitoring of GIE: A Review. High Volt. 2021, 6, 955–966. [Google Scholar] [CrossRef]
- Zeng, F.P.; Feng, X.X.; Chen, X.Y.; Yao, Q.; Miao, Y.L.; Dai, L.J.; Li, Y.; Tang, J. First-principles analysis of Ti3C2Tx MXene as a promising candidate for SF6 decomposition characteristic components sensor. Appl. Surf. Sci. 2022, 578, 152020. [Google Scholar] [CrossRef]
- Yuan, J.H.; Yu, N.N.; Wang, J.F.; Xue, K.H.; Miao, X.S. Design lateral heterostructure of monolayer ZrS2 and HfS2 from first principles calculations. Appl. Surf. Sci. 2018, 436, 919–926. [Google Scholar] [CrossRef]
- Kanazawa, T.; Amemiya, T.; Ishikawa, A.; Upadhyaya, V.; Tsuruta, K.; Tanaka, T.; Miyamoto, Y. Few-layer HfS2 transistors. Sci. Rep. 2016, 6, 22277. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Adsorption Configuration | D(Å) | ||
|---|---|---|---|
| HfS2/CO | C-S 3.568 | −0.180 | 0.012 |
| Ag-HfS2/CO | C-S 1.589 | −0.815 | 0.283 |
| Au-HfS2/CO | C-S 1.717 | 2.142 | −0.336 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, G.; Dai, W.; Zhou, F.; Ma, H.; Wang, S.; Hu, J.; Zhou, Q. Adsorption Characteristics of Carbon Monoxide on Ag- and Au-Doped HfS2 Monolayers Based on Density Functional Theory. Chemosensors 2022, 10, 82. https://doi.org/10.3390/chemosensors10020082
Qian G, Dai W, Zhou F, Ma H, Wang S, Hu J, Zhou Q. Adsorption Characteristics of Carbon Monoxide on Ag- and Au-Doped HfS2 Monolayers Based on Density Functional Theory. Chemosensors. 2022; 10(2):82. https://doi.org/10.3390/chemosensors10020082
Chicago/Turabian StyleQian, Guochao, Weiju Dai, Fangrong Zhou, Hongming Ma, Shan Wang, Jin Hu, and Qu Zhou. 2022. "Adsorption Characteristics of Carbon Monoxide on Ag- and Au-Doped HfS2 Monolayers Based on Density Functional Theory" Chemosensors 10, no. 2: 82. https://doi.org/10.3390/chemosensors10020082
APA StyleQian, G., Dai, W., Zhou, F., Ma, H., Wang, S., Hu, J., & Zhou, Q. (2022). Adsorption Characteristics of Carbon Monoxide on Ag- and Au-Doped HfS2 Monolayers Based on Density Functional Theory. Chemosensors, 10(2), 82. https://doi.org/10.3390/chemosensors10020082