
Cucurbit[8]uril-Based Potentiometric Sensor Coupled to HPLC for Determination of Tetracycline Residues in Milk Samples

 , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Experimental Section
2.1. Chemicals and Solutions
2.2. Sample Preparation
2.3. Apparatus
2.4. Chromatographic Conditions
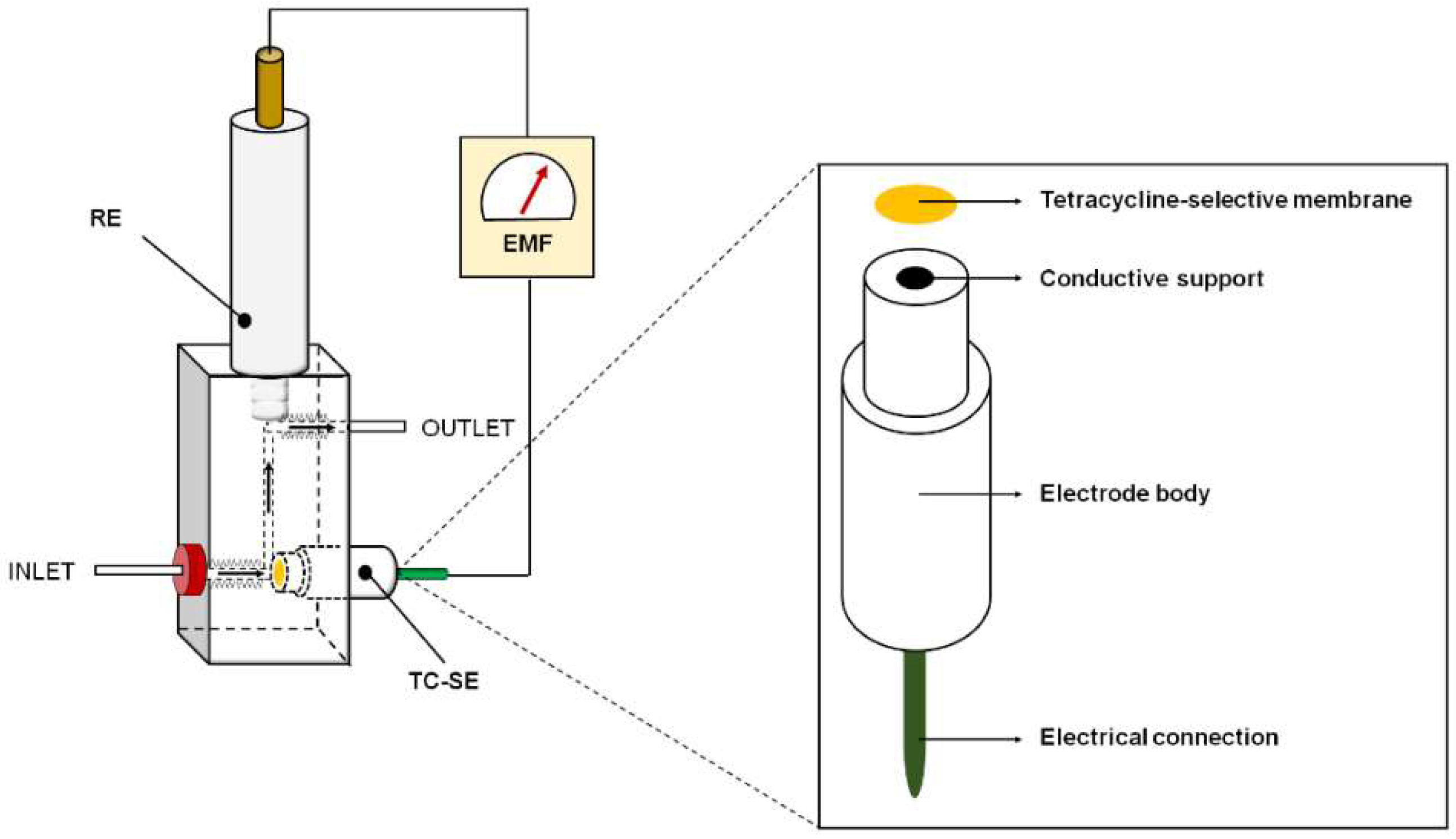
2.5. Detection Cell
2.6. Preparation of the TC-Selective Electrodes
3. Results and Discussion
3.1. Evaluation of the Conventionally Shaped TC-Selective Electrodes
3.2. Evaluation of the Miniaturized TC-Selective Electrode Coupled to HPLC System
3.2.1. Optimization of Mobile Phase Composition
3.2.2. Optimization of Hydrodynamic Conditions
3.2.3. Effect of Carbon Nanotubes
3.3. Method Validation
3.3.1. Analytical Figures of Merit
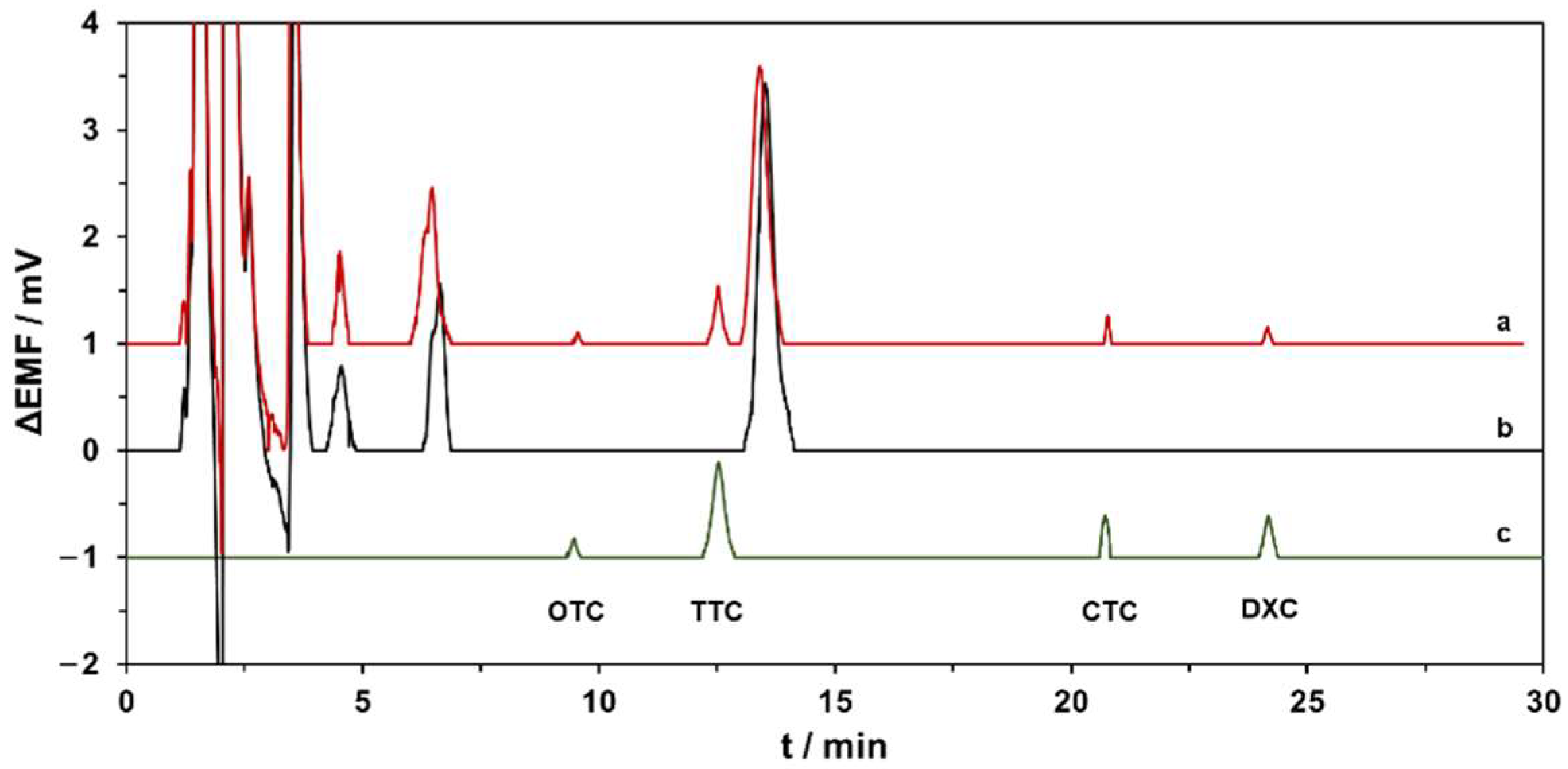
3.3.2. Application in Real Sample Analysis
3.4. Comparison of the Proposed Method with Other Conventional Methods Reported in the Literature
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chopra, I.; Roberts, M. Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Tao, Y.; Chen, D.; Wang, Y.; Yuan, Z. Development of an HPLC–UV method for the simultaneous determination of tetracyclines in muscle and liver of porcine, chicken and bovine with accelerated solvent extraction. Food Chem. 2011, 124, 1131–1138. [Google Scholar] [CrossRef]
- Sczesny, S.; Nau, H.; Hamscher, G. Residue Analysis of Tetracyclines and Their Metabolites in Eggs and in the Environment by HPLC Coupled with a Microbiological Assay and Tandem Mass Spectrometry. J. Agric. Food Chem. 2003, 51, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Zheng, N.; Yu, Z.; Wang, J.; Xu, X.; Qu, X.; Li, S.; Zhang, Y. Simultaneous determination of 38 veterinary antibiotic residues in raw milk by UPLC–MS/MS. Food Chem. 2015, 181, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Mookantsa, S.; Dube, S.; Nindi, M. Development and application of a dispersive liquid–liquid microextraction method for the determination of tetracyclines in beef by liquid chromatography mass spectrometry. Talanta 2016, 148, 321–328. [Google Scholar] [CrossRef]
- Phillips, I. Withdrawal of growth-promoting antibiotics in Europe and its effects in relation to human health. Int. J. Antimicrob. Agents 2007, 30, 101–107. [Google Scholar] [CrossRef]
- Food and Drug Administration. Code of Federal Regulations: Title 21 Food and Drugs; FDA: Silver Spring, MD, USA, 2021; Part 556, Sections 150, 500, and 720. [Google Scholar]
- Codex Alimentarius Commission of the FAO/WHO. Maximum Residue Limits for Veterinary Drugs in Foods CX/MRL; FAO/WHO: Rome, Italy, 2018; pp. 1–46. [Google Scholar]
- Önal, A. Overview on liquid chromatographic analysis of tetracycline residues in food matrices. Food Chem. 2011, 127, 197–203. [Google Scholar] [CrossRef]
- Kargin, I.D.; Sokolova, L.S.; Pirogov, A.V.; Shpigun, O.A. HPLC determination of tetracycline antibiotics in milk with post-column derivatization and fluorescence detection. Inorg. Mater. 2016, 52, 1365–1369. [Google Scholar] [CrossRef]
- Wei, D.; Wu, S.; Zhu, Y. Magnetic solid phase extraction based on graphene oxide/nanoscale zero-valent iron for the determination of tetracyclines in water and milk by using HPLC-MS/MS. RSC Adv. 2017, 7, 44578–44586. [Google Scholar] [CrossRef] [Green Version]
- Igualada, C.; Giraldo, J.; Font, G.; Yusà, V. Validation of a multi-residue UHPLC-HRMS method for antibiotics screening in milk, fresh cheese, and whey. J. Food Compos. Anal. 2022, 106, 104265. [Google Scholar] [CrossRef]
- Zhang, L.; Shi, L.; He, Q.; Li, Y. A rapid multiclass method for antibiotic residues in goat dairy products by UPLC-quadrupole/electrostatic field orbitrap high-resolution mass spectrometry. J. Anal. Sci. Technol. 2021, 12, 14. [Google Scholar] [CrossRef]
- Bobacka, J.; Ivaska, A.; Lewenstam, A. Potentiometric Ion Sensors. Chem. Rev. 2008, 108, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Zdrachek, E.; Bakker, E. Potentiometric Sensing. Anal. Chem. 2018, 91, 2–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, P.W.; Haddad, P.R.; Trojanowicz, M. Potentiometric detection in ion chromatography using a metallic copper indicator electrode. Chromatographia 1985, 20, 179–184. [Google Scholar] [CrossRef]
- Haddad, P.; Alexander, P.; Trojanowicz, M. Ion chromatography of Mg, Ca, Sr and Ba ions using a metallic copper electrode as a potentiometric detector. J. Chromatogr. A 1984, 294, 397–402. [Google Scholar] [CrossRef]
- Haddad, P.; Alexander, P.; Trojanowicz, M. Ion chromatography of inorganic anions with potentiometric detection using a metallic copper electrode. J. Chromatogr. A 1985, 321, 363–374. [Google Scholar] [CrossRef]
- Zielinska, D.; Gil, A.; Pietraszkiewicz, M.; Van De Vijver, D.; Nagels, L. Podand and macrocyclic amine receptors with urea functionalities for potentiometric detection of organic acids in HPLC. Anal. Chim. Acta 2004, 523, 177–184. [Google Scholar] [CrossRef]
- Bazylak, G.; Nagels, L.J. Simultaneous high-throughput determination of clenbuterol, ambroxol and bromhexine in pharmaceutical formulations by HPLC with potentiometric detection. J. Pharm. Biomed. Anal. 2003, 32, 887–903. [Google Scholar] [CrossRef]
- Vissers, B.; Bohets, H.; Everaert, J.; Cool, P.; Vansant, E.; Du Prez, F.; Kauffmann, J.; Nagels, L. Characteristics of new composite- and classical potentiometric sensors for the determination of pharmaceutical drugs. Electrochim. Acta 2006, 51, 5062–5069. [Google Scholar] [CrossRef]
- Daems, D.; Van Camp, G.; Fernandez, M.; Guisez, Y.; Prinsen, E.; Nagels, L. Use of potentiometric detection in (ultra) high performance liquid chromatography and modelling with adsorption/desorption binding kinetics. Anal. Chim. Acta 2013, 777, 25–31. [Google Scholar] [CrossRef]
- Füglein, R.; Bräuchle, C.; Hampp, N. Ion-Selective Electrodes for the Determination of the Antibiotic Drug Chlortetracycline. Anal. Sci. 1994, 10, 959–962. [Google Scholar] [CrossRef] [Green Version]
- El-Ansary, A.; Issa, Y.; Tag-Eldin, A. Tetracycline Sensitive Membrane Electrodes Based on Poly (Vinyl Chloride) Matrices and their use in Drug Analysis. Anal. Lett. 1999, 32, 2177–2190. [Google Scholar] [CrossRef]
- Moreira, F.T.; Kamel, A.H.; Guerreiro, J.R.; Sales, M.G.F. Man-tailored biomimetic sensor of molecularly imprinted materials for the potentiometric measurement of oxytetracycline. Biosens. Bioelectron. 2010, 26, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Moreira, F.T.C.; Guerreiro, J.R.L.; Azevedo, V.L.; Kamel, A.H.; Sales, M.G.F. New biomimetic sensors for the determination of tetracycline in biological samples: Batch and flow mode operations. Anal. Methods 2010, 2, 2039–2045. [Google Scholar] [CrossRef] [Green Version]
- Gai, P.; Guo, Z.; Yang, F.; Duan, J.; Hao, T.; Wang, S. Highly-sensitive ion selective electrode based on molecularly imprinted polymer particles for determination of tetracycline in aqueous samples. Russ. J. Electrochem. 2011, 47, 940–947. [Google Scholar] [CrossRef]
- Amorim, C.; Araújo, A.; Montenegro, M.; Silva, V. Cyclodextrin-based potentiometric sensors for midazolam and diazepam. J. Pharm. Biomed. Anal. 2008, 48, 1064–1069. [Google Scholar] [CrossRef] [PubMed]
- Shishkanova, T.; Sykora, D.; Sessler, J.; Král, V. Potentiometric response and mechanism of anionic recognition of heterocalixarene-based ion selective electrodes. Anal. Chim. Acta 2007, 587, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Ali, T.A.; Ali, T.A.; Mohamed, G.G.; Mohamed, G.G.; El-Sonbati, A.Z.; El-Sonbati, A.Z.; Diab, M.A.; Diab, M.A.; Elkfass, A.M.; Elkfass, A.M. A Potentiometric Sensor for Determination of Doxycycline Hydrochloride in Pharmaceutical Preparation and Biological Fluids. Russ. J. Electrochem. 2018, 54, 1081–1095. [Google Scholar] [CrossRef]
- Cunha, C.O.; Silva, R.C.R.; Amorim, C.G.; Júnior, S.A.; Araújo, A.N.; Montenegro, M.C.B.S.M.; Silva, V.L. Tetracycline Potentiometric Sensor Based on Cyclodextrin for Pharmaceuticals and Waste Water Analysis. Electroanalysis 2010, 22, 2967–2972. [Google Scholar] [CrossRef]
- Barrow, S.J.; Kasera, S.; Rowland, M.J.; del Barrio, J.; Scherman, O.A. Cucurbituril-Based Molecular Recognition. Chem. Rev. 2015, 115, 12320–12406. [Google Scholar] [CrossRef] [Green Version]
- Lagona, J.; Mukhopadhyay, P.; Chakrabarti, S.; Isaacs, L. The Cucurbit[n]uril Family. Angew. Chem. Int. Ed. 2005, 44, 4844–4870. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Assaf, K.I.; Nau, W.M. Applications of Cucurbiturils in Medicinal Chemistry and Chemical Biology. Front. Chem. 2019, 7, 619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.-X.; Qiu, Y.-Q.; Du, L.-M.; Li, C.-F.; Guo, M. Determination of ranitidine, nizatidine, and cimetidine by a sensitive fluorescent probe. Analyst 2011, 136, 4168–4173. [Google Scholar] [CrossRef] [PubMed]
- Occello, V.N.S.; Veglia, A.V. Cucurbit[6]uril nanocavity as an enhanced spectrofluorimetric method for the determination of pyrene. Anal. Chim. Acta 2011, 689, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Guo, J.; Shi, L. A ultrastable cucurbit[6]uril-based multifunctional supramolecular assembly for efficient detection of nitroaromatic compounds and antibiotics. New J. Chem. 2021, 45, 18221–18228. [Google Scholar] [CrossRef]
- del Pozo, M.; Mejías, J.; Hernández, P.; Quintana, C. Cucurbit[8]uril-based electrochemical sensors as detectors in flow injection analysis. Application to dopamine determination in serum samples. Sens. Actuators B Chem. 2014, 193, 62–69. [Google Scholar] [CrossRef]
- Liu, J.; Lambert, H.; Zhang, Y.-W.; Lee, T.-C. Rapid Estimation of Binding Constants for Cucurbit[8]uril Ternary Complexes Using Electrochemistry. Anal. Chem. 2021, 93, 4223–4230. [Google Scholar] [CrossRef]
- Cheng, W.; Ma, J.; Kong, D.; Zhang, Z.; Khan, A.; Yi, C.; Hu, K.; Yi, Y.; Li, J. One step electrochemical detection for matrix metalloproteinase 2 based on anodic stripping of silver nanoparticles mediated by host-guest interactions. Sens. Actuators B Chem. 2020, 330, 129379. [Google Scholar] [CrossRef]
- Amorim, C.G.; Araújo, A.; Montenegro, M.D.C. Use of Cucurbit[6]uril as Ionophore in Ion Selective Electrodes for Etilefrine Determination in Pharmaceuticals. Electroanalysis 2019, 31, 2171–2178. [Google Scholar] [CrossRef]
- Ferreira, C.; Palmeira, A.; Sousa, E.; Amorim, C.G.; Araújo, A.N.; Montenegro, M.C. Supramolecular Atropine Potentiometric Sensor. Sensors 2021, 21, 5879. [Google Scholar] [CrossRef]
- Gil, R.L.; Amorim, C.G.; Montenegro, M.C.; Araújo, A.N. Determination of biogenic amines in tomato by ion-pair chromatography coupled to an amine-selective potentiometric detector. Electrochim. Acta 2021, 378, 138134. [Google Scholar] [CrossRef]
- Gil, R.L.; Amorim, C.G.; Montenegro, M.C.; Araújo, A.N. HPLC-potentiometric method for determination of biogenic amines in alcoholic beverages: A reliable approach for food quality control. Food Chem. 2021, 372, 131288. [Google Scholar] [CrossRef] [PubMed]
- Waters. 2013. Available online: https://www.waters.com/webassets/cms/library/docs/720004582en.pdf (accessed on 1 July 2021).
- Wen, Y.; Wang, Y.; Feng, Y.-Q. Simultaneous residue monitoring of four tetracycline antibiotics in fish muscle by in-tube solid-phase microextraction coupled with high-performance liquid chromatography. Talanta 2006, 70, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Cuartero, M.; Amorim, C.G.; Araújo, A.N.; Ortuño, J.A.; Montenegro, M.C. A SO2-selective electrode based on a Zn-porphyrin for wine analysis. Anal. Chim. Acta 2013, 787, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Tóth, K.; Fucskó, J.; Lindner, E.; Fehér, Z.; Pungor, E. Potentiometric detection in flow analysis. Anal. Chim. Acta 1986, 179, 359–370. [Google Scholar] [CrossRef]
- Buck, R.P.; Lindner, E. Recommendations for nomenclature of ion-selective electrodes (IUPAC Recommendations 1994). Pure Appl. Chem. 1994, 66, 2527–2536. [Google Scholar] [CrossRef]
- Poels, I.; Nagels, L. Potentiometric detection of amines in ion chromatography using macrocycle-based liquid membrane electrodes. Anal. Chim. Acta 2001, 440, 89–98. [Google Scholar] [CrossRef]
- Zielinska, D.; Poels, I.; Pietraszkiewicz, M.; Radecki, J.; Geise, H.; Nagels, L. Potentiometric detection of organic acids in liquid chromatography using polymeric liquid membrane electrodes incorporating macrocyclic hexaamines. J. Chromatogr. A 2001, 915, 25–33. [Google Scholar] [CrossRef]
- Amorim, C.; Souza, R.; Araújo, A.; Montenegro, M.; Silva, V. SI lab-on-valve analysis of histamine using potentiometric detection for food quality control. Food Chem. 2010, 122, 871–876. [Google Scholar] [CrossRef]
- Tóth, K.; Stulík, K.; Kutner, W.; Fehér, Z.; Lindner, E. Electrochemical detection in liquid flow analytical techniques: Characterization and classification (IUPAC Technical Report). Pure Appl. Chem. 2004, 76, 1119–1138. [Google Scholar] [CrossRef]
- Montenegro, M.C.B.S.M.; Araújo, A.N. Flow Potentiometry. In Advances in Flow Analysis; Trojanowicz, M., Ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Amorim, C.G.; Araújo, A.N.; Montenegro, M.C.B.S.M.; Silva, V.L. Sequential Injection Lab-on-Valve Procedure for the Determination of Amantadine Using Potentiometric Methods. Electroanalysis 2007, 19, 2227–2233. [Google Scholar] [CrossRef]
- Ahammad, A.J.S.; Lee, J.-J.; Rahman, A. Electrochemical Sensors Based on Carbon Nanotubes. Sensors 2009, 9, 2289–2319. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Anthis, A.H.C.; Ghahraman Afshar, M.; Pankratova, N.; Cuartero, M.; Crespo, G.A.; Bakker, E. All-Solid-State Potentiometric Sensors with a Multiwalled Carbon Nanotube Inner Transducing Layer for Anion Detection in Environmental Samples. Anal. Chem. 2015, 87, 8640–8645. [Google Scholar] [CrossRef] [PubMed]
- Crespo, G.A.; Macho, S.; Rius, F.X. Ion-Selective Electrodes Using Carbon Nanotubes as Ion-to-Electron Transducers. Anal. Chem. 2008, 80, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Desimoni, E.; Brunetti, B. About Estimating the Limit of Detection by the Signal to Noise Approach. Pharm. Anal. Acta 2015, 6, 4. [Google Scholar] [CrossRef]
- Daems, D.; De Wael, K.; Vissenberg, K.; Van Camp, G.; Nagels, L. Potentiometric sensors doped with biomolecules as a new approach to small molecule/biomolecule binding kinetics analysis. Biosens. Bioelectron. 2014, 54, 515–520. [Google Scholar] [CrossRef]
- ICH. ICH Harmonized Tripartite Guideline: Validation of Analytical Procedures: Text and Methodology Q2 (R1); ICH: Geneva, Switzerland, 2005. [Google Scholar]
- Eurpean Commission. Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Communities 2002, 221, 8–36. [Google Scholar]
- Sekula, J.; Everaert, J.; Bohets, H.; Vissers, B.; Pietraszkiewicz, M.; Pietraszkiewicz, O.; Du Prez, F.; Vanhoutte, K.; Prus, P.; Nagels, L.J. Coated Wire Potentiometric Detection for Capillary Electrophoresis Studied Using Organic Amines, Drugs, and Biogenic Amines. Anal. Chem. 2006, 78, 3772–3779. [Google Scholar] [CrossRef]
- Rossi, R.; Saluti, G.; Moretti, S.; Diamanti, I.; Giusepponi, D.; Galarini, R. Multiclass methods for the analysis of antibiotic residues in milk by liquid chromatography coupled to mass spectrometry: A review. Food Addit. Contam. Part A 2017, 35, 241–257. [Google Scholar] [CrossRef]
- Vuran, B.; Ulusoy, H.I.; Sarp, G.; Yilmaz, E.; Morgül, U.; Kabir, A.; Tartaglia, A.; Locatelli, M.; Soylak, M. Determination of chloramphenicol and tetracycline residues in milk samples by means of nanofiber coated magnetic particles prior to high-performance liquid chromatography-diode array detection. Talanta 2021, 230, 122307. [Google Scholar] [CrossRef]
- Tang, H.-Z.; Wang, Y.-H.; Li, S.; Wu, J.; Gao, Z.-X.; Zhou, H.-Y. Development and application of magnetic solid phase extraction in tandem with liquid–liquid extraction method for determination of four tetracyclines by HPLC with UV detection. J. Food Sci. Technol. 2020, 57, 2884–2893. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Qiu, Y.; Liu, X.; Ji, C. Determination of Multi-Residues of Tetracyclines and Their Metabolites in Milk by High Performance Liquid Chromatography-Tandem Positive-ion Electrospray Ionization Mass Spectrometry. Chin. J. Anal. Chem. 2006, 34, 1255–1259. [Google Scholar] [CrossRef]
- Mamani, M.C.V.; Reyes, F.; Rath, S. Multiresidue determination of tetracyclines, sulphonamides and chloramphenicol in bovine milk using HPLC-DAD. Food Chem. 2009, 117, 545–552. [Google Scholar] [CrossRef]
- Kaynaker, M.; Antep, M.; Merdivan, M. Determination of Tetracyclines in Milk, Eggs and Honey Using in-situ Ionic Liquid Based Dispersive Liquid–Liquid Microextraction. J. Anal. Chem. 2018, 73, 23–29. [Google Scholar] [CrossRef]
- Al-Afy, N.; Sereshti, H.; Hijazi, A.; Nodeh, H.R. Determination of three tetracyclines in bovine milk using magnetic solid phase extraction in tandem with dispersive liquid-liquid microextraction coupled with HPLC. J. Chromatogr. B 2018, 1092, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, J.; Li, C.; Chen, L. Analysis of tetracyclines from milk powder by molecularly imprinted solid-phase dispersion based on a metal-organic framework followed by ultra high performance liquid chromatography with tandem mass spectrometry. J. Sep. Sci. 2018, 41, 2604–2612. [Google Scholar] [CrossRef]
- de Faria, H.; Rosa, M.A.; Silveira, A.T.; Figueiredo, E.C. Direct extraction of tetracyclines from bovine milk using restricted access carbon nanotubes in a column switching liquid chromatography system. Food Chem. 2017, 225, 98–106. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
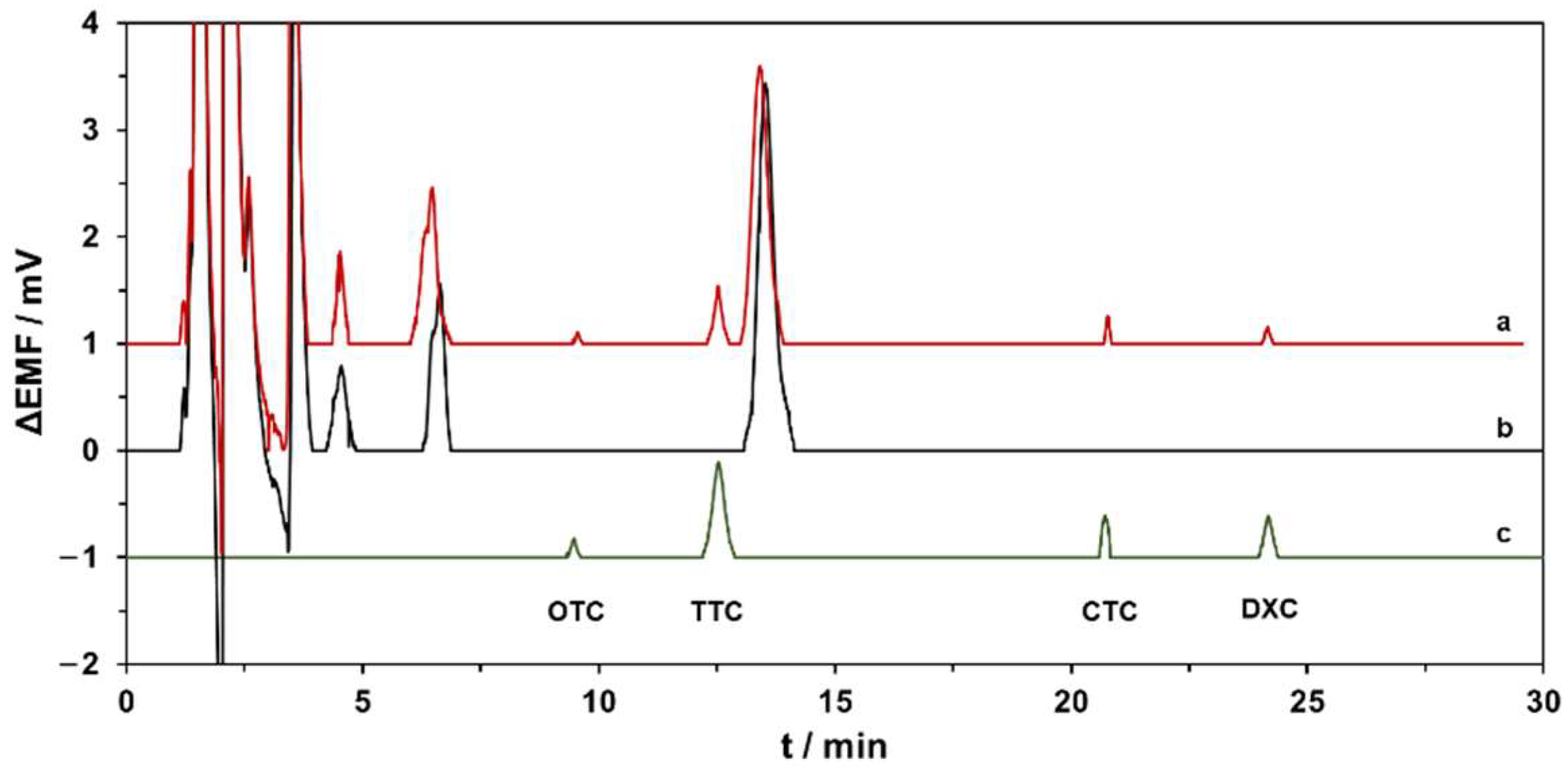{kind=link}
| Components in the ISM (wt%) | |||||
|---|---|---|---|---|---|
| ISM | PVC | FNDPE | CB[8] | TCPB | MWCNTs |
| A | 31.00 | 68.97 | – | 0.03 | – |
| B | 31.00 | 68.07 | 0.90 | 0.03 | – |
| C | 31.00 | 67.07 | 0.90 | 0.03 | 1.00 |
| D | 31.00 | 66.07 | 0.90 | 0.03 | 2.00 |
| Calibration Parameter | ISM | CTC | DXC | OTC | TTC |
|---|---|---|---|---|---|
| Slope a, mV dec−1 | A | 62.5 ± 1.1 | 58.6 ± 0.9 | 54.0 ± 0.6 | 52.9 ± 1.6 |
| B | 63.4 ± 2.1 | 59.0 ± 0.1 | 56.2 ± 0.9 | 53.8 ± 2.2 | |
| Linear response range, M | A | 3.0 × 10−7–1.0 × 10−3 | 3.0 × 10−7–1.0 × 10−3 | 6.0 × 10−6–1.0 × 10−3 | 1.0 × 10−6–1.0 × 10−3 |
| B | 3.0 × 10−7–1.0 × 10−3 | 3.0 × 10−7–1.0 × 10−3 | 6.0 × 10−6–1.0 × 10−3 | 1.0 × 10−6–1.0 × 10−3 | |
| Limit of detection a, M | A | (1.3 ± 0.6) × 10−7 | (1.7 ± 0.3) × 10−7 | (2.9 ± 0.0) × 10−6 | (3.6 ± 0.2) × 10−7 |
| B | (1.5 ± 1.3) × 10−7 | (1.8 ± 0.7) × 10−7 | (2.5 ± 0.0) × 10−6 | (4.3 ± 0.5) × 10−7 |
| Calibration Parameter | ISM | MWCNTs (wt%) | OTC | TTC | CTC | DXC |
|---|---|---|---|---|---|---|
| Slope a, mV dec−1 | A | 0.0 | 11.2 ± 0.3 | 24.9 ± 5.8 | 34.7 ± 6.2 | 21.0 ± 7.0 |
| B | 1.0 | 14.5 ± 0.3 | 31.0 ± 3.4 | 53.1 ± 5.5 | 41.3 ± 1.4 | |
| C | 2.0 | 13.9 ± 2.3 | 33.0 ± 4.5 | 52.4 ± 6.5 | 41.5 ± 3.0 | |
| Limit of detection b, M | A | 0.0 | 1.0 × 10−7 | 1.0 × 10−7 | 1.0 × 10−7 | 1.0 × 10−7 |
| B | 1.0 | 1.0 × 10−7 | 3.0 × 10−8 | 3.0 × 10−8 | 3.0 × 10−8 | |
| C | 2.0 | 1.0 × 10−7 | 1.0 × 10−7 | 1.0 × 10−7 | 1.0 × 10−7 | |
| Precision c, RSD% | A | 0.0 | 0.4 | 18.6 | 19.7 | 42.1 |
| B | 1.0 | 5.8 | 0.2 | 3.4 | 1.8 | |
| C | 2.0 | 4.8 | 2.5 | 5.2 | 1.7 |
| Validation Parameter | OTC | TTC | CTC | DXC |
|---|---|---|---|---|
| Calibration curve | tR vs. [TC] µM | |||
| Linear range, M | 2.0 × 10−7–1.0 × 10−5 | 1.0 × 10−7–1.0 × 10−5 | 1.0 × 10−7–1.0 × 10−5 | 1.0 × 10−7–1.0 × 10−5 |
| Slope | 63.4 ± 2.1 | 59.0 ± 0.1 | 56.2 ± 0.9 | 53.8 ± 2.2 |
| Intercept | 0.9987 ± 0.0003 | 0.9988 ± 0.0010 | 0.9993 ± 0.0001 | 0.9991 ± 0.0000 |
| R2 | 0.9987 ± 0.0012 | 0.9986 ± 0.0006 | 0.9973 ± 0.0026 | 0.9983 ± 0.0027 |
| LOD, M (μg L−1) | 1.0 × 10−7 (46.0) | 3.0 × 10−8 (13.3) | 3.0 × 10−8 (14.4) | 3.0 × 10−8 (13.3) |
| LOQ, M (μg L−1) | 2.0 × 10−7 (92.1) | 1.0 × 10−7 (44.4) | 1.0 × 10−7 (47.9) | 1.0 × 10−7 (44.4) |
| Precision, RSD% | ||||
| Intra-day | ||||
| 3.0 × 10−7 M | 10.2 | 7.9 | 12.5 | 7.4 |
| 1.0 × 10−6 M | 6.0 | 8.3 | 6.7 | 6.6 |
| 1.0 × 10−5 M | 6.6 | 4.1 | 7.1 | 4.9 |
| Inter-day | ||||
| 3.0 × 10−7 M | 4.4 | 11.7 | 13.1 | 13.5 |
| 1.0 × 10−6 M | 1.6 | 10.0 | 7.2 | 4.9 |
| 1.0 × 10−5 M | 1.7 | 3.7 | 5.1 | 7.2 |
| Inter-electrode | ||||
| 3.0 × 10−7 M | 12.0 | 10.7 | 5.1 | 2.9 |
| 1.0 × 10−6 M | 5.9 | 9.4 | 8.3 | 12.9 |
| 1.0 × 10−5 M | 6.2 | 3.7 | 8.9 | 6.2 |
| Analytes | Added μg L−1 | UHT Skimmed Milk | UHT Semi-Skimmed Milk | Fresh Semi-Skimmed Milk | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Found μg L−1 | RSD % | RV % | Found μg L−1 | RSD % | RV % | Found μg L−1 | RSD % | RV % | ||
| OTC | 0 | N.D. | – | – | N.D. | – | – | N.D. | – | – |
| 50 | <LOQ | – | – | <LOQ | – | – | <LOQ | – | – | |
| 100 | 100.1 ± 3.2 | 3.2 | 100.1 | 103.3 ± 4.9 | 4.8 | 103.3 | 98.0 ± 6.9 | 7.1 | 98.0 | |
| 200 | 204.6 ± 10.7 | 5.2 | 102.3 | 178.0 ± 6.2 | 3.5 | 89.0 | 180.4 ± 15.0 | 8.3 | 90.2 | |
| TTC | 0 | N.D | – | – | N.D | – | – | N.D | – | – |
| 50 | 49.1 ± 4.2 | 8.6 | 98.3 | 50.1 ± 1.4 | 2.8 | 100.1 | 51.9 ± 1.6 | 3.1 | 103.8 | |
| 100 | 91.0 ± 6.1 | 6.7 | 91.0 | 87.7 ± 5.3 | 6.0 | 87.7 | 84.1 ± 3.2 | 3.8 | 84.1 | |
| 200 | 174.3 ± 16.8 | 9.6 | 87.1 | 194.4 ± 8.6 | 4.4 | 97.2 | 183.1 ± 13.9 | 7.6 | 91.5 | |
| CTC | 0 | N.D | – | – | N.D | – | – | N.D | – | – |
| 50 | 46.9 ± 3.0 | 6.4 | 93.8 | 47.6 ± 1.2 | 2.6 | 95.2 | 49.2 ± 3.0 | 6.1 | 98.4 | |
| 100 | 88.5 ± 7.9 | 8.9 | 88.5 | 88.7 ± 2.0 | 2.3 | 88.7 | 103.9 ± 4.5 | 4.3 | 103.9 | |
| 200 | 197.0 ± 15.4 | 7.8 | 98.5 | 192.4 ± 5.6 | 2.9 | 96.2 | 185.4 ± 11.5 | 6.2 | 92.7 | |
| DXC | 0 | N.D | – | – | N.D | – | – | N.D | – | – |
| 50 | 45.9 ± 3.2 | 6.9 | 91.8 | 48.5 ± 2.1 | 4.3 | 96.9 | 44.1 ± 1.9 | 4.3 | 88.2 | |
| 100 | 95.6 ± 2.0 | 2.1 | 95.6 | 108.5 ± 2.1 | 1.9 | 108.5 | 88.1 ± 3.1 | 3.5 | 88.1 | |
| 200 | 190.1 ± 12.2 | 6.4 | 95.0 | 162.7 ± 6.8 | 4.2 | 81.3 | 177.4 ± 10.7 | 6.0 | 88.7 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil, R.L.; Amorim, C.M.P.G.; Montenegro, M.d.C.B.S.M.; Araújo, A.N. Cucurbit[8]uril-Based Potentiometric Sensor Coupled to HPLC for Determination of Tetracycline Residues in Milk Samples. Chemosensors 2022, 10, 98. https://doi.org/10.3390/chemosensors10030098
Gil RL, Amorim CMPG, Montenegro MdCBSM, Araújo AN. Cucurbit[8]uril-Based Potentiometric Sensor Coupled to HPLC for Determination of Tetracycline Residues in Milk Samples. Chemosensors. 2022; 10(3):98. https://doi.org/10.3390/chemosensors10030098
Chicago/Turabian StyleGil, Renato L., Célia M. P. G. Amorim, Maria da Conceição B. S. M. Montenegro, and Alberto N. Araújo. 2022. "Cucurbit[8]uril-Based Potentiometric Sensor Coupled to HPLC for Determination of Tetracycline Residues in Milk Samples" Chemosensors 10, no. 3: 98. https://doi.org/10.3390/chemosensors10030098
APA StyleGil, R. L., Amorim, C. M. P. G., Montenegro, M. d. C. B. S. M., & Araújo, A. N. (2022). Cucurbit[8]uril-Based Potentiometric Sensor Coupled to HPLC for Determination of Tetracycline Residues in Milk Samples. Chemosensors, 10(3), 98. https://doi.org/10.3390/chemosensors10030098