

Ultrafast Electrochemical Self-Doping of Anodic Titanium Dioxide Nanotubes for Enhanced Electroanalytical and Photocatalytic Performance
 ,
,  , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. TiO2 Nanotubes Production
- Heating at 30 °C/min until reaching 420 °C.
- Heating at 5 °C/min until reaching 450 °C.
- Maintaining an isothermal condition at 450 °C for 1 h.
- The heat treatment was conducted in a quartz tubular furnace in the presence of air. Following calcination, the samples were allowed to cool naturally within the furnace and were removed once they reached room temperature.
- The entire procedure to fabricate TiO2 was optimized in a previous work [36]. As-prepared materials’ results mechanically stable and no issues were related to air exposure.
2.2. Electrochemical Self-Doping of TiO2 Nanotubes and Preliminary Characterization
2.3. Electrochemical Characterization
- F (Faraday’s constant) = 96,485 C/mol;
- D (diffusion coefficient) = 7.6 × 10−6 cm2/s;
- R (gas constant) = 8.314 J/K × mol;
- T (temperature) = 298.15 K;
- n = Number of electrons exchanged;
- v = Scan rate (mV/s).
2.4. Determination of H2O2 Content through Voltammetric Measurements
2.5. Photocatalytic Test
3. Results and Discussion
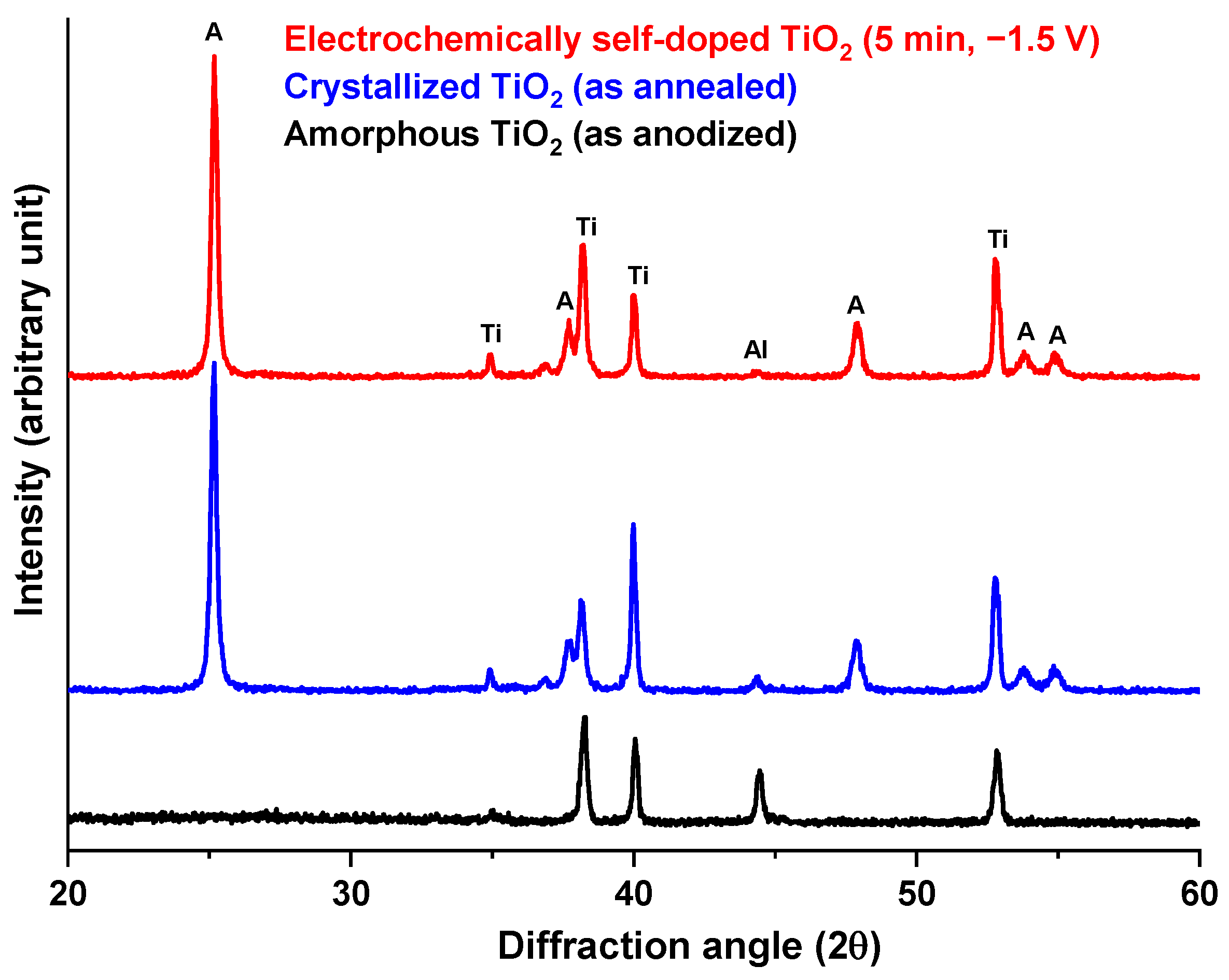
3.1. Anodic TiO2 Nanotubes Fabrication and Morphological Characterization
3.2. Electrochemical Self-Doping of TiO2 Nanotubes
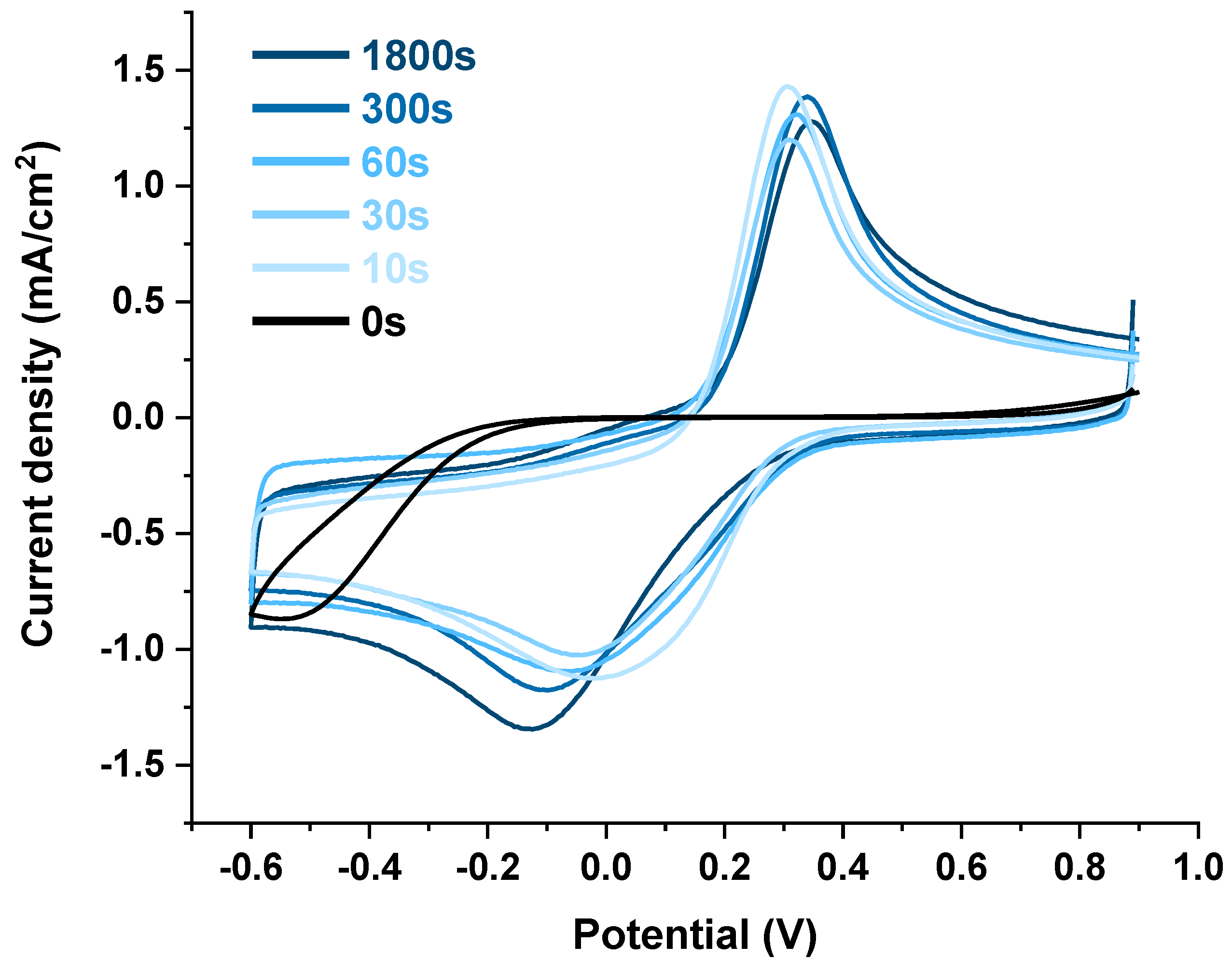
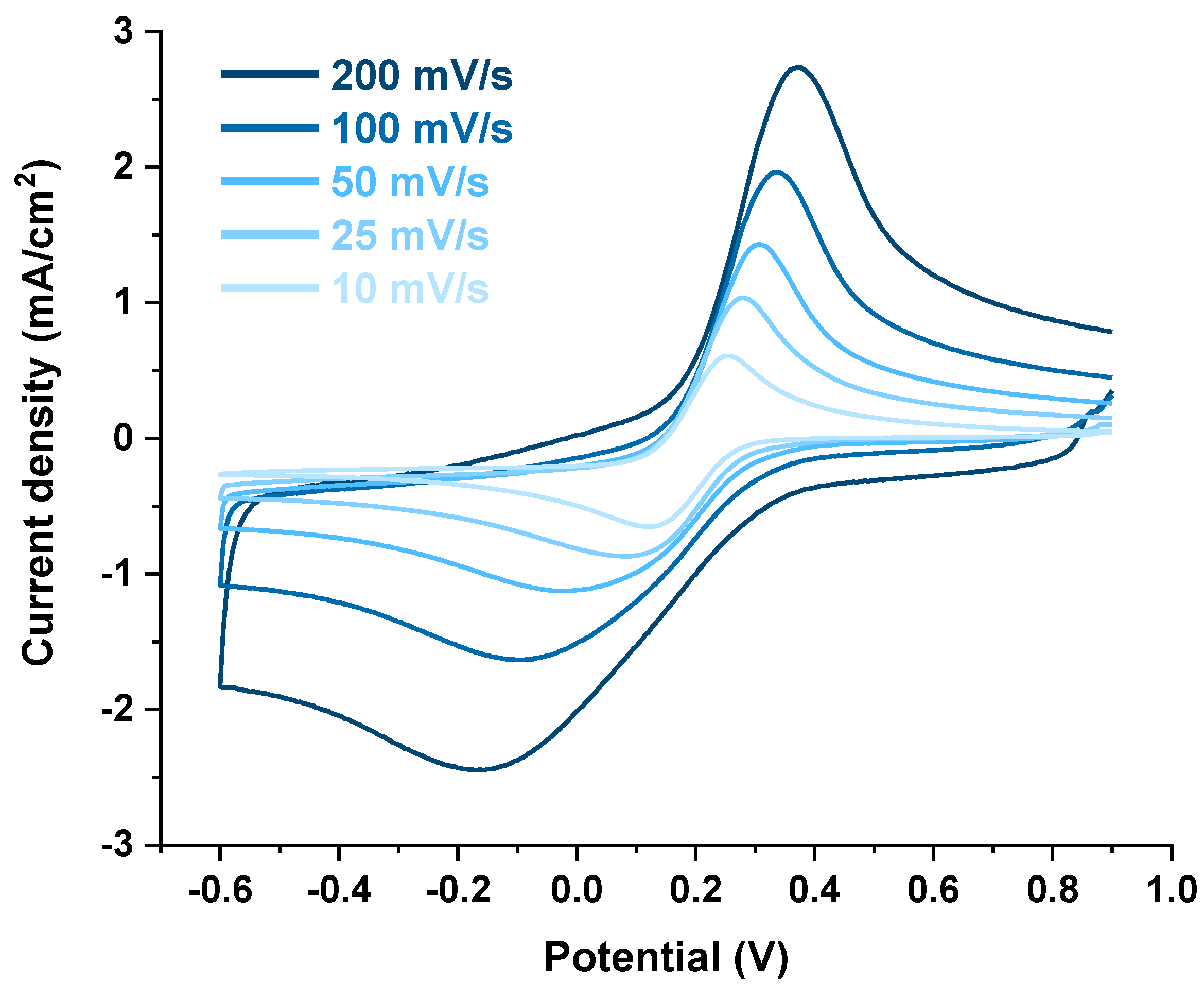
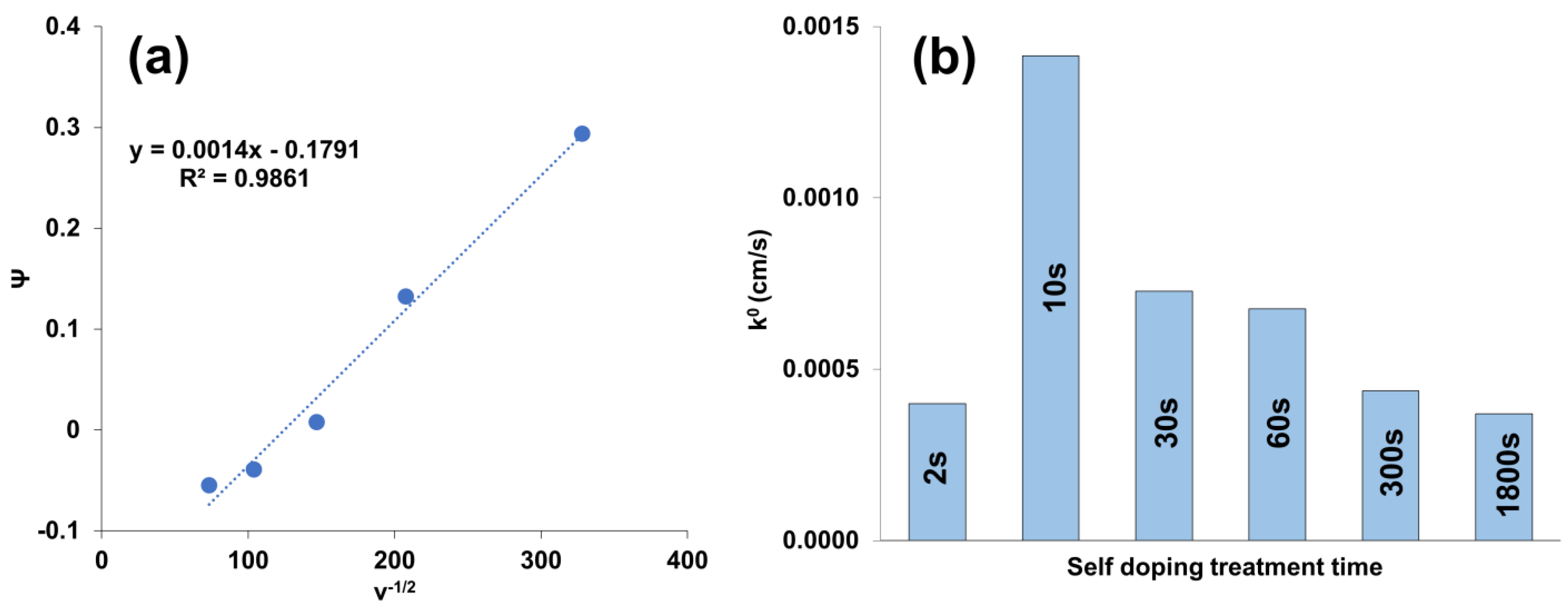
3.3. Electrochemical Characterization
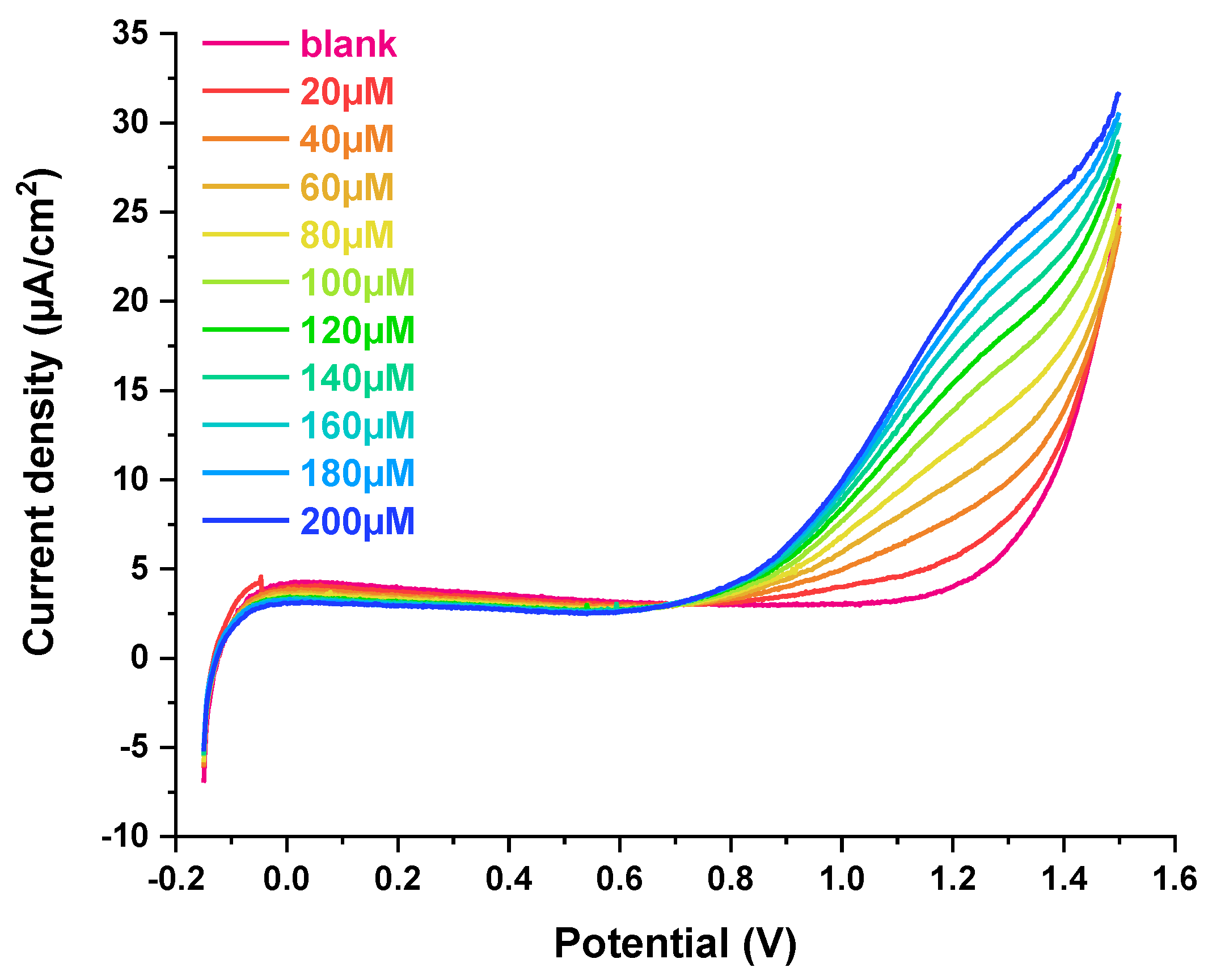
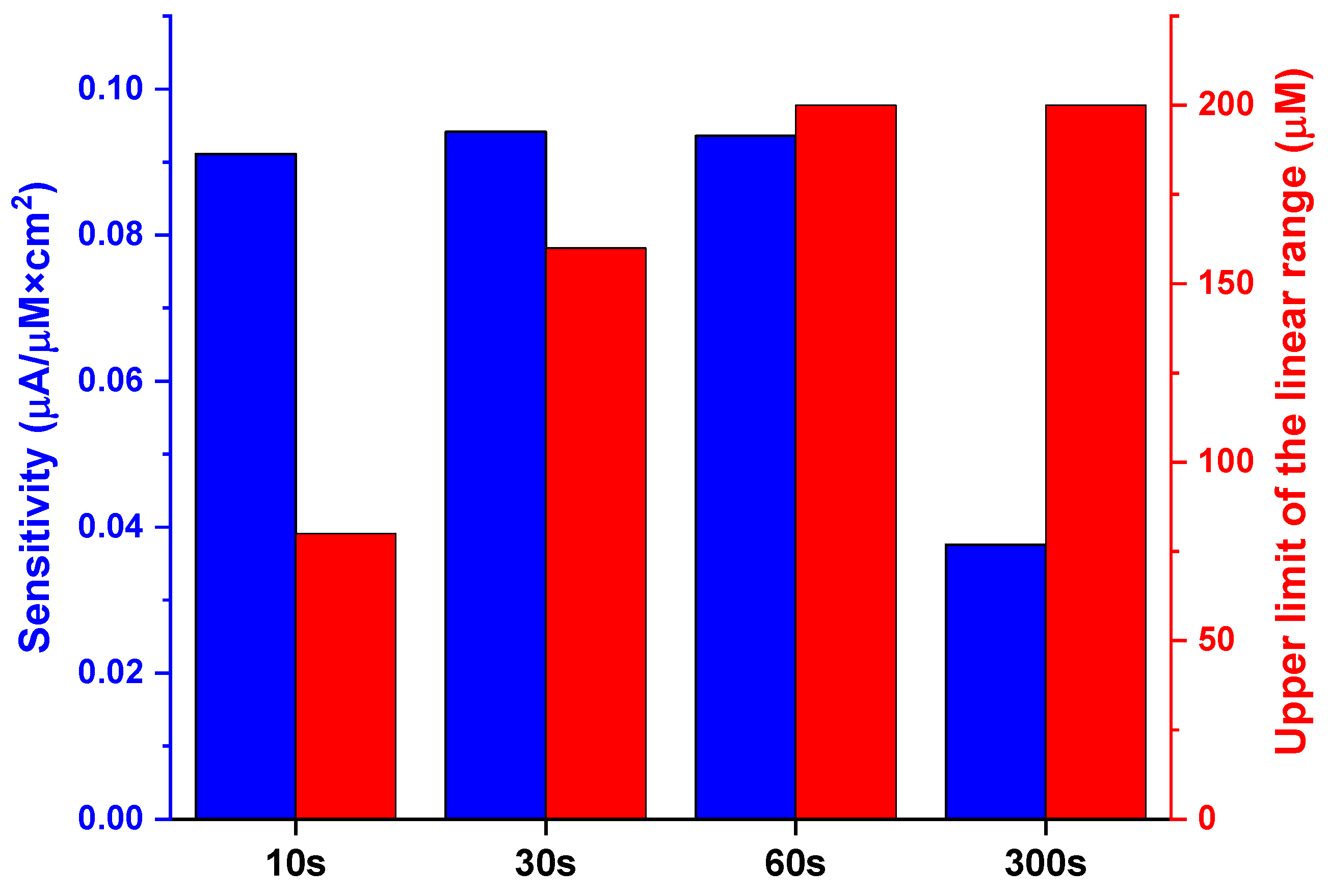
3.4. Voltametric Detection of H2O2
3.5. Photocatalytic Methylene Blue Degradation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, H.; Song, S.; Xu, X.; Dai, J.; Yu, J.; Zhou, W.; Shao, Z.; Jung, W. Recent Progress on Structurally Ordered Materials for Electrocatalysis. Adv. Energy Mater. 2021, 11, 2101937. [Google Scholar] [CrossRef]
- Tong, H.; Ouyang, S.; Bi, Y.; Umezawa, N.; Oshikiri, M.; Ye, J. Nano-photocatalytic Materials: Possibilities and Challenges. Adv. Mater. 2012, 24, 229–251. [Google Scholar] [CrossRef] [PubMed]
- Riboni, F.; Nguyen, N.T.; So, S.; Schmuki, P. Aligned metal oxide nanotube arrays: Key-aspects of anodic TiO2 nanotube formation and properties. Nanoscale Horiz. 2016, 1, 445–466. [Google Scholar] [CrossRef]
- Kowalski, D.; Kim, D.; Schmuki, P. TiO2 nanotubes, nanochannels and mesosponge: Self-organized formation and applications. Nano Today 2013, 8, 235–264. [Google Scholar] [CrossRef]
- Paramasivam, I.; Jha, H.; Liu, N.; Schmuki, P. A Review of Photocatalysis using Self-organized TiO2 Nanotubes and Other Ordered Oxide Nanostructures. Small 2012, 8, 3073–3103. [Google Scholar] [CrossRef] [PubMed]
- Valeeva, A.A.; Dorosheva, I.B.; Kozlova, E.A.; Sushnikova, A.A.; Kurenkova, A.Y.; Saraev, A.A.; Schroettner, H.; Rempel, A.A. Solar photocatalysts based on titanium dioxide nanotubes for hydrogen evolution from aqueous solutions of ethanol. Int. J. Hydrogen Energy 2021, 46, 16917–16924. [Google Scholar] [CrossRef]
- Ge, M.; Li, Q.; Cao, C.; Huang, J.; Li, S.; Zhang, S.; Chen, Z.; Zhang, K.; Al-Deyab, S.S.; Lai, Y. One-dimensional TiO2 Nanotube Photocatalysts for Solar Water Splitting. Adv. Sci. 2017, 4, 1600152. [Google Scholar] [CrossRef] [PubMed]
- Pinna, M.; Wei, A.W.W.; Spanu, D.; Will, J.; Yokosawa, T.; Spiecker, E.; Recchia, S.; Schmuki, P.; Altomare, M. Amorphous NiCu Thin Films Sputtered on TiO2 Nanotube Arrays: A Noble-Metal Free Photocatalyst for Hydrogen Evolution. ChemCatChem 2022, 14, e202201052. [Google Scholar] [CrossRef]
- Hou, X.; Aitola, K.; Jiang, H.; Lund, P.D.; Li, Y. Reduced TiO2 nanotube array as an excellent cathode for hydrogen evolution reaction in alkaline solution. Catal. Today 2022, 402, 3–9. [Google Scholar] [CrossRef]
- Spanu, D.; Bestetti, A.; Hildebrand, H.; Schmuki, P.; Altomare, M.; Recchia, S. Photocatalytic reduction and scavenging of Hg(II) over templated-dewetted Au on TiO2 nanotubes. Photochem. Photobiol. Sci. 2019, 18, 1046–1055. [Google Scholar] [CrossRef]
- Alsheheri, S.Z. Nanocomposites containing titanium dioxide for environmental remediation. Des. Monomers Polym. 2021, 24, 22–45. [Google Scholar] [CrossRef] [PubMed]
- Weon, S.; Choi, W. TiO2 Nanotubes with Open Channels as Deactivation-Resistant Photocatalyst for the Degradation of Volatile Organic Compounds. Environ. Sci. Technol. 2016, 50, 2556–2563. [Google Scholar] [CrossRef] [PubMed]
- Raj, C.C.; Prasanth, R. Review—Advent of TiO2 Nanotubes as Supercapacitor Electrode. J. Electrochem. Soc. 2018, 165, E345–E358. [Google Scholar] [CrossRef]
- Galstyan, V.; Comini, E.; Faglia, G.; Sberveglieri, G. TiO2 Nanotubes: Recent Advances in Synthesis and Gas Sensing Properties. Sensors 2013, 13, 14813–14838. [Google Scholar] [CrossRef]
- Kwon, Y.; Kim, H.; Lee, S.; Chin, I.-J.; Seong, T.-Y.; Lee, W.I.; Lee, C. Enhanced ethanol sensing properties of TiO2 nanotube sensors. Sens. Actuators B Chem. 2012, 173, 441–446. [Google Scholar] [CrossRef]
- Nah, Y.; Paramasivam, I.; Schmuki, P. Doped TiO2 and TiO2 Nanotubes: Synthesis and Applications. ChemPhysChem 2010, 11, 2698–2713. [Google Scholar] [CrossRef]
- Malati, M.A.; Wong, W.K. Doping TiO2 for solar energy applications. Surf. Technol. 1984, 22, 305–322. [Google Scholar] [CrossRef]
- Dozzi, M.V.; Selli, E. Doping TiO2 with p-block elements: Effects on photocatalytic activity. J. Photochem. Photobiol. C Photochem. Rev. 2013, 14, 13–28. [Google Scholar] [CrossRef]
- Karlsson, R.K.B.; Cornell, A.; Pettersson, L.G.M. The electrocatalytic properties of doped TiO2. Electrochim. Acta 2015, 180, 514–527. [Google Scholar] [CrossRef]
- Lai, Y.-K.; Huang, J.-Y.; Zhang, H.-F.; Subramaniam, V.-P.; Tang, Y.-X.; Gong, D.-G.; Sundar, L.; Sun, L.; Chen, Z.; Lin, C.-J. Nitrogen-doped TiO2 nanotube array films with enhanced photocatalytic activity under various light sources. J. Hazard. Mater. 2010, 184, 855–863. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, S.; Bard, A.J. Novel Carbon-Doped TiO2 Nanotube Arrays with High Aspect Ratios for Efficient Solar Water Splitting. Nano Lett. 2006, 6, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Aijo John, K.; Naduvath, J.; Remillard, S.K.; Shaji, S.; DeYoung, P.A.; Kellner, Z.T.; Mallick, S.; Thankamoniamma, M.; Okram, G.S.; Philip, R.R. A simple method to fabricate metal doped TiO2 nanotubes. Chem. Phys. 2019, 523, 198–204. [Google Scholar] [CrossRef]
- Na, S.; Seo, S.; Lee, H. Recent Developments of Advanced Ti3+-Self-Doped TiO2 for Efficient Visible-Light-Driven Photocatalysis. Catalysts 2020, 10, 679. [Google Scholar] [CrossRef]
- Zhu, G.; Shan, Y.; Lin, T.; Zhao, W.; Xu, J.; Tian, Z.; Zhang, H.; Zheng, C.; Huang, F. Hydrogenated blue titania with high solar absorption and greatly improved photocatalysis. Nanoscale 2016, 8, 4705–4712. [Google Scholar] [CrossRef]
- Zhu, Q.; Peng, Y.; Lin, L.; Fan, C.-M.; Gao, G.-Q.; Wang, R.-X.; Xu, A.-W. Stable blue TiO2−x nanoparticles for efficient visible light photocatalysts. J. Mater. Chem. A 2014, 2, 4429. [Google Scholar] [CrossRef]
- Yin, H.; Lin, T.; Yang, C.; Wang, Z.; Zhu, G.; Xu, T.; Xie, X.; Huang, F.; Jiang, M. Gray TiO2 Nanowires Synthesized by Aluminum-Mediated Reduction and Their Excellent Photocatalytic Activity for Water Cleaning. Chem.—A Eur. J. 2013, 19, 13313–13316. [Google Scholar] [CrossRef]
- Bessegato, G.G.; Hudari, F.F.; Zanoni, M.V.B. Self-doped TiO2 nanotube electrodes: A powerful tool as a sensor platform for electroanalytical applications. Electrochim. Acta 2017, 235, 527–533. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, Y. Electrochemically Self-Doped TiO2 Nanotube Arrays for Supercapacitors. J. Phys. Chem. C 2014, 118, 5626–5636. [Google Scholar] [CrossRef]
- Liao, W.; Yang, J.; Zhou, H.; Murugananthan, M.; Zhang, Y. Electrochemically Self-Doped TiO2 Nanotube Arrays for Efficient Visible Light Photoelectrocatalytic Degradation of Contaminants. Electrochim. Acta 2014, 136, 310–317. [Google Scholar] [CrossRef]
- Gan, L.; Wu, Y.; Song, H.; Lu, C.; Zhang, S.; Li, A. Self-doped TiO2 nanotube arrays for electrochemical mineralization of phenols. Chemosphere 2019, 226, 329–339. [Google Scholar] [CrossRef]
- Pinto, V.L.; Cervantes, T.N.M.; Soto, P.C.; Sarto, G.; Bessegato, G.G.; de Almeida, L.C. Multivariate optimization of methylene blue dye degradation using electro-Fenton process with self-doped TiO2 nanotube anode. Chemosphere 2023, 344, 140336. [Google Scholar] [CrossRef] [PubMed]
- Soto, P.C.; Salamanca-Neto, C.A.R.; Moraes, J.T.; Sartori, E.R.; Bessegato, G.G.; Lopes, F.; Almeida, L.C. A novel sensing platform based on self-doped TiO2 nanotubes for methylene blue dye electrochemical monitoring during its electro-Fenton degradation. J. Solid State Electrochem. 2020, 24, 1951–1959. [Google Scholar] [CrossRef]
- Zhang, Z.; Hedhili, M.N.; Zhu, H.; Wang, P. Electrochemical reduction induced self-doping of Ti3+ for efficient water splitting performance on TiO2 based photoelectrodes. Phys. Chem. Chem. Phys. 2013, 15, 15637. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.-D.; Wang, C.-W.; Chen, J.-B.; Li, Y.; Wang, J. Enhanced field emission from Ti3+ self-doped TiO2 nanotube arrays synthesized by a facile cathodic reduction process. Appl. Surf. Sci. 2014, 301, 525–529. [Google Scholar] [CrossRef]
- Hong, S.P.; Kim, S.; Kim, N.; Yoon, J.; Kim, C. A short review on electrochemically self-doped TiO2 nanotube arrays: Synthesis and applications. Korean J. Chem. Eng. 2019, 36, 1753–1766. [Google Scholar] [CrossRef]
- Pinna, M.; Binda, G.; Altomare, M.; Marelli, M.; Dossi, C.; Monticelli, D.; Spanu, D.; Recchia, S. Biochar Nanoparticles over TiO2 Nanotube Arrays: A Green Co-Catalyst to Boost the Photocatalytic Degradation of Organic Pollutants. Catalysts 2021, 11, 1048. [Google Scholar] [CrossRef]
- Lavagnini, I.; Antiochia, R.; Magno, F. An Extended Method for the Practical Evaluation of the Standard Rate Constant from Cyclic Voltammetric Data. Electroanalysis 2004, 16, 505–506. [Google Scholar] [CrossRef]
- Chen, W.; Cai, S.; Ren, Q.-Q.; Wen, W.; Zhao, Y.-D. Recent advances in electrochemical sensing for hydrogen peroxide: A review. Analyst 2012, 137, 49–58. [Google Scholar] [CrossRef]
- Cha, G.; Schmuki, P.; Altomare, M. Anodic TiO2 nanotube membranes: Site-selective Pt-activation and photocatalytic H2 evolution. Electrochim. Acta 2017, 258, 302–310. [Google Scholar] [CrossRef]
- Rao, B.M.; Roy, S.C. Anatase TiO2 nanotube arrays with high temperature stability. RSC Adv. 2014, 4, 38133–38139. [Google Scholar] [CrossRef]
- Spanu, D.; Minguzzi, A.; Recchia, S.; Shahvardanfard, F.; Tomanec, O.; Zboril, R.; Schmuki, P.; Ghigna, P.; Altomare, M. An Operando X-ray Absorption Spectroscopy Study of a NiCu−TiO2 Photocatalyst for H2 Evolution. ACS Catal. 2020, 10, 8293–8302. [Google Scholar] [CrossRef]
- Macak, J.M.; Gong, B.G.; Hueppe, M.; Schmuki, P. Filling of TiO2 Nanotubes by Self-Doping and Electrodeposition. Adv. Mater. 2007, 19, 3027–3031. [Google Scholar] [CrossRef]
- Thi Thanh Thuy, M.; Thi Van Anh, N.; Thi Xuan, M.; Vinh, T.Q.; Thi Binh, P. Study on the photoelectrocatalytic activity of reduced TiO2 nanotube films for removal of methyl orange. Green Process. Synth. 2023, 12, 20228159. [Google Scholar] [CrossRef]
- Klingler, R.J.; Kochi, J.K. Electron-transfer kinetics from cyclic voltammetry. Quantitative description of electrochemical reversibility. J. Phys. Chem. 1981, 85, 1731–1741. [Google Scholar] [CrossRef]
- AlHoshan, M.S.; BaQais, A.A.; Al-Hazza, M.I.; Al-Mayouf, A.M. Heat treatment and electrochemical activation of titanium oxide nanotubes: The effect of hydrogen doping on electrochemical behavior. Electrochim. Acta 2012, 62, 390–395. [Google Scholar] [CrossRef]
- Takata, T.; Domen, K. Defect Engineering of Photocatalysts by Doping of Aliovalent Metal Cations for Efficient Water Splitting. J. Phys. Chem. C 2009, 113, 19386–19388. [Google Scholar] [CrossRef]
- Cheng, C.; Long, R. Charge-Compensated Doping Extends Carrier Lifetimes in SrTiO3 by Passivating Oxygen Vacancy Defects. J. Phys. Chem. Lett. 2021, 12, 12040–12047. [Google Scholar] [CrossRef]
- Goudon, T.; Miljanović, V.; Schmeiser, C. On the Shockley–Read–Hall Model: Generation-Recombination in Semiconductors. SIAM J. Appl. Math. 2007, 67, 1183–1201. [Google Scholar] [CrossRef]
- Trachioti, M.G.; Lazanas, A.C.; Prodromidis, M.I. Shedding light on the calculation of electrode electroactive area and heterogeneous electron transfer rate constants at graphite screen-printed electrodes. Microchim. Acta 2023, 190, 251. [Google Scholar] [CrossRef]
- Wang, J. Electrochemical Sensing of Explosives. Electroanalysis 2007, 19, 415–423. [Google Scholar] [CrossRef]
- Murugan, P.; Nagarajan, R.D.; Sundramoorthy, A.K.; Ganapathy, D.; Atchudan, R.; Nallaswamy, D.; Khosla, A. Electrochemical Detection of H2O2 Using an Activated Glassy Carbon Electrode. ECS Sens. Plus 2022, 1, 034401. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, X.; Guo, J. Functionalized Carbon-Based Electrochemical Sensors for Food and Alcoholic Beverage Safety. Appl. Sci. 2022, 12, 9082. [Google Scholar] [CrossRef]
- Wang, T.; Zhu, H.; Zhuo, J.; Zhu, Z.; Papakonstantinou, P.; Lubarsky, G.; Lin, J.; Li, M. Biosensor Based on Ultrasmall MoS2 Nanoparticles for Electrochemical Detection of H2O2 Released by Cells at the Nanomolar Level. Anal. Chem. 2013, 85, 10289–10295. [Google Scholar] [CrossRef]
- Patella, B.; Buscetta, M.; Di Vincenzo, S.; Ferraro, M.; Aiello, G.; Sunseri, C.; Pace, E.; Inguanta, R.; Cipollina, C. Electrochemical sensor based on rGO/Au nanoparticles for monitoring H2O2 released by human macrophages. Sens. Actuators B Chem. 2021, 327, 128901. [Google Scholar] [CrossRef]
- Baig, N.; Sajid, M. Applications of layered double hydroxides based electrochemical sensors for determination of environmental pollutants: A review. Trends Environ. Anal. Chem. 2017, 16, 1–15. [Google Scholar] [CrossRef]
- Sanford, A.L.; Morton, S.W.; Whitehouse, K.L.; Oara, H.M.; Lugo-Morales, L.Z.; Roberts, J.G.; Sombers, L.A. Voltammetric Detection of Hydrogen Peroxide at Carbon Fiber Microelectrodes. Anal. Chem. 2010, 82, 5205–5210. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, R.H.; Gonçalves, D.A.; dos Reis, D.D. TiO2/MWCNT/Nafion-Modified Glassy Carbon Electrode as a Sensitive Voltammetric Sensor for the Determination of Hydrogen Peroxide. Sensors 2023, 23, 7732. [Google Scholar] [CrossRef]
- Dutta, A.K.; Maji, S.K.; Srivastava, D.N.; Mondal, A.; Biswas, P.; Paul, P.; Adhikary, B. Peroxidase-like activity and amperometric sensing of hydrogen peroxide by Fe2O3 and Prussian Blue-modified Fe2O3 nanoparticles. J. Mol. Catal. A Chem. 2012, 360, 71–77. [Google Scholar] [CrossRef]
- Majumder, S.; Saha, B.; Dey, S.; Mondal, R.; Kumar, S.; Banerjee, S. A highly sensitive non-enzymatic hydrogen peroxide and hydrazine electrochemical sensor based on 3D micro-snowflake architectures of α-Fe2O3. RSC Adv. 2016, 6, 59907–59918. [Google Scholar] [CrossRef]
- Cai, J.; Ding, S.; Chen, G.; Sun, Y.; Xie, Q. In situ electrodeposition of mesoporous aligned α-Fe2O3 nanoflakes for highly sensitive nonenzymatic H2O2 sensor. Appl. Surf. Sci. 2018, 456, 302–306. [Google Scholar] [CrossRef]
- Sun, X.; Wang, C.; Su, D.; Wang, G.; Zhong, Y. Application of Photocatalytic Materials in Sensors. Adv. Mater. Technol. 2020, 5, 1900993. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Self-Doping Time | Electroactive Surface Area (cm2) |
|---|---|
| 10 s | 0.09 |
| 30 s | 0.19 |
| 60 s | 0.20 |
| 300 s | 0.18 |
| 1800 s | 0.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spanu, D.; Dhahri, A.; Binda, G.; Monticelli, D.; Pinna, M.; Recchia, S. Ultrafast Electrochemical Self-Doping of Anodic Titanium Dioxide Nanotubes for Enhanced Electroanalytical and Photocatalytic Performance. Chemosensors 2023, 11, 560. https://doi.org/10.3390/chemosensors11110560
Spanu D, Dhahri A, Binda G, Monticelli D, Pinna M, Recchia S. Ultrafast Electrochemical Self-Doping of Anodic Titanium Dioxide Nanotubes for Enhanced Electroanalytical and Photocatalytic Performance. Chemosensors. 2023; 11(11):560. https://doi.org/10.3390/chemosensors11110560
Chicago/Turabian StyleSpanu, Davide, Aicha Dhahri, Gilberto Binda, Damiano Monticelli, Marco Pinna, and Sandro Recchia. 2023. "Ultrafast Electrochemical Self-Doping of Anodic Titanium Dioxide Nanotubes for Enhanced Electroanalytical and Photocatalytic Performance" Chemosensors 11, no. 11: 560. https://doi.org/10.3390/chemosensors11110560