Abstract
Amine groups are common constituents of organic dyes and play important roles in tuning fluorescence properties. In particular, intensive research works have demonstrated the tendency and capabilities of amines in influencing chromophore brightness. Such properties have been explained by multiple mechanisms spanning from twisted intramolecular charge transfer (TICT) to the energy gap law and beyond, which introduce additional nonradiative energy dissipation pathways. In this review, we aim to provide a focused overview of the mechanistic insights mainly for the TICT mechanism, accompanied by a few other less common or influential fluorescence quenching mechanisms in the amine-containing fluorescent molecules. Various aspects of current scientific findings including the rational design and synthesis of organic chromophores, theoretical calculations, steady-state and time-resolved electronic and vibrational spectroscopies are reviewed. These in-depth understandings of how the amine groups with diverse chemical structures at various atomic sites affect excited-state nonradiative decay pathways will facilitate the strategic and targeted development of fluorophores with desired emission properties as versatile chemosensors for broad applications.
1. Introduction
Light-emitting dyes with specific photophysical properties such as color and fluorescence quantum efficiency are pivotal to the advancement of modern chemical and biological imaging and sensing applications across many disciplines [1,2,3,4,5]. The amine groups play a special role in these regards due to their pronounced electron-donating capabilities with a lone electron pair that yield interesting electronic and geometrical properties. For example, the amine group is a common constituent of red/near-infrared dyes because it promotes intramolecular charge transfer (ICT) in the electronic excited state [2] which is mechanistically more efficient in reducing the electronic transition energy gaps than expanding π-conjugation in hydrocarbons. Besides the effects on color, recent decades of research efforts on flexible amines (prone to rotation around the amine-backbone bond) have revealed another aspect of their properties: the fluorescence quantum yield (FQY, directly proportional to the molecular brightness that is the product of FQY and extinction coefficient) can be markedly influenced by the amine group. One significant underpinning is twisted intramolecular charge transfer (TICT) that suggests the formation of a charge transfer (CT) state with a perpendicular conformation via intramolecular rotation around a chemical bond [6,7,8,9,10,11,12]. The interest in TICT stemmed from the discovery of unusual dual fluorescence of 4-dimethyaminobenzonitrile (DMABN) in polar solvents by Lippert et al. in the 1960s [13]. The pertinent study then motivated intensive investigations with debates about the origin of dual fluorescence bands [14,15,16,17,18,19,20,21,22,23,24,25,26,27]. After extensive experimental and theoretical efforts, the TICT mechanism has gained the most popularity to explain the nature of the long-wavelength emission band in DMABN. Consequently, the short- and unusual long-wavelength bands have been attributed to the locally excited (LE) and TICT states in view of their different sensitivities to solvent polarity and geometries (Figure 1a) [6].
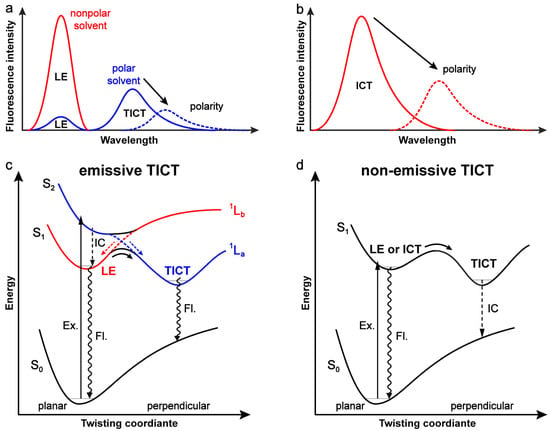
Figure 1.
Schematics for emission properties and potential energy curves along the amine-twisting coordinate with an (a,c) emissive or (b,d) non-emissive TICT state. In (a) and (b), the relative fluorescence intensities in solvents of different polarities are illustrative and not exactly based on experimental data. In (c) and (d), the transitions of excitation (Ex.), internal conversion (IC), and fluorescence (Fl.) are indicated by solid, dashed, and wavy arrows, respectively. In case of an emissive TICT state, blue and red curves represent the presumable 1La and 1Lb states, respectively, in dual-emission molecules such as DMABN. The crossing of two potential energy curves of 1La and 1Lb states is depicted by dashed lines. S1 and S2 potential energy curves are shown as an avoided crossing to demonstrate the precursor-successor relationship between LE and TICT states.
In recent decades, researchers have expanded attention to more molecular scaffolds and examined a wider array of structures, particularly with the amine group as a twisting unit, to elucidate the properties of TICT [7]. In contrast to DMABN and its close analogues, most of these amine-containing chromophores exhibit a single emission band with certain degrees of intramolecular charge transfer (ICT) character and marked solvent polarity-dependent FQYs (Figure 1b) [28,29,30,31,32,33,34,35]. Both experiments and computations support the idea that TICT occurs in these molecules and is responsible for the observed fluorescence dependence on the solvent polarity [7]. However, the TICT states in these cases have been shown to be completely dark due to the zero orbital overlap at the perpendicular geometry between the amine group and the aromatic backbone. The difference between the molecules with an emissive and non-emissive TICT state is also associated with their excited-state dynamics as illustrated by the potential energy curves along the amine-twisting coordinate (Figure 1c,d).
The dark-state attribute of TICT is very useful in modulating the fluorescence of small organic dyes as fluorescent labels and probes in bioimaging and biosensing applications. For example, suppressing TICT in rhodamines with strategic amine groups can significantly boost the FQY and increase the chromophore photostability, which greatly benefits the super-resolution imaging [36,37,38]. Though the molecules with an emissive TICT state such as DMABN do not directly connect to practical applications, they have served as molecular prototypes and provided important insights into the TICT process.
Moreover, the studies on DMABN and other dual-fluorescence analogues have revealed major fluorescence quenching pathways including intersystem crossing (ISC) and excited-to-ground-state internal conversion (IC), which account for their low FQYs (both LE and TICT fluorescence). These radiationless pathways have seldom been discussed for rigid amine-containing organic fluorophores without major nuclear motion-induced deactivation pathways including TICT. Many of these fluorophores only exhibit moderate FQYs, indicative of the presence of other nonradiative pathways such as those governed by the energy gap law. A comprehensive understanding of all possible pathways and their influences is crucial for the rational development of ideal fluorescent molecules and materials for diverse applications.
Although some related topics, particularly TICT, have been reviewed in the past decades, a comprehensive review solely on the amine-containing chromophores highlighting the role of amine groups is still lacking. Our current review aims to (1) provide an overview of key results for validation of the TICT mechanism by both experimental and computational studies, including systems with emissive and non-emissive TICT states; (2) summarize the factors affecting TICT based on the large pool of reported fluorophores; and (3) briefly introduce some other fluorescence quenching mechanisms, which are universal but may be much less influential than TICT. We will focus our discussion on the amine-containing molecules in which there are no other major rotating units (i.e., negligible contributions to the nonradiative decay pathways) beside the amine group or amine twisting is also inhibited.
2. Insights into the Chromophores with an Emissive TICT State
The establishment and validation of the TICT model as the prevailing mechanism to explain the prototypical dual-fluorescence molecule DMABN are based on decades of intensive experimental and theoretical efforts. Lippert et al. explained the dual fluorescence by two excited states, namely, 1La and 1Lb [13]. Due to the measured large permanent (electric) dipole moment and pronounced solvatochromism, the long-wavelength band (1La) is attributed to the lower-lying CT state (A band), while the short-wavelength band (1Lb) is the higher-lying LE state (B band) as shown in Figure 1c. Lippert et al. ascribed the dual fluorescence bands to the inversion of 1La and 1Lb states during the reorientational relaxation in a polar solvent. However, this interpretation contradicts the polarization experiments and fluorescence properties in viscous solvents such as glycerol [39]. Therefore, Grabowski et al. later proposed that the CT emission band results from twisting of the dimethyl amino group (–NMe2) against the benzonitrile plane that produces a perpendicular configuration [39] and hence was termed the TICT state, in contrast to the LE state where the molecule stays planar. The presence of the TICT state was then supported by a large number of DMABN derivatives with strategic structural modifications. Computational perspectives were also rapidly expanding at the same time to shed light on the dual-fluorescence phenomenon. Different natures of the CT state from TICT were also proposed along the way, resulting in a significant debate. The advancement in ultrafast electronic and vibrational spectroscopies provided further evidence in favor of the TICT model.
2.1. Evidence for TICT
2.1.1. Strategic Modifications of Chemical Structure
The TICT model highlights the twisting of the amine group in generating a highly polar CT state. A rigidified structure constraining the emission state at a planar or twisted geometry is the straightforward evidence for TICT. Alkyl cyclization of DMABN (1a, Figure 2) to confine the amine group at a relatively planar geometry is supposed to suppress the twisting. As a result, 1b and 1c exhibit only LE fluorescence with similar wavelengths to the LE band of DMABN (Figure 3a,b) [15,40]. However, the seven-membered heterocyclic ring (1d) still gives rise to a CT fluorescence band (Figure 3b, bottom panel). This result was attributed to the increased flexibility of a large cyclic ring that remains sufficient to enable the TICT process [15]. It is expected that the flexibility should decrease with the smaller ring size (i.e., 1d > 1c > 1b). Notably, the six-membered heterocyclic ring is still flexible to some extent; therefore, when the driving force of TICT becomes larger, such as that in 1e due to the increased electron-donating capability and steric hindrance of a tert-butyl group [41], dual fluorescence is clearly observed across a wide temperature range (Figure 3c, see LE and ICT labels) [25,42] and the redder component within the emission band is attributable to a TICT state as confirmed by theoretical calculations [26].
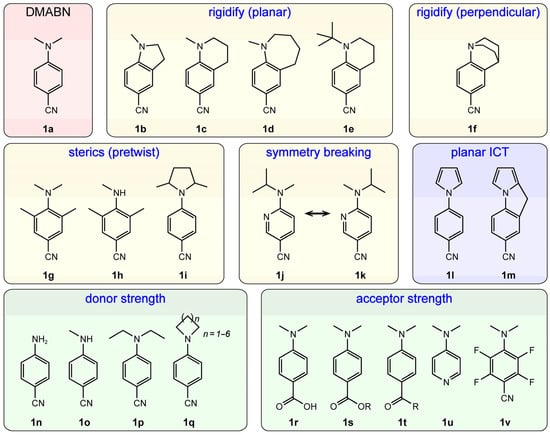
Figure 2.
Structural modifications of DMABN for elucidation of the TICT mechanism. The various series of chromophores are grouped according to the blue labels in each colored box.
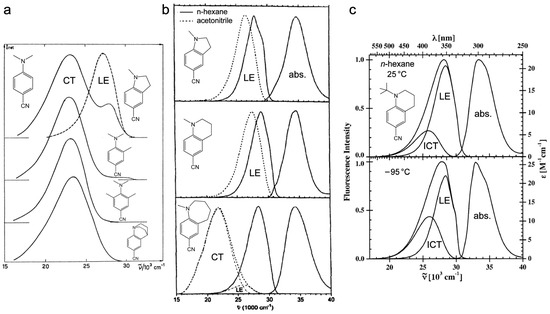
Figure 3.
Steady-state electronic spectra for DMABN derivatives with various geometrical confinements. (a) Emission spectra of 1a, 1b, 1f, and 1g in CH2Cl2 at room temperature. The 3-methyl-4-dimethylaminobenzonitrile (second row from the top) shown has a single CT emission band as well, which is similar to 1g. We only use the latter as an example to demonstrate the steric effect. Adapted with permission from Ref. [40]. Copyright 1979 Elsevier B.V. (b) Absorption (abs.) and emission (LE and CT) spectra of 1b–d in n-hexane (solid lines) and acetonitrile (dotted lines) at 25 °C. Adapted with permission from Ref. [15]. Copyright 1997 Elsevier B.V. (c) Absorption (abs.) and emission spectra of 1e in n-hexane at 25 °C (top panel) and −95 °C (bottom panel) with the LE and ICT bands denoted separately underneath the broad emission band. Adapted with permission from Ref. [25]. Copyright 2007 American Chemical Society.
Meanwhile, 1f is another example to evince TICT which shows exclusively a polar CT band (Figure 3a) with a large Stokes shift and pronounced solvatochromism [40]. The quinuclidine group forces the lone electron pair of nitrogen to lie within the conjugated benzonitrile plane, which is a close structure to the TICT state of DMABN. In the case of DMABN, the –NMe2 group is perpendicular to the aromatic plane in the TICT state which results in zero overlap between the nitrogen lone pair and aromatic orbitals. For comparison, 1f has a weak absorption band at the red edge of the major π → π* transition (S2), and this weak (likely S1) absorption band has been ascribed to the n → π* transition that promotes a nitrogen lone pair electron to the aromatic π* orbital [43,44]. The small but non-zero oscillator strength is owing to the slight structural deformation in the quinuclidine cage despite the large conformational rigidity, which results in a small orbital overlap. This insight has been supported by the crystal structure and semiempirical calculations [45,46].
DMABN derivatives such as 1g, 1h and 1i provide similar molecular insights via steric hindrance [14,40,43,47]. The presence of methyl groups at the ortho sites on the benzene ring (1g and 1h) or at the cyclic amine ring (1i), particularly in the former case, causes strong steric hindrance between the two moieties and hence a twisted LE state (e.g., ~60° between the –NMe2 and the benzene ring plane in 1g) [40,43]. The pretwist essentially results in a shallow LE state minimum (i.e., an elevated energy) compared to the planar LE state in DMABN and therefore a smaller transition energy barrier toward the TICT state, substantiated by the enhanced emission and formation rate of TICT [47].
Other structural modifications have also been reported to support TICT. The amino-cyanopyridine analogue 1j and its conformational isomer 1k show dual fluorescence bands, similar to those in DMABN [48]. The symmetry breaking of the aromatic ring and the amine group with respect to DMABN predicts the formation of an isomeric product upon TICT, which can be discerned by NMR. With a low temperature at around −90 °C to slow down thermal equilibration between the initial 1j and product 1k in the ground state, the 1H-NMR difference spectrum exhibits the appearance of 1k, essentially confirming the amine-twisting process [49].
The pyrrole derivatives 1l and 1m also exhibit dual fluorescence (LE and CT) similar to DMABN but the conformational locking in 1m indicates a different origin for the long-wavelength CT band which must have a planar structure [22,50]. Calculations showed that the donor π-orbital of 1m is clearly different from that in DMABN whose orbital includes the nitrogen lone pair [23,24,51]. Therefore, it is not a suitable example as evidence against TICT.
In addition, dual fluorescence was found to be sensitive to the structure of the amine group that acts as the electron donor. For example, the primary amine (1n) and simple secondary amine such as 1o only display LE emission [27]. In contrast, the diethylamino derivative 1p exhibits enhanced CT fluorescence [52]. The appearance of a CT band in azacylic derivatives 1q depends on the ring size, and a larger ring (likely with more conformational flexibilities as mentioned above) favors the formation of a CT state [53]. These observations have been associated with the electron-donating strength of the donor group. This finding is in agreement with the TICT model that suggests a complete charge transfer from the amine group [21]. Likewise, strong electron acceptors would also promote the generation of CT/TICT fluorescence: the derivatives with substitutions of –CN (1a) by –COOH (1r), –COOR (1s), and –COR (1t) indeed show dual fluorescence [6,54,55]. Changing the benzonitrile moiety with pyridine (1u) also results in dual fluorescence [56]. Notably, the increase of electron acceptor strength such as a tetrafluorinated benzonitrile moiety (e.g., 1v) leads to exclusive CT/TICT fluorescence even when the primary amine (–NH2) is the donor [27], substantiating the dominant electronic effect (“push-pull”) in this molecular system.
2.1.2. Time-Resolved Electronic and Vibrational Spectroscopies
During the TICT model development, a few other models have also been proposed to explain the CT emission: PICT (planar ICT) and RICT (rehybridized ICT) as shown in Figure 4a, as well as WICT (wagged ICT). In the WICT model, it was proposed that the amine nitrogen undergoes rehybridization from sp2 to sp3 (pyramidalization or wagging), thus decoupling the lone pair from the benzonitrile and increasing the charge separation [14,15]. However, the wagging motion was shown by calculations to be unable to generate a highly polar state [16,17,18]. The RICT state featuring rehybridization of the nitrile group from sp to sp2 (Figure 4a) is found as an excited-state minimum but experiences a large barrier from the LE state [21]. Another RICT model that characterizes a πσ* state as an intermediate state in the LE-to-TICT reaction was recently proposed on the basis of time-resolved transient absorption spectroscopy [57,58,59,60]. However, it has been questioned by other researchers [61,62]. Therefore, a major debate remained between the TICT and PICT models, which had prompted many experimental efforts over the years, particularly those from ultrafast time-resolved electronic and vibrational spectroscopies in addition to computational studies.
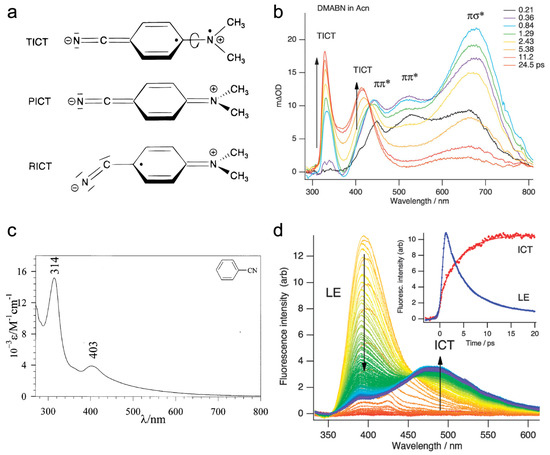
Figure 4.
Theoretical models for the CT emission of DMABN and time-resolved excited-state electronic spectra for DMABN in solution. (a) TICT, PICT, and RICT models to explain the CT state structure of DMABN. Reproduced with permission from Ref. [21]. Copyright 2000 American Chemical Society. (b) Femtosecond time-resolved excited-state absorption spectra of DMABN in acetonitrile (Acn) at room temperature with 305 nm excitation. Peak intensity evolutions are indicated by black arrows and the transient species are shown as ππ *, πσ*, and TICT. Reproduced with permission from Ref. [60]. Copyright 2011 The Royal Society of Chemistry. (c) Steady-state absorption spectrum of the radical anion of benzonitrile in 0.1 M tetrabutylammonium tetrafluoroborate (TBABF4) + DMF solution. Reproduced with permission from Ref. [63]. Copyright 1998 Elsevier B.V. (d) Picosecond time-resolved fluorescence spectra of DMABN in acetonitrile at room temperature obtained with the optical Kerr-gating method with 267 nm excitation. The intensity evolutions of two marker bands (LE and ICT) are indicated by arrows. The temporal profiles of band-integrated LE and CT fluorescence are shown as inset. Reproduced with permission from Ref. [60]. Copyright 2011 The Royal Society of Chemistry.
Notably, the PICT state of DMABN is characterized by a planar quinoidal resonance structure in which the amine group is strongly coupled with the benzonitrile moiety [21]. The TICT model, on the other hand, corresponds to the complete decoupling of these moieties due to twisting of the amine group (see Figure 4a). This key difference implies that (1) the CT state should have similar properties to the radical anion of benzonitrile; (2) the frequency of phenyl–N stretch of the CT state should red-shift for TICT but blue-shift for PICT (i.e., acquiring more double-bond character) from the ground to excited state [21]. Both TICT and PICT models predict the redshift of the nitrile CN stretch frequency as a result of change from triple to double bond character via electron redistribution (resonance effect).
For DMABN in acetonitrile, femtosecond transient absorption (fs-TA) spectroscopy shows multiple excited-state absorption (ESA) bands of the LE state across the UV and visible wavelengths at early time (several picoseconds from the time zero of photoexcitation), followed by a rise of the absorption bands at ~330 and 420 nm on the 4–5 ps timescale (Figure 4b) [60,64]. These two nascent ESA bands can be readily assigned to the product state due to the rise dynamics, and their resemblance to the absorption of the benzonitrile radical anion (Figure 4c) [63,65] is consistent with the TICT model that predicts the radical-like properties (decoupled electron clouds) for the perpendicular conformation of the amine group in DMABN. The pertinent LE-to-ICT transition was also monitored by time-resolved fluorescence measurements up to ~25 ps with clearly correlated decay and rise dynamics of the LE and ICT bands, respectively (Figure 4d) [57,60].
Compared to electronic spectroscopy that provides holistic information for reaction coordinates, vibrational spectroscopy is inherently sensitive to and can capture subtle structural changes along these reaction paths. Umapathy et al. identified the characteristic phenyl–N stretch mode in the ground and excited electronic states of DMABN in methanol using resonance Raman and time-resolved transient resonance Raman (TR2), respectively [66]. The mode frequency was found to undergo a ~96 cm−1 redshift from the ground (1377 cm−1) to the CT state (1281 cm−1, at the time delay of 50 ps when the CT state is prominent), in accord with the aforementioned TICT process (Figure 4a). The assignment is further corroborated by isotopic labeling of the amine nitrogen and methyl-group hydrogens (see Figure 5a,b). These results provide direct support for the TICT model that predicts a weakened C–N single bond due to electronic decoupling.
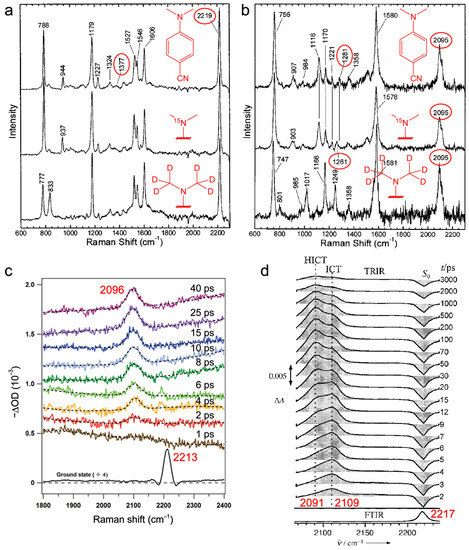
Figure 5.
Time-resolved excited-state Raman and IR spectra of DMABN in solution. (a) Ground-state resonance Raman spectra of DMABN, DMABN-15N, and DMABN-d6 in methanol with 330 nm excitation. Adapted with permission from Ref. [66]. Copyright 2001 American Chemical Society. (b) Transient resonance Raman (TR2) spectra of DMABN, DMABN-15N, and DMABN-d6 in methanol at 50 ps after 330 nm excitation. Adapted with permission from Ref. [66]. Copyright 2001 American Chemical Society. (c) Femtosecond stimulated Raman spectra of DMABN in methanol with a 330 nm Raman pump. Adapted with permission from Ref. [67]. Copyright 2012 American Chemical Society. (d) Ground-state Fourier transform IR (FTIR) and time-resolved excited-state IR spectra (TRIR) of DMABN in methanol after 267 nm excitation. Adapted with permission from Ref. [68]. Copyright 2003 Wiley-VCH.
Meanwhile, the nitrile CN stretch exhibits a frequency redshift from the ground state (2219 cm−1) to the CT state (2095 cm−1) as circled in Figure 5a,b. A similar frequency shift has also been observed via femtosecond stimulated Raman spectroscopy (FSRS) [67] and time-resolved IR spectroscopy (TRIR) [68,69,70] with higher temporal resolutions as shown in Figure 5c,d, respectively. For the latter case, the nitrile stretch region is finely resolved as two peaks at 2109 and 2091 cm−1 which correspond to the interchanging “free” ICT and hydrogen-bonded ICT (HICT) states, respectively (Figure 5d) [68,70]. However, this mode assignment was later questioned by theoretical calculations which showed that hydrogen bonding interactions with C≡N should blue-shift its mode frequency [71]. Nevertheless, the overall mode frequency redshift is consistently observed by different spectroscopic techniques. This result alone cannot distinguish PICT and TICT (see Figure 4a, top two structures) because both models suggest a redshift in the mode frequency, but the red-shifted frequency at ~2095 cm−1 (Figure 5b–d) matches the CN stretch of the benzonitrile radical anion [72,73]. Therefore, it rules out the RICT model (Figure 4a, bottom structure) which predicts the sp2-hybridized CN stretch frequency to be ~1550–1650 cm−1 [21,74]. Besides the nitrile stretch, many other Raman modes have shown remarkable resemblance to the benzonitrile radical anion in IR measurements [66], strongly suggesting a full charge transfer from the amine to benzonitrile moieties in the TICT model.
2.2. Potential Energy Surface for TICT in DMABN
For DMABN, various computational methods have established that the ground state has a planar structure and the initially accessed state is 1La state with CT character which is the second excited singlet state (S2). The 1Lb state energetically lies below the 1La state as S1 at the planar geometry. When the amine group twists by 90° to reach a perpendicular geometry against the benzonitrile moiety, the two energy states flip in order and the 1La state becomes the lower state which is stabilized by polar solvents (Figure 1c) [75,76]. After the Franck–Condon relaxation in S2 and subsequent radiationless S2 → S1 internal conversion (IC), the LE emission occurs at the 1Lb state minimum. The CT emission takes place at the global minimum of the 1La state with a perpendicular geometry (Figure 1c).
Many theoretical works have agreed on the crossing of 1La and 1Lb states along the amine-twisting coordinate [24,74,75,76,77]. A conical intersection (CI) has thus been proposed in nonadiabatically channeling the formation of the TICT state. One opinion suggests that the photoexcited 1La state first evolves along the amine-twisting coordinate and then passes through a CI, thereby branching into the LE and TICT states on the S1 potential energy surface (PES, see red and blue dashed arrows in Figure 1c). Therefore, a single rise time constant is sufficient to describe the process [78]. However, an adiabatic reaction between the LE and TICT states (avoided crossing, see solid S1 and S2 curves in Figure 1c) has been validated by a large amount of kinetic results from time-resolved spectroscopic techniques such as fs-TA and time-resolved fluorescence spectroscopy (Figure 4b,d) [57,60,79,80,81]. For example, the broadband fluorescence spectra using optical Kerr gating show the decay/rise dynamics with the same time constant for the LE/TICT fluorescence bands (Figure 4d), clearly indicative of their precursor-successor relationship [60].
The nonadiabatic and adiabatic processes were reconciled by Robb et al. via an extended CI seam between S2 and S1 on a multidimensional PES (Figure 6) [82]. The initially excited S2 state first relaxes to its minimum from the Franck–Condon region and then evolves toward the minimum energy point of a nearby CI seam (see the red curve in Figure 6), followed by nonadiabatic decay pathways to S1 states. The S2 minimum (labeled as S2-(P)ICT in Figure 6) is shallow and the CI seam can be easily accessed, in agreement with the ultrafast rise of S1-LE emission as experimentally observed. The S2 → S1 IC can occur at any amine-twisting angle at the CI seam but the direct IC to S1-LE should be favored because the structure at the minimum energy point on the CI seam is not twisted (i.e., a still-planar geometry) according to the calculations. This inference could explain why S1-LE behaves kinetically as a precursor for the TICT state. Finally, the adiabatic LE-to-TICT reaction occurs on the S1 PES (see blue curved arrows in Figure 6) prior to the TICT emission from the S1-(T)ICT state.
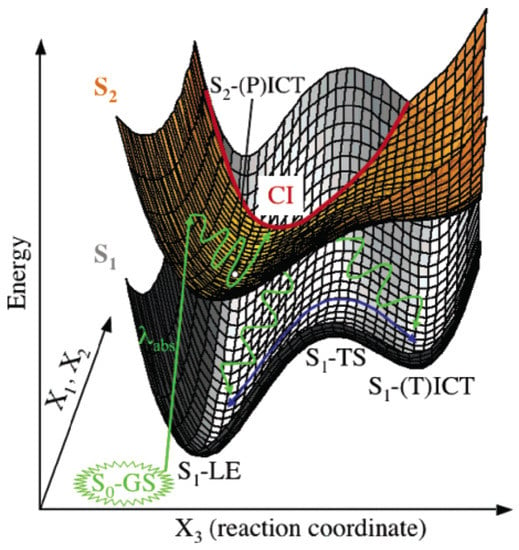
Figure 6.
Schematic S1 and S2 potential energy surfaces of DMABN in vacuo. The amine group twisting is the reaction coordinate X3. The conical intersection (CI) seam is depicted by the thick red curve. The S2 state energy minimum is marked by a white dot and the label of S2-(P)ICT close to the planar (P) geometry. The green and blue curved arrows indicate the nonadiabatic and adiabatic pathways, respectively. Reproduced with permission from Ref. [82]. Copyright 2005 American Chemical Society.
The calculations showed that the S1-TICT state of DMABN in vacuo has a higher energy than the S1-LE state, in accord with the absence of TICT fluorescence in the gas phase [6]. In solution, the TICT state should be notably stabilized by the solvent which leads to more efficient TICT and the associated fluorescence. For DMABN analogues that do not undergo TICT such as 4-aminobenzonitrile (ABN), the TICT state lies at a much higher energy than the LE state and thus cannot be stabilized to a substantial extent to exhibit TICT fluorescence in solvents [82].
3. Insights into Chromophores with a Non-Emissive TICT State
Recent studies have found more chromophore frameworks that display TICT properties when a flexible amine group is incorporated as the electron donor, suggesting the generality of flexible amines in promoting TICT. Note that we use “amine” here to generally refer to functional groups that contain nitrogen with a lone pair. These molecular frameworks include coumarin, rhodamine, naphthalimide, naphthalene, oxazine, benzoxadiazole, maleimide, and locked benzylidene-imidazolinone (green fluorescent protein or GFP chromophore), etc., as grouped in Figure 7 [28,29,30,31,33,35,83,84,85,86,87]. The presence of TICT in these systems can be rationalized experimentally and computationally which further reveals the difference from the TICT in DMABN and its analogues (e.g., Figure 2).
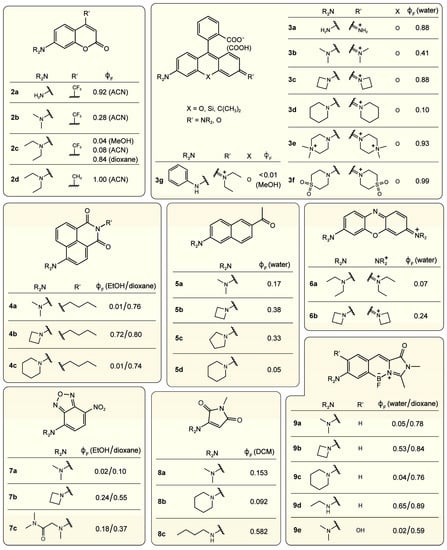
Figure 7.
Fluorophore scaffolds and common amines in modulating fluorescence quantum yield (FQY) by affecting the TICT efficiency. The FQY data () for various derivatives in certain solvents (in parentheses) listed are taken from Refs. [28,88] for coumarin (2a–d), see main text for rhodamine (3a–g), Ref. [31] for naphthalimide (4a–c), Ref. [84] for naphthalene (5a–d), Ref. [85] for oxazine (6a,b), Ref. [87] for benzoxadiazole (7a–c), Ref. [86] for maleimide (8a–c), and Refs. [34,35,89] for benzylidene-imidazolinone (9a–e).
3.1. Rationalization of TICT
First, flexible amines such as dialkylamine lead to low FQYs in polar solvents, similar to DMABN. For example, derivatives of coumarin 2c [88], naphthalimide 4a [31], oxazine 6a [85], benzoxadiazole 7a [87], and locked benzylidene-imidazolinone 9a [34] exhibit low FQYs of 0.01–0.07 in water or alcohols (Figure 7). The low FQYs indicate that major nonradiative decay pathways exist for these chromophores. Meanwhile, solvent polarity-dependent FQY is also characteristic of TICT-capable chromophores. As examples, the molecules 2c, 4a, 4c, 7a, 9a, 9c, and 9e show marked FQY difference in water/EtOH and dioxane (see Figure 7) [31,34,35,87,88,89]. Note that such fluorescence quenching and solvent sensitivity also depend on the amine structure of the chromophore as denoted in Figure 7, which will be elaborated in Section 4 below.
Second, the structural inhibition of twisting motions of the amine group gives rise to a high FQY. Figure 8 shows the alkyl cyclization of the amine moiety for various scaffolds in the generation of bright fluorophores. The six-membered fused ring is expected to force the amine to stay planar, hence inhibiting TICT. The increase of FQY is indeed observed for derivatives of coumarin (2c), locked benzylidene-imidazolinone (9a and 9e), and rhodamine (10a) in polar solvent where TICT is efficient and can thus be effectively hindered by the conformational locking (see “cyclization” in Figure 8) [28,34,35,89,90]. It is noteworthy that the julolidine derivatives (2e, 9f, and 10b) are less fluorescent than the tetrahydroquinoline derivatives (2f, 9g, 9h, and 10c) in polar solvents. This pattern may arise from the moderate rigidity of the six-membered ring that cannot completely overpower the increased donor strength in julolidine derivatives which would increase the driving force for TICT (see Section 4.2 for more details).
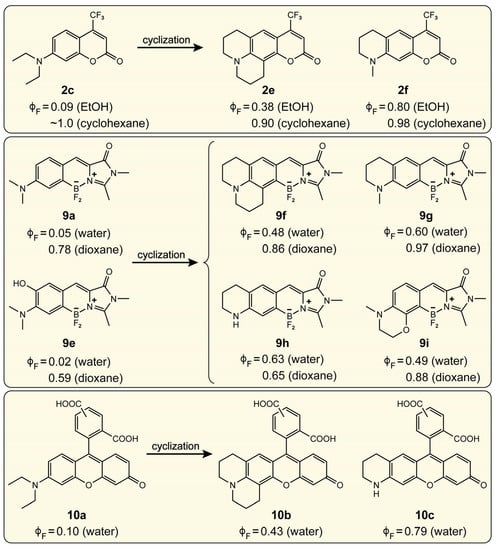
Figure 8.
Inhibition of TICT by structurally constraining the amine group twist for various amine-containing chromophores. The FQY data () are taken from Ref. [28] for coumarin, Refs. [34,35,89] for benzylidene-imidazolinone, and Ref. [90] for rhodamine derivatives.
Theoretical calculations support the presence of a non-emissive TICT state in these systems. Liu et al. has comprehensively explored and compared the functionals and solvation models using time-dependent density functional theory (TDDFT) in providing reliable descriptions of the TICT formation [84]. It was found that the qualitatively correct modeling of the TICT path strongly depends on the percentage of Hartree–Fock (HF) exchange in the hybrid functional, which compensates for the underestimation of excitation energies of “pure” functionals for CT states. Global hybrid functionals with high HF percentage (e.g., M062X) and range-separated functions (e.g., CAM-B3LYP and ωB97XD) yield reasonable twisting barriers, while pure and global hybrid functionals (e.g., PBE and B3LYP with 0–20% HF) severely underestimate the de-excitation energy of the TICT state and mistakenly predict the TICT tendency in various solvents (using Coumarin 152 or 2b in this review as an example, see Figure 9a). The solvation model (SMD) was also found to impact the TICT modeling. The correlated linear response (cLR)-SMD produces consistent energy barriers in different solvents with experimental observations (Figure 9a). The linear response (LR)-SMD fails to describe the increased TICT tendency with increasing solvent polarity (e.g., in water, Figure 9b). The state-specific (SS)-SMD overpredicts the TICT formation in low-polarity solvents such as cyclohexane (Figure 9c), contradictory to the observed near-unity FQY (0.97 in cyclohexane). Therefore, it was concluded that the combination of CAM-B3LYP (or M062X, ωB97XD, etc.) with the cLR solvation model can reasonably map the TICT PES of the amine-containing chromophore in various solvents. Similar insights have also been provided by other researchers [91].
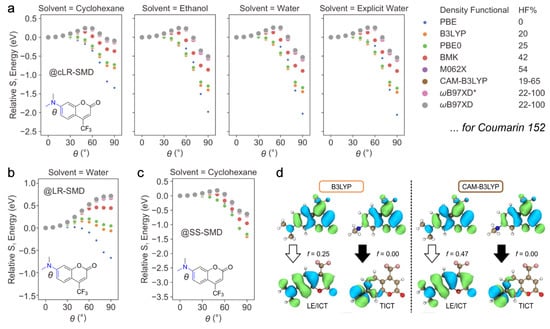
Figure 9.
Theoretical modeling of TICT in Coumarin 152 by TDDFT method. Relative S1 energy path along the amine-twisting coordinate (as a function of θ in degree unit) using various functionals and (a) cLR-SMD solvent formulism in four different solvents, (b) LR-SMD solvent formulism in water, and (c) SS-SMD solvent formulism in cyclohexane. (d) The electron density distributions of hole and electron natural transition orbitals (NTOs) of LE/ICT and TICT states as calculated at the B3LYP (left panels) and CAM-B3LYP (right panels) level of theory in vacuo. The transition oscillator strength () is listed by the downward block arrows (white: emissive; dark: non-emissive). Adapted with permission from Ref. [84]. Copyright 2020 Wiley-VCH.
3.2. Comparison with TICT in DMABN
Due to the twisting process and electronic donor–acceptor decoupling in the twisted state, the terminology “TICT” originating from the dual-fluorescence DMABN has often been used to describe a similar process in many other molecular systems such as those listed in Figure 7 and Figure 8. The TICT processes in these two types of systems do share some similarities besides the structures. For example, in both cases, the TICT formation is sensitive to solvent polarity: a greater solvent polarity enhances TICT efficiency (Figure 1a,b). This property has been attributed to the larger permanent (electric) dipole moment of the TICT state compared to its precursor LE/ICT state.
However, the difference is also significant between these two scenarios which should call for more in-depth analysis. The first obvious difference is that the TICT states in DMABN and its many analogues are emissive while those in other studied systems are non-emissive. The latter case is understandable because the decoupling of the donor and acceptor when the amine twists to 90° leads to zero orbital overlap and hence no transition probability (e.g., Coumarin 152 in Figure 9d) [84]. The unusual TICT fluorescence in DMABN clearly is not qualified by the complete electronic donor–acceptor decoupling and zero orbital overlap as confirmed by many theoretical and spectroscopic studies (see Section 2 above). This mystery was also the source of significant debates over whether the CT state is twisted. Nevertheless, the TICT mechanism remains the most reasonable and consistent model, with extensive experimental and computational support. In view of the temperature dependence of the radiative decay rate () for the TICT state, the observed TICT fluorescence has been attributed to the emission from higher-lying vibrational levels and hence “hot” fluorescence (see Figure 1c) [92]. For systems with a non-emissive TICT state, the zero orbital overlap results in a forbidden transition and it is often spectrally invisible even for the excited-state electronic absorption (e.g., fs-TA) spectroscopy [35,88]. Another possibility is that the S1 to Sn transition bands are out of the normal probe wavelength window, which could motivate future studies.
The excited-state PES is also quite different. For systems with an emissive TICT state such as DMABN, it has been established that two excited states (1Lb and 1La) are involved to yield the two S1 emission states (LE and TICT, respectively) intimately associated with the amine-twisting coordinate (Figure 1c). Furthermore, the state crossing and adiabatic reaction between LE and TICT states can be described by a multidimensional PES as shown in Figure 6 [82]. The systems with a non-emissive TICT state, on the other hand, have been shown to undergo an adiabatic reaction from the planar/near-planar LE/ICT state to the TICT state solely in S1 (i.e., without the involvement of a higher-lying state such as S2, see Figure 1d). In the emissive TICT case, photoexcitation initially accesses S2 or 1La state at the planar geometry, whereas in the non-emissive TICT case, S1 is the initially accessed state [35,84,91]. Moreover, these two types of systems differ in the degree of CT for the planar emission state. For DMABN and its analogues, the planar emission state was considered an LE state and its absorption and emission wavelengths are largely insensitive to solvent polarity [6]. In contrast, the planar emission state in dark-TICT systems typically has LE/ICT character [7], which could be seen from the solvatochromic results. For example, rhodamines usually exhibit very small changes in absorption and emission wavelengths with varying solvent polarity [93]; therefore, it can be largely viewed as an LE state which is corroborated by the electron density distribution [84]. Many other dyes such as amino derivatives of coumarin, naphthalimide, and locked benzylidene-imidazolinone (Figure 7) exhibit marked solvatochromism [31,34,35,88]; the planar emission states thus have more pronounced ICT character in nature. However, the ICT in the planar state of these chromophores remains weak (i.e., smaller in magnitude) in comparison to that in the TICT state which essentially leads to electronic decoupling between the donor and acceptor moieties (e.g., Coumarin 152, see Figure 9d).
4. Factors Affecting TICT
From the two-state kinetic model established for the LE and TICT states, it is conceivable that any factors that change the reaction barrier or energetics between LE and TICT states would affect the TICT efficiency. To this end, Rettig et al. put forward Equations (1) and (2) to guide and predict possible TICT systems [92,94]:
where and are the energies of the LE and TICT states, respectively; and represent the ionization (or oxidation) potential and electron affinity (or reduction potential) of the donor and acceptor, respectively; C denotes the Coulombic attraction between the radical cation and anion produced by the decoupled donor and acceptor in the TICT state; and as a typically negative value represents the solvent stabilization energy.
The two equations summarize the dominant factors that affect TICT. The donor IP and acceptor EA values describe their respective electron-donating and -accepting capabilities. C is dependent upon the donor and acceptor’s chemical structures. is directly associated with the solvent polarity. Besides, can be affected by applying steric hindrance (see above).
4.1. Donor and Acceptor Strengths
Since the TICT is characterized by the donor–acceptor electronic decoupling event, i.e., a complete electron transfer to form the radical cation and anion, greater strengths of the donor and acceptor would increase the driving force for the charge separation. The electron-donating and -accepting capabilities can be evaluated by the respective IP and EA as used in Equation (2) above.
On the donor end, the IP of amines decreases in the order of primary > secondary > tertiary amines [95,96]. As a result, the primary (–NH2) amine typically cannot enable TICT as observed from the single LE band in 4-aminobenzonitrile (ABN, 1n in Figure 2) [27] and high FQYs of the amino derivatives of dark-TICT chromophores (e.g., 2a and 3a in Figure 7) even in polar solvents [28,85]. These results agree with calculations showing that the TICT state of ABN lies at a very high energy that is difficult to be populated [82]. Simple secondary alkyl amines (–NHR) do not easily promote TICT either (e.g., 7c, 8c, and 9d in Figure 7) [86,87,89] but as the IP is lowered TICT can occur (e.g., –NH-phenyl in 3g of Figure 7) [97]. In comparison, tertiary such as dialkyl amines have lower IP values and can promote TICT more effectively (e.g., low FQYs in dark-TICT chromophores 2c, 4a, 6a, 7a, and 9a in Figure 7). Lengthening the alkyl chain of the dialkylamine group decreases IP(D) and can enhance the TICT access. For example, the TICT fluorescence in amino-benzonitrile is enhanced from dimethyl to didecyl amine (Figure 10a, top to bottom panels) in the same solvent due to the emissive TICT [52]. Meanwhile, the FQY of aminocoumarin decreases from –NH2 to –NEt2 in acetonitrile due to the non-emissive TICT being more populated (i.e., 0.92 to 0.08, from 2a to 2c in Figure 7) [28].
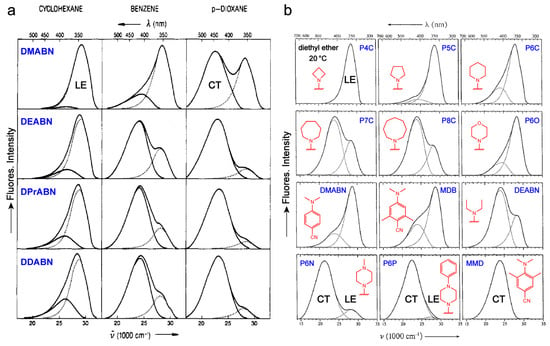
Figure 10.
Effects of donor structure on TICT fluorescence. (a) Fluorescence spectra of DMABN and its derivatives with the increasing alkyl chain length of amine: DMABN (methyl), DEABN (ethyl), DPrABN (n-propyl), and DDABN (n-decyl) at 25 °C in cyclohexane (left), benzene (middle), and 1,4-dioxane (right panels). The fluorescence spectra are decomposed into two bands (LE, dotted; and TICT, solid curves). The frequency (wavenumber) and wavelength axes are shown below and above the spectral data panels, respectively. Adapted with permission from Ref. [52]. Copyright 1992 American Chemical Society. (b) Fluorescence spectra of a series of DMABN derivatives at 20 °C in diethyl ether. The fluorescence spectra are decomposed into LE and/or TICT bands (dotted curves). The chemical structures (red) are depicted in each figure inset. Adapted with permission from Ref. [53]. Copyright 1995 Royal Netherlands Chemical Society.
A similar effect can be observed in chromophores with azacyclic substituents. When the ring size increases, TICT is facilitated due to a lower IP which leads to enhanced TICT fluorescence from azetidinyl to azocanyl group in amino-benzonitrile (i.e., P4C to P8C in Figure 10b) or lower FQYs for dark-TICT chromophores (e.g., azetidinyl versus piperidinyl, Figure 7) [53,83,85]. The four-membered azetidinyl group instead of open-chain dialkylamines has thus often been used to improve the FQY for fluorophores [83,85,98]. Fine tuning of IP or the electron-donating capability of the cyclic amine by introducing heteroatoms is also useful in modulating the TICT efficiency and hence FQYs. For example, changing piperidinyl (P6C) to piperazinyl (P6N) and phenyl-piperazinyl (P6P) groups in amino-benzonitrile decreases the IP and enhances the “bright” TICT fluorescence (Figure 10b) [53]. The morpholinyl group (P6O) has a similar effect on TICT as piperidinyl (P6C) due to their close IP. Inversely, increasing the IP of the cyclic amine hinders TICT and hence improves the FQY for chromophores with “dark” TICT states. As examples, the incorporation of electron-withdrawing groups such as the positively charged ammonium (NMe2+) or sulfone (SO2) to the “donor” piperazinyl group in rhodamine effectively inhibits TICT and enables an exceptional near-unity FQY even in water (3e and 3f, Figure 7) [37,99]. The strategy has recently been adopted to optimize various fluorescent probes for bioimaging applications [37,99,100,101].
On the acceptor end, strong electron-withdrawing groups (large EA) can aid the donor amine to promote TICT. The carboxylic acid, ester, carbonyl, pyridine derivatives of DMABN (1r–1u, Figure 2) also exhibit dual fluorescence, while the tetrafluorinated derivative (1v, Figure 2) only shows the CT emission, though the authors disagreed with the TICT assignment [6,27]. These observations interestingly rule out the RICT model for DMABN that suggests the rehybridization of the nitrile group (Figure 4a). Notably, for these aminobenzene derivatives, it has been shown that very strong acceptors may reverse the energy ordering of 1La and 1Lb type states (Figure 1c) and lead to the LE emission from 1La state such as 1v (Figure 2) instead of 1Lb that occurs in DMABN [102]. Meanwhile, the –CH3 and –CF3 substitutions of diethylamino-coumarin (2d and 2c, Figure 7) clearly indicate the importance of acceptor strength in the occurrence of TICT. Due to the weak electron-withdrawing capability of –CH3, 2d has a FQY of ~1.0 in acetonitrile, much larger than 2c (FQY ≈ 0.08) with a strong electron-withdrawing –CF3 group as the acceptor to promote the access to a non-emissive TICT state (Figure 1d) [28].
4.2. Steric Hindrance and Structural Constraint
Besides electronic factors, the energetics and reaction barrier toward TICT can also be modified by steric hindrance and structural constraint. These two approaches are essentially equivalent in that they destabilize one or the other (i.e., LE/ICT or TICT) states.
The common steric hindrance applied is to incorporate bulky groups (larger than hydrogen) near the twisting amine N–Ph bond such as ortho-dimethylated DMABN (1g, Figure 2) and 2,5-dimethylpyrrolidine-benzonitrile (1i, Figure 2). Due to steric hindrance, the methyl groups cause a deviation from coplanarity between the amine and benzonitrile moieties, resulting in a pre-twisted structure in the ground state [40,43]. In consequence, the LE state also has a twisted structure, leading to a higher energy than the planar geometry without methyl groups. Meanwhile, the methyl groups have little steric influence on the TICT state. As a result, the energy barrier to the TICT state is reduced as seen from the increased LE-to-TICT transition rate [47].
This type of steric hindrance essentially increases the driving force for TICT [103]. A good example is 3,5-dimethyl-4-methylamino-benzonitrile (1h, Figure 2) that shows TICT fluorescence in polar solvents such as acetonitrile while 4-methylamino-benzonitrile (1o, Figure 2) does not [14,27]. The high IP of the secondary amine (–NHMe) is compensated by steric hindrance caused by two methyl groups on the phenyl ring, hence favoring the TICT state formation in 1h. In dark-TICT systems, a bulkier ortho-substituent causes the FQY drop with respect to the unsubstituted one under the same condition. For example, the benzylidene-imidazolinone derivative 9e (0.02, see Figure 7) has a lower FQY than 9a (0.05) in water due to steric hindrance of the ortho–OH group, evinced by ~20° deviation from coplanarity between the –NMe2 group and benzylidene-imidazolinone backbone [34,35]. The steric effect is also present for the heterocyclic amine groups. As the ring becomes larger, the enhanced TICT results from not only the increased donor strength (lower IP) but also increased steric hindrance between the ring and aromatic backbone (e.g., from azetidinyl to piperidinyl groups, see Figure 2 and Figure 7).
The structural constraint/confinement such as the fused cyclization of amine can greatly destabilize the TICT state and hence inhibits TICT (e.g., compounds in Figure 2 and Figure 8). The caveat, however, is that the cycle size needs to be strategically selected to overcome the increased donor strength. Large rings with lower IPs have less rigidity than smaller ones and to certain extents would allow TICT to occur. For example, with the other arm maintained with a methyl group, the seven-membered ring (1d in Figure 2) can still give rise to TICT emission while the five- and six-membered rings (1b and 1c) cannot [15]. However, when the other arm is changed to a tert-butyl group (1e) which notably increases the electron-donating capability of amine, the six-membered ring is no longer rigid enough to suppress TICT [25].
Similar results were also observed for the rhodamine derivatives wherein the donor amine consists of a phenyl group [97]. The strong electron-donating capability makes the secondary amine (3g, see Figure 11) undergo efficient non-emissive TICT formation, thus showing a very low FQY of <0.01 in methanol (Figure 7 and Figure 11). Moreover, the large driving force for TICT cannot be overpowered by the conformational rigidity applied by the six-membered ring (3h) in view of the still-low FQY (0.01) in methanol. Instead, the more rigid and compact five-membered ring effectively inhibits the “dark” TICT and results in a high FQY (0.43) as summarized in Figure 11.
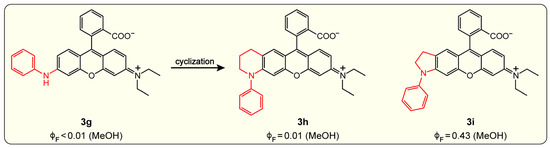
Figure 11.
Effects of fused cyclization of amine groups on the FQY of rhodamine. The FQY data () are taken from Ref. [97].
Another structural constraint has been commonly seen in the julolidine derivatives. The double six-membered rings have been considered rigid to suppress TICT, yet their FQYs are outperformed by a single six-membered ring (Figure 8) [28,34,35,90]. Though it has not been definitively confirmed experimentally or computationally, the aforementioned insights point to a similar origin for the lower FQY in julolidine derivatives. The six-membered ring is not rigid enough to completely inhibit TICT as the donor strength is enhanced by the second ring with respect to methyl in the case of a single six-membered ring. This point is substantiated particularly by the observation that the FQY increases as the donor strength decreases from a six-membered ring to –Me and –H for the locked benzylidene-imidazolinone derivatives (i.e., from 9f to 9g and 9h, Figure 8) in water (i.e., ≈ 0.48 to 0.60 and 0.63) [34,35].
4.3. Solvent Polarity and Viscosity
The solvent polarity dependence is characteristic of the TICT state and LE-to-TICT reaction. This property has been attributed to the large permanent dipole moment of the TICT state, which is much larger than the LE state, on the basis of works on DMABN and its derivatives. Therefore, a larger solvent polarity (i.e., larger in Equation (2)) favors the TICT state formation both thermodynamically and kinetically. On the other hand, the solvent viscosity dependence usually provides convincing evidence for conformational twisting motions which is particularly valuable for the TICT model; however, the pertinent studies in solvents often suffer from the interference by polarity that is difficult to be separated or deconvolved from viscosity [104].
Recently, Chen et al. have proposed an analytical method that quantitatively separates the contributions from solvent polarity and viscosity [35,88,105] to the LE/ICT→TICT reaction rate. The method, as described by Equation (3) below, can be applied to molecular systems wherein TICT is the dominant nonradiative pathway such that :
where and are the total nonradiative decay and isomerization (twisting) rate constants, respectively; represents the twisting rate constant in the absence of solute–solvent interactions; the solvatochromic Kamlet–Taft parameters , , and * describe the solvent H-bond donating, H-bond accepting, and dipolar capabilities, respectively; and is the solvent viscosity. Notably, this approach not only separates solvent polarity and viscosity but also more finely differentiates the specific H-bonding interactions ( and ) from the nonspecific dipolar interactions (*) as fundamental sources for solvent polarity. The latter factor has often been overlooked in previous solvent polarity studies, which could lead to inaccurate or incomplete conclusions.
In practice, the dependence and contribution (coefficients of , , , in Equation (3)) by each parameter can be extracted via rigorous linear regression analysis by sampling a sufficient number of solvents with diverse properties. The experimental value can be calculated by the FQY () and the excited state (i.e., fluorescent state or FS of the chromophore) decay rate constant () via Equations (4) and (5):
where is the radiative decay rate constant. can be obtained from time-resolved spectroscopy such as fs-TA (e.g., see Figure 4b above) while can be measured by steady-state electronic spectroscopy using the relative method with a known standard [106].
The analysis has been successfully applied to the “dark” TICT-capable coumarin and locked benzylidene-imidazolinone derivatives (2c and 9e, Figure 7) and yielded interesting results. For 2c (Coumarin 481), the TICT formation is governed by both H-bonding and dipolar solute–solvent interactions as well as solvent viscosity [88]. The viscosity dependence supports the presence of twisting motions and hence TICT. For 9e, only H-bonding interactions were found to dictate the TICT formation [35]. The comparison between 2c and 9e results implies that, depending on the chromophore structure, the seemingly uniform solvent polarity dependence might be caused by different interactions between the chromophore and solvent molecules (i.e., H-bonding and/or dipolar factors). Though the solvent parameters (, , *) are empirical, the good linear correlations obtained with Equation (3) for chromophores 2c and 9e have provided deep insights into the solute–solvent interactions in controlling the TICT formation. On a related note, the single empirical polarity parameter ET(30) and solvent polarity function Δ based on the Lippert–Mataga equation should be used with caution for spectral data analysis [88,107,108]. The former parameter does not distinguish “specific” H-bonding and “non-specific” dipolar interactions, while the latter parameter calculates the dipole moment difference between the ground and excited states (i.e., only orientational dipole-dipole interaction is considered while all other interactions are omitted) for which H-bonding solvents with the solute should not be included.
4.4. Site Specificity of Donor and Acceptor
The large amount of studies on amine-containing TICT-capable molecules seem to give the impression that TICT can occur in the presence of a flexible amine group as long as the conditions such as donor and acceptor strengths as well as sterics are met. However, the relative positioning of the donor and acceptor groups in aromatic systems is also important for the TICT occurrence. This factor has often been overlooked, leading to some incorrect interpretations for the observed spectral data [109]. Figure 12 lists a series of examples showing such effects on the FQY for various molecular frameworks in solution.
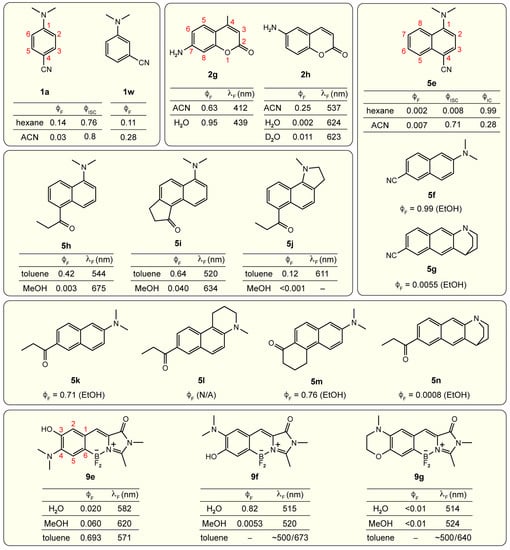
Figure 12.
Positional effects of donor and acceptor on TICT. The available fluorescence (), intersystem crossing (), internal conversion () quantum yield and fluorescence wavelength data for various chromophore derivatives listed are taken from Refs. [27,110] for aminobenzonitrile (1a and 1w), see main text for coumarin (2g and 2h), naphthalene (5e–5n), and the locked benzylidene-imidazolinone (9e–9g). Solvent abbreviations are: ACN, acetonitrile; MeOH, methanol; and EtOH, ethanol.
For the aminobenzene scaffold, the para-substituted derivatives such as DMABN (1a) or others shown in Figure 2 are prone to TICT in medium- to high-polarity solvents; therefore, dual fluorescence bands are often observed. The meta-cyano derivative 1w, however, only displays a single emission band in various solvents [80,110,111,112]. This band also exhibits pronounced solvatochromism (spectral wavelength or color) and fluorogenicity (FQY or brightness), indicative of the ICT nature of the pertinent electronic state [113]. A similar meta effect has also been reported for other di-substituted benzenes [114,115,116]. The role of TICT for these meta-aminobenzonitrile derivatives has not received definitive support due to the limited attention, which can motivate future studies.
Likewise, 6-aminocoumarin (2h, Figure 12) differs from 7-aminocoumarin (2g, with a methyl group at position 4) and the dialkylaminocoumarins (e.g., 2b and 2c in Figure 7) in the extent of solvatochromism and fluorogenicity [117,118,119,120]. The properties of 2h were once ascribed to TICT [109] but this has been recently refuted by theoretical calculations performed on the dimethyl derivative which suggested a different mechanism [121], similar to the meta effect in a benzene ring. For 2h, the highest occupied molecular orbital (HOMO) is drastically destabilized with respect to 2g due to better π-delocalization in the latter by the 7-position electron-donating amino group. This result can be generally understood from the resonance theory as shown in Figure 13 (top middle panel). In contrast to the ground state, the 6-position amino group does not block the π-delocalization in the excited state and hence results in substantial ICT, which accounts for the marked solvatochromism (see 2h data in Figure 12). However, the origin for the observed solvent polarity-governed fluorogenicity remains elusive while TICT was rejected by the energy scan along the amine-twisting coordinate of 2h. One hypothesis suggested that 2h has a lower transition oscillator strength than 2g due to the larger degree of ICT and hence less orbital overlap; it then corresponds to a smaller radiative decay rate constant () which would make competing nonradiative pathways more effective in reducing the FQY in various solvents [121]. Other mechanisms including H-bonding-facilitated IC and triplet state formation have also been proposed [120,122,123].
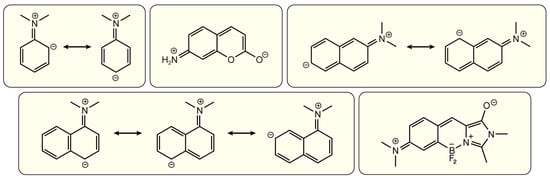
Figure 13.
Representative resonance structures of the aminated chromophore scaffolds listed in Figure 12. The negative signs on certain ring sites denote the specific locations (see the atomic numbering in Figure 12) to position an electron-withdrawing group and thus achieve the improved electron delocalization and energy stabilization.
The naphthalene framework represents another good example to demonstrate the site specificity of donor and acceptor for TICT. The resonance structures of amino-naphthalene (see Figure 13) show that the incorporation of electron-withdrawing groups at positions 4, 5, and 7 for 1-amino-naphthalene (Figure 13, bottom left panel) and positions 6, 8 for 2-amino-naphthalene (Figure 13, top right panel) would give rise to a better electron-delocalized structure, thereby more likely enabling TICT based on the insights gained from aminobenzonitrile and coumarin as mentioned above.
If the acceptor is –CN, the 1,4-subsituted derivative 5e exhibits dual fluorescence (LE and TICT) in polar solvents such as acetonitrile [124] while the 2,6-substituted derivative 5f has a near-unity FQY (0.99) in EtOH (Figure 12) [125]. The twisted chromophore derivative 5g, locked at a perpendicular geometry between the amine donor and naphthalene ring by a quinuclidine moiety, exhibits a minuscule FQY (0.0055) in EtOH. The dramatically contrasting FQY for 5f versus 5g suggests that a non-emissive TICT exists in 5f but is hardly accessible due to a limited driving force, and the near-unity FQY of 5f in EtOH can be considered as LE/ICT fluorescence.
If the acceptor is a carbonyl, the 1,5-substituted derivative 5h shows striking solvent polarity-dependent FQY and fluorescence peak wavelength [126,127]. Though the properties are similar to those of TICT molecules, the observation that the structurally rigidified derivatives 5i and 5j have almost the same solvent dependence (Figure 12) indicates that twisting of neither the amine nor the carbonyl is plausible; therefore, TICT can be excluded [126,127,128]. The origin of the observed intriguing fluorogenicity still requires more investigations. These results suggest that the good electron delocalization by selective positioning of the donor and acceptor is not exclusively responsible or a robust predictor for TICT. For comparison, the 2,6-substituted derivative 5k (also well-known as Prodan dye) with a propionyl group (–COEt) as the acceptor is known for its sensitivity to solvent polarity. Structural modifications of 5k such as confinement for a planar donor (5l) or planar acceptor (5m) and perpendicular donor (5n) have provided direct evidence for a planar structure as the emissive ICT state [125,129,130]. The extremely low FQY of 5n (0.0008 in ethanol) implies the presence of a non-emissive TICT state in 5k if the amine group can freely rotate. This point has been validated by TD-DFT calculations for a similar molecule 5a with an acetyl group (–COMe, see Figure 7) as the acceptor [84]. The smaller FQY variation upon changing the donor or acceptor strength compared to other frameworks (Figure 7) indicates that the barrier for TICT formation is relatively high for the 2,6-subsituted naphthalene (5a–5c in Figure 7, and 5k versus 5f in Figure 12).
Furthermore, the benzylidene-imidazolinone derivatives 9e, 9f, and 9g (see Figure 12, bottom panel) are examples with recently available experimental data from our lab to showcase the site specificity for TICT [35,131], similar to coumarin derivatives 2g and 2h. The derivative 9e is capable of TICT with characteristic polarity-dependent FQYs due to the 4-position –NMe2 group that facilitates electron delocalization (Figure 12 and Figure 13). In sharp contrast, the 3-position –NMe2 group in 9f results in significant fluorescence quenching in alcohols (e.g., 0.0053 in MeOH). The peculiar high FQY in water (0.82) was attributed to the formation of a water-mediated intermolecular H-bonding network between the –NMe2 and 4-position –OH groups of the chromophore, with an intervening solvent (water) molecule. The TICT mechanism was deemed inapplicable for 9f based on quantum calculations as well as the locked amino derivative 9g that exhibits low FQYs in both water and alcohols. In toluene, both 9f and 9g exhibit dual fluorescence (the redder emission band is much stronger than the bluer emission band) but the origin remains elusive (Figure 12), which constitutes a potentially engaging and revealing future research project.
5. Brief Insights into Other Fluorescence Quenching Mechanisms
Besides TICT that can effectively deactivate the excited state, some other processes or mechanisms could also lead to fluorescence quenching in amine-containing molecules. Liu et al. proposed a twisted intramolecular charge shuttle (TICS) mechanism which involves the twisting of the amine group [132]. TICS differs from TICT in that the electron donor and acceptor moieties dynamically switch their roles during the twisting events in the excited state. This mechanism has so far only been proposed and demonstrated in boron-dipyrromethene (BODIPY) derivatives. Photoinduced electron transfer (PET) and Förster resonance energy transfer (FRET) can also cause dramatic fluorescence quenching [133,134]. While the intermolecular PET and FRET have been well studied, the intramolecular ones, particularly the former, are less exhaustively explored [133,135,136,137,138,139,140,141]. For example, intramolecular PET has not received compelling evidence from time-resolved spectroscopy to validate the presence of radical ions, especially for compact molecules. Thus, they are much less common when the aforementioned discussions are limited to compact amine-containing chromophores.
In this section, we will review and discuss some other mechanisms besides TICT that are universal. These energy dissipation/relaxation pathways can readily occur in amine-containing fluorescent molecules, and quench fluorescence to various extents. Note that these mechanisms are usually not exclusive of each other for the same chromophore.
5.1. Intersystem Crossing
Although intersystem crossing (ISC) is not directly caused by amine groups, it can occur in many amine-containing chromophores and significantly affect FQY (). For example, DMABN (1a) has high ISC quantum yields () to enter the lowest triplet state (T1) in various solvents (>70%, see Figure 12 top left panel); that is, ISC is the most important decay pathway [27]. It has been shown that T1 in DMABN is preceded by the TICT state and has a non-twisted structure with no clear CT character [27,69,142]. The prominent ISC is also present for many DMABN derivatives. The naphthalene derivative 5e shows solvent-dependent (Figure 12 top right panel), indicative of the polar excited states (singlet and/or triplet) [143,144].
Though the theory for ISC has advanced from the purely electronic spin-orbital coupling described by El-Sayed’s rule to spin-vibronic coupling with the vibrational contribution outlined by the energy gap law [145,146], an accurate prediction of ISC given a chromophore structure remains challenging. However, some insights into ISC may become practically useful in reducing for a higher FQY.
First, the spin-orbital coupling reveals the heavy-atom effect. Besides metal–organic complexes, halogens are known to promote ISC for organic chromophores such as coumarin and naphthalene (see Section 3 above). For example, the incorporation of iodine at position 3 of Coumarin 1 (7-diethylamino-4-methylcoumarin) causes efficient ISC and leads to a much lower FQY (0.026) than Coumarin 1 (0.375, 2d in Figure 7) in MeOH [147]. Though this trend is not universal, it is noteworthy when low FQYs occur in chromophores that contain halogens (particularly Br and I).
Second, the vibrational aspects of ISC can be described by the energy gap law that formulates the radiationless transition probability of ISC such as S1 → T1 [148]. The weak and strong coupling limits have been discussed to distinguish cases of a small or large displacement between PESs of the two electronic states involved in the transition of interest, respectively. In the weak coupling limit, the transition probability decreases exponentially with increasing energy gap; while in the strong coupling limit, the dependence exhibits a gaussian profile. However, the energy gap law might not be conclusive in attempting to inhibit S1 → T1 transition due to the typically small S1–T1 energy gap. In this case, the acceptor vibrational modes do not necessarily need to be of high frequency such as C–H stretch compared to S1 → S0 and T1 → S0 transitions with large energy gaps (see below in Section 5.2). Lower-frequency modes with relatively high quanta can still be resonant with the S1–T1 energy gap to promote ISC [149,150,151]. Therefore, despite the expected intramolecular isotopic effect as observed for some molecules, the deuteration of C–H with lower vibrational frequencies for different chromophore structures may not lower [152,153,154]. A trial-and-error approach is likely still needed to modify the chromophore structure for effective inhibitions of ISC and achievement of a higher FQY.
5.2. Energy Gap Law
Besides ISC such as S1 → T1 transition, the energy gap law (EGL) is also applicable to the S1 → S0 radiationless transition as internal conversion (IC) that lowers FQY [155,156]. Such IC efficiency usually increases with smaller transition gaps; as a result, the FQY decreases for red-emitting chromophores. Besides the weak coupling limit (regular EGL), the EGL predicts a gaussian dependence of the transition probability on , wherein is the transition energy gap and is the reorganization energy (related to the Stokes shift), in the strong coupling limit where the nuclear displacement between the two PESs (e.g., S1 and S0) is large. In the inverted region, the transition probability increases with a larger [145,146,148]. The fact that many chromophores with large S1–S0 displacements exhibit regular EGL (i.e., a greater transition probability with a decreasing ) is likely due to the much larger electronic transition gap (, in the range of tens of thousands of cm−1) with respect to the Stokes shift (usually thousands of cm−1).
For large transition gaps, high-frequency modes such as C–H stretch are favorable as the energy acceptor modes to promote IC since less quanta are needed with respect to the low-frequency ones (see Section 5.1 above). Therefore, the S1 → S0 IC typically exhibits a pronounced isotopic effect upon deuteration of C–H in the chromophore because C–D has a much lower frequency [149,157,158,159]. The same insight has been manifested by the lengthening of phosphorescence lifetime upon C–H deuteration due to the relatively large T1–S0 gap. The amine group is commonly seen in red-emitting donor–acceptor chromophores due to its strong electron-donating capability while the EGL may play a role in promoting IC and lowering FQY, which can be verified by the deuterium isotope effect.
5.3. Solvent-Assisted Electronic-to-Vibrational FRET
Similar to the EGL that describes the intramolecular electronic-to-vibrational resonance energy transfer (EVRET), the solute-to-solvent intermolecular EVRET (SS-EVRET) has also been found to affect the S1 → S0 IC and hence FQY of the solute chromophore [160,161,162,163,164]. The high-frequency vibrational modes of the solvent such as O–H stretch can act as the acceptors so appreciable deuterium isotope effects usually exist for the chromophores in solvents. For example, water/H2O is a common fluorescence quencher and in many cases the quenching is caused by the SS-EVRET [165,166,167]. Heavy water/D2O can mitigate the quenching by a factor of up to ~3 as reported [167]. The SS-EVRET in water follows an EGL-like trend and the quenching becomes more efficient as the chromophore goes into redder regions. This is because the overtone and combination transitions of the high-frequency O–H stretching modes (symmetric and asymmetric) have weak absorption in the visible red region [168], thus enabling the FRET-type quenching. D2O diminishes this absorption due to the lower O–D stretch frequency. The SS-EVRET has also been found operative in alcohols and solvents with C–H bonds (for some chromophores), which is weaker than that in water [167].
5.4. H-Bonding-Induced Fluorescence Quenching
The H-bonding-induced IC is a commonly discussed mechanism for fluorescence quenching. Many chromophores have been shown to demonstrate the FQY dependence on the strength of solute–solvent H-bonds as studied by both experiments and theoretical calculations, and the influence could be significant [119,169,170,171,172,173,174,175,176,177,178,179,180]. However, the fundamental origin of such H-bonding-assisted nonradiative deactivation is not clearly understood. Moreover, other quenching mechanisms were often neglected in these studies such as ISC, EGL, etc. (see Section 5.1 and Section 5.2 above), so the observed fluorescence quenching was all ascribed to the H-bonding interaction, essentially leading to overinterpretation. It is more likely the case when H-bonding can dramatically red-shift the absorption and emission wavelengths in protic solvents, the other mechanisms such as EGL (Section 5.2) and SS-EVRET (Section 5.3) can also come into play.
On the other hand, the magnitude of FQY reduction upon H-bonding is very large in some cases. Such a result is particularly common for meta-conjugated donor–acceptor chromophores which are highly solvatochromic and fluorogenic with solvent polarity (e.g., 1w and 2h in Figure 12 and others in literature) [114,122]. Other non-meta-conjugated chromophores such as 5h–5j (Figure 12) may also exhibit drastic H-bonding/polarity-dependent FQY [126,127]. These results can hardly be explained by mechanisms such as SS-EVRET which usually results in up to a few folds (<3) of FQY enhancement upon solvent deuteration [167], yet the prominence of mechanisms such as ISC or others still needs to be examined by suitable experimental and computational methods. Nevertheless, the H-bonding-induced fluorescence quenching remains an underexplored mechanism and thus requires further studies to dissect the underlying contributing factors at the molecular level with sufficient spectral and temporal resolutions.
6. Conclusions and Perspectives
In summary, we have reviewed common fluorescence-modulating mechanisms that are directly or indirectly caused by the amine groups which are important constituents for a myriad of organic chromophores such as chemosensors, biosensors, and beyond. The major player, twisted intramolecular charge transfer (TICT), was discussed in detail. We focused on the fundamental aspects of TICT and aimed to provide a comprehensive understanding of TICT including objective evidence, current debates, and key controlling factors. Two types of systems with active TICT state formations, either emissive or non-emissive, are present because of twisting of the amine group. The TICT mechanism is supported by both experimental and computational evidence: (1) structural modifications such as rigidified amines suggest the necessity of the amine-twisting motion in producing a lower-energy CT state in dual-fluorescence chromophores; (2) time-resolved electronic and vibrational spectroscopic data validate the donor–acceptor electronic decoupling upon TICT; (3) theoretical calculations reveal that the twisted conformation is an energy minimum on the excited-state PES. Debates on the assignment of the lower-energy CT emission to a TICT state for dual-fluorescence systems have involved a few other models including PICT (planar), RICT (rehybridized), and WICT (wagged). These models, however, were mostly refuted by either time-resolved spectroscopic or computational studies. TICT remains the most popular and accepted mechanism in explaining the dual fluorescence bands in the family of amino-benzonitriles as well as the significant solvent polarity-dependent fluorescence quenching for many other amine-containing fluorescent molecules. The controlling factors for TICT generation and access were also summarized. In particular, donor and acceptor strengths, steric hindrance and structural constraint, solvent polarity and viscosity, as well as site specificity of donor and acceptor, are shown to be crucial in controlling the occurrence and efficiency of TICT.
Besides TICT, we have also outlined other mechanisms that can contribute to fluorescence quenching pathways for the amine-containing chromophores, especially for those compact ones. Except for PET and FRET, which usually occur in relatively large molecules, intersystem crossing (ISC), energy gap law (EGL), solvent-assisted electronic-to-vibrational FRET, and H-bonding-induced fluorescence quenching constitute several common processes that can influence the fluorescence efficiency.
Practically speaking, organic fluorophores with very high FQYs or environment-sensitive FQYs are desirable for diverse bioimaging and biosensing applications across disciplines. Understanding the structure–energy–function (fluorescence) relationships with fundamental knowledge of possible excited-state quenching pathways is critical for the rational improvement of bright chemosensors and fluorescence-based probes/labels. Since researchers usually focus on one mechanism and overlook the others within one study while the contributing factors are not mutually exclusive, most engineered fluorophores cannot reach unity or near-unity FQYs. Consequently, the research of comprehensively exploring fluorescence quenching pathways for the same system followed by the corresponding molecular engineering, or focusing on one mechanism while disabling the others are expected to be feasible and impactful in guiding the broad communities to develop super-bright fluorophores with suitable emission colors (typically “redder is better” in bioimaging applications) for relevant applications. For example, to study the H-bonding-induced fluorescence quenching, one should ideally ensure beforehand that TICT, ISC, EGL, and SS-EVRET are effectively suppressed/inoperative and thus make marginal interference. Accordingly, one can structurally constrain the amine group, select a fluorophore with no appreciable ISC, use a perdeuterated fluorophore, and use perdeuterated solvents. The conclusion obtained this way would be quantitatively more meaningful in understanding the H-bonding-induced fluorescence quenching, and drawing stronger molecular insights with more predictive power.
Through this review, we hope that the correlated mechanistic insights into TICT as well as general knowledge of other common fluorescence quenching mechanisms could help broad communities to improve the fundamental understandings of typical energy dissipation pathways for the amine-containing chromophores. As the mechanisms such as EGL, SS-EVRET, and H-bonding-induced fluorescence quenching remain underexplored compared to TICT and ISC, more in-depth investigations (from advanced spectroscopic techniques to high-level calculations) for these excited-state non-equilibrium processes in action are welcome in the future. We envision that the comprehensive portrait of radiative and nonradiative decay pathways will substantially boost the efficiency and efficacy in the rational development of bright and colorful fluorescent molecules, materials, sensors, probes, and a versatile group of functional nanomachines in general.
Author Contributions
Conceptualization, C.C. and C.F.; methodology, C.C. and C.F.; validation, C.C. and C.F.; formal analysis, C.C.; investigation, C.C.; resources, C.F.; data curation, C.C.; writing—original draft preparation, C.C.; writing—review and editing, C.F.; visualization, C.C. and C.F.; supervision, C.F.; project administration, C.F.; funding acquisition, C.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by U.S. National Science Foundation (NSF) grant CHE-2003550 to C.F.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mei, J.; Leung, N.L.C.; Kwok, R.T.K.; Lam, J.W.Y.; Tang, B.Z. Aggregation-induced emission: Together we shine, united we soar! Chem. Rev. 2015, 115, 11718–11940. [Google Scholar] [CrossRef]
- Yuan, L.; Lin, W.; Zheng, K.; He, L.; Huang, W. Far-red to near infrared analyte-responsive fluorescent probes based on organic fluorophore platforms for fluorescence imaging. Chem. Soc. Rev. 2013, 42, 622–661. [Google Scholar] [CrossRef]
- Xiao, H.; Li, P.; Tang, B. Recent progresses in fluorescent probes for detection of polarity. Coord. Chem. Rev. 2021, 427, 213582. [Google Scholar] [CrossRef]
- Qin, X.; Yang, X.; Du, L.; Li, M. Polarity-based fluorescence probes: Properties and applications. RSC Med. Chem. 2021, 12, 1826–1838. [Google Scholar] [CrossRef]
- Sasaki, S.; Drummen, G.P.C.; Konishi, G.-i. Recent advances in twisted intramolecular charge transfer (TICT) fluorescence and related phenomena in materials chemistry. J. Mater. Chem. C 2016, 4, 2731–2743. [Google Scholar] [CrossRef]
- Grabowski, Z.R.; Rotkiewicz, K.; Rettig, W. Structural changes accompanying intramolecular electron transfer: Focus on twisted intramolecular charge-transfer states and structures. Chem. Rev. 2003, 103, 3899–4032. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chi, W.; Qiao, Q.; Tan, D.; Xu, Z.; Liu, X. Twisted intramolecular charge transfer (TICT) and twists beyond TICT: From mechanisms to rational designs of bright and sensitive fluorophores. Chem. Soc. Rev. 2021, 50, 12656–12678. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, R.; Batsanov, A.S.; Pander, P.; Hsu, Y.-T.; Chi, Z.; Dias, F.B.; Bryce, M.R. Intramolecular charge transfer controls switching between room temperature phosphorescence and thermally activated delayed fluorescence. Angew. Chem. Int. Ed. 2018, 57, 16407–16411. [Google Scholar] [CrossRef] [PubMed]
- Haberhauer, G.; Gleiter, R.; Burkhart, C. Planarized intramolecular charge transfer: A concept for fluorophores with both large Stokes shifts and high fluorescence quantum yields. Chem. Eur. J. 2016, 22, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Dereka, B.; Svechkarev, D.; Rosspeintner, A.; Tromayer, M.; Liska, R.; Mohs, A.M.; Vauthey, E. Direct observation of a photochemical alkyne–allene reaction and of a twisted and rehybridized intramolecular charge-transfer state in a donor–acceptor dyad. J. Am. Chem. Soc. 2017, 139, 16885–16893. [Google Scholar] [CrossRef]
- Stallhofer, K.; Nuber, M.; Schüppel, F.; Thumser, S.; Iglev, H.; de Vivie-Riedle, R.; Zinth, W.; Dube, H. Electronic and geometric characterization of TICT formation in hemithioindigo photoswitches by picosecond infrared spectroscopy. J. Phys. Chem. A 2021, 125, 4390–4400. [Google Scholar] [CrossRef] [PubMed]
- Rout, Y.; Montanari, C.; Pasciucco, E.; Misra, R.; Carlotti, B. Tuning the fluorescence and the intramolecular charge transfer of phenothiazine dipolar and quadrupolar derivatives by oxygen functionalization. J. Am. Chem. Soc. 2021, 143, 9933–9943. [Google Scholar] [CrossRef] [PubMed]
- Lippert, E.; Lüder, W.; Moll, F.; Nägele, W.; Boos, H.; Prigge, H.; Seibold–Blankenstein, I. Umwandlung von elektronenanregungsenergie. Angew. Chem. 1961, 73, 695–706. [Google Scholar] [CrossRef]
- Zachariasse, K.A.; von der Haar, T.; Hebecker, A.; Leinhos, U.; Kuhnle, W. Intramolecular charge transfer in aminobenzonitriles: Requirements for dual fluorescence. Pure Appl. Chem. 1993, 65, 1745–1750. [Google Scholar] [CrossRef]
- Zachariasse, K.A.; Grobys, M.; von der Haar, T.; Hebecker, A.; Il’ichev, Y.V.; Morawski, O.; Rückert, I.; Kühnle, W. Photo-induced intramolecular charge transfer and internal conversion in molecules with a small energy gap between S1 and S2. Dynamics and structure. J. Photochem. Photobiol. A 1997, 105, 373–383. [Google Scholar] [CrossRef]
- Gorse, A.-D.; Pesquer, M. Intramolecular charge transfer excited state relaxation processes in para-substituted N,N-dimethylaniline: A theoretical study including solvent effects. J. Phys. Chem. 1995, 99, 4039–4049. [Google Scholar] [CrossRef]
- Parusel, A.B.J.; Köhler, G.; Grimme, S. Density functional study of excited charge transfer state formation in 4-(N,N-dimethylamino)benzonitrile. J. Phys. Chem. A 1998, 102, 6297–6306. [Google Scholar] [CrossRef]
- Sudholt, W.; Sobolewski, A.L.; Domcke, W. Ab initio study of the amino group twisting and wagging reaction paths in the intramolecular charge transfer of 4-(N,N-dimethylamino)benzonitrile. Chem. Phys. 1999, 240, 9–18. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W. Charge transfer in aminobenzonitriles: Do they twist? Chem. Phys. Lett. 1996, 250, 428–436. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W. Promotion of intramolecular charge transfer in dimethylamino derivatives: Twisting versus acceptor-group rehybridization. Chem. Phys. Lett. 1996, 259, 119–127. [Google Scholar] [CrossRef]
- Dreyer, J.; Kummrow, A. Shedding light on excited-state structures by theoretical analysis of femtosecond transient infrared spectra: Intramolecular charge transfer in 4-(dimethylamino)benzonitrile. J. Am. Chem. Soc. 2000, 122, 2577–2585. [Google Scholar] [CrossRef]
- Yoshihara, T.; Druzhinin, S.I.; Zachariasse, K.A. Fast intramolecular charge transfer with a planar rigidized electron donor/acceptor molecule. J. Am. Chem. Soc. 2004, 126, 8535–8539. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Cao, Z.; Zhang, Q. Computational characterization of low-lying states and intramolecular charge transfers in N-phenylpyrrole and the planar-rigidized fluorazene. J. Phys. Chem. A 2006, 110, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Cogan, S.; Zilberg, S.; Haas, Y. The electronic origin of the dual fluorescence in donor−acceptor substituted benzene derivatives. J. Am. Chem. Soc. 2006, 128, 3335–3345. [Google Scholar] [CrossRef] [PubMed]
- Druzhinin, S.I.; Kovalenko, S.A.; Senyushkina, T.; Zachariasse, K.A. Dynamics of ultrafast intramolecular charge transfer with 1-tert-butyl-6-cyano-1,2,3,4-tetrahydroquinoline (NTC6) in n-hexane and acetonitrile. J. Phys. Chem. A 2007, 111, 12878–12890. [Google Scholar] [CrossRef] [PubMed]
- Hättig, C.; Hellweg, A.; Köhn, A. Intramolecular charge-transfer mechanism in quinolidines: The role of the amino twist angle. J. Am. Chem. Soc. 2006, 128, 15672–15682. [Google Scholar] [CrossRef]
- Galievsky, V.A.; Druzhinin, S.I.; Demeter, A.; Jiang, Y.-B.; Kovalenko, S.A.; Pérez Lustres, L.; Venugopal, K.; Ernsting, N.P.; Allonas, X.; Noltemeyer, M.; et al. Ultrafast intramolecular charge transfer and internal conversion with tetrafluoro-aminobenzonitriles. ChemPhysChem 2005, 6, 2307–2323. [Google Scholar] [CrossRef]
- Jones, G., II; Jackson, W.R.; Choi, C.Y.; Bergmark, W.R. Solvent effects on emission yield and lifetime for coumarin laser dyes. Requirements for a rotatory decay mechanism. J. Phys. Chem. 1985, 89, 294–300. [Google Scholar] [CrossRef]
- Vogel, M.; Rettig, W.; Sens, R.; Drexhage, K.H. Structural relaxation of rhodamine dyes with different N-substitution patterns: A study of fluorescence decay times and quantum yields. Chem. Phys. Lett. 1988, 147, 452–460. [Google Scholar] [CrossRef]
- Peng, T.; Yang, D. Construction of a library of rhodol fluorophores for developing new fluorescent probes. Org. Lett. 2010, 12, 496–499. [Google Scholar] [CrossRef]
- Saha, S.; Samanta, A. Influence of the structure of the amino group and polarity of the medium on the photophysical behavior of 4-amino-1,8-naphthalimide derivatives. J. Phys. Chem. A 2002, 106, 4763–4771. [Google Scholar] [CrossRef]
- Nad, S.; Kumbhakar, M.; Pal, H. Photophysical properties of coumarin-152 and coumarin-481 dyes: Unusual behavior in nonpolar and in higher polarity solvents. J. Phys. Chem. A 2003, 107, 4808–4816. [Google Scholar] [CrossRef]
- Soujanya, T.; Fessenden, R.W.; Samanta, A. Role of nonfluorescent twisted intramolecular charge transfer state on the photophysical behavior of aminophthalimide dyes. J. Phys. Chem. 1996, 100, 3507–3512. [Google Scholar] [CrossRef]
- Baranov, M.S.; Solntsev, K.M.; Baleeva, N.S.; Mishin, A.S.; Lukyanov, S.A.; Lukyanov, K.A.; Yampolsky, I.V. Red-shifted fluorescent aminated derivatives of a conformationally locked GFP chromophore. Chem. Eur. J. 2014, 20, 13234–13241. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Boulanger, S.A.; Sokolov, A.I.; Baranov, M.S.; Fang, C. A novel dialkylamino GFP chromophore as an environment-polarity sensor reveals the role of twisted intramolecular charge transfer. Chemosensors 2021, 9, 234. [Google Scholar] [CrossRef]
- Chi, W.; Qiao, Q.; Wang, C.; Zheng, J.; Zhou, W.; Xu, N.; Wu, X.; Jiang, X.; Tan, D.; Xu, Z.; et al. Descriptor ΔGc-o enables the quantitative design of spontaneously blinking rhodamines for live-cell super-resolution imaging. Angew. Chem. Int. Ed. 2020, 59, 20215–20223. [Google Scholar] [CrossRef]
- Ye, Z.; Yang, W.; Wang, C.; Zheng, Y.; Chi, W.; Liu, X.; Huang, Z.; Li, X.; Xiao, Y. Quaternary piperazine-substituted rhodamines with enhanced brightness for super-resolution imaging. J. Am. Chem. Soc. 2019, 141, 14491–14495. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Ren, T.-B.; D’Este, E.; Xiong, M.; Xiong, B.; Johnsson, K.; Zhang, X.-B.; Wang, L.; Yuan, L. A synergistic strategy to develop photostable and bright dyes with long Stokes shift for nanoscopy. Nat. Commun. 2022, 13, 2264. [Google Scholar] [CrossRef]
- Rotkiewicz, K.; Grellmann, K.H.; Grabowski, Z.R. Reinterpretation of the anomalous fluorescense of p-N,N-dimethylamino-benzonitrile. Chem. Phys. Lett. 1973, 19, 315–318. [Google Scholar] [CrossRef]
- Grabowski, Z.R.; Rotkiewicz, K.; Siemiarczuk, A. Dual fluorescence of donor-acceptor molecules and the twisted intramolecular charge transfer (TICT) states. J. Lumin. 1979, 18-19, 420–424. [Google Scholar] [CrossRef]
- Demeter, A.; Druzhinin, S.; George, M.; Haselbach, E.; Roulin, J.-L.; Zachariasse, K.A. Dual fluorescence and fast intramolecular charge transfer with 4-(diisopropylamino)benzonitrile in alkane solvents. Chem. Phys. Lett. 2000, 323, 351–360. [Google Scholar] [CrossRef]
- Zachariasse, K.A.; Druzhinin, S.I.; Bosch, W.; Machinek, R. Intramolecular charge transfer with the planarized 4-aminobenzonitrile 1-tert-butyl-6-cyano-1,2,3,4-tetrahydroquinoline (NTC6). J. Am. Chem. Soc. 2004, 126, 1705–1715. [Google Scholar] [CrossRef]
- Rotkiewicz, K.; Rubaszewska, W. Intramolecular charge transfer state and unusual fluorescence from an upper excited singlet of a nonplanar derivative of p-cyano-N, N-dimethylaniline. J. Lumin. 1982, 27, 221–230. [Google Scholar] [CrossRef]
- Rotkiewicz, K.; Rubaszewska, W.x. Intramolecular electron-transfer excited state in 6-cyanobenzquinuclidine. Chem. Phys. Lett. 1980, 70, 444–448. [Google Scholar] [CrossRef]
- Köhler, G.; Rechthaler, K.; Grabner, G.; Luboradzki, R.; Suwińska, K.; Rotkiewicz, K. Structure of cage amines as models for twisted intramolecular charge-transfer states. J. Phys. Chem. A 1997, 101, 8518–8525. [Google Scholar] [CrossRef]
- Bulliard, C.; Allan, M.; Wirtz, G.; Haselbach, E.; Zachariasse, K.A.; Detzer, N.; Grimme, S. Electron energy loss and DFT/SCI study of the singlet and triplet excited states of aminobenzonitriles and benzoquinuclidines: Role of the amino group twist angle. J. Phys. Chem. A 1999, 103, 7766–7772. [Google Scholar] [CrossRef]
- Rettig, W.; Gleiter, R. Dependence of intramolecular rotation in p-cyano-N,N-dialkylanilines on the twist angle. A fluorescence, UV absorption, and photoelectron spectroscopic study. J. Phys. Chem. 1985, 89, 4676–4680. [Google Scholar] [CrossRef]
- Dobkowski, J.; Michl, J.; Waluk, J. Electronic spectroscopy and photophysics of 2-(N-methyl-N-isopropylamino)-5-cyanopyridine and related compounds. Phys. Chem. Chem. Phys. 2003, 5, 1027–1031. [Google Scholar] [CrossRef]
- Dobkowski, J.; Wójcik, J.; Koźmiński, W.; Kołos, R.; Waluk, J.; Michl, J. An experimental test of C−N bond twisting in the TICT state: Syn−anti photoisomerization in 2-(N-methyl-N-isopropylamino)-5-cyanopyridine. J. Am. Chem. Soc. 2002, 124, 2406–2407. [Google Scholar] [CrossRef]
- Cornelissen-Gude, C.; Rettig, W. Dual fluorescence and multiple charge transfer nature in derivatives of N-pyrrolobenzonitrile. J. Phys. Chem. A 1998, 102, 7754–7760. [Google Scholar] [CrossRef]
- Bohnwagner, M.V.; Burghardt, I.; Dreuw, A. Solvent polarity tunes the barrier height for twisted intramolecular charge transfer in N-pyrrolobenzonitrile (PBN). J. Phys. Chem. A 2016, 120, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Schuddeboom, W.; Jonker, S.A.; Warman, J.M.; Leinhos, U.; Kuehnle, W.; Zachariasse, K.A. Excited-state dipole moments of dual fluorescent 4-(dialkylamino)benzonitriles: Influence of alkyl chain length and effective solvent polarity. J. Phys. Chem. 1992, 96, 10809–10819. [Google Scholar] [CrossRef]
- von Der Haar, T.; Hebecker, A.; Il’Ichev, Y.; Jiang, Y.-B.; Kühnle, W.; Zachariasse, K.A. Excited-state intramolecular charge transfer in donor/acceptor-substituted aromatic hydrocarbons and in biaryls. The significance of the redox potentials of the D/A subsystems. Recl. Trav. Chim. Pays-Bas 1995, 114, 430–442. [Google Scholar] [CrossRef]
- Cowley, D.J.; Peoples, A.H. Rotational isomerism and dual luminescence in dipolar dialkylamino-compounds. J. Chem. Soc., Chem. Commun. 1977, 352–353. [Google Scholar] [CrossRef]
- Grabowski, Z.R.; Dobkowski, J.; Kühnle, W. Model compounds in study of the photophysical behaviour of carbonyl derivatives of N,N-dimethylaniline. J. Mol. Struct. 1984, 114, 93–100. [Google Scholar] [CrossRef]
- Herbich, J.; Waluk, J. Excited charge transfer states in 4-aminopyrimidines, 4-(dimethylanilino)pyrimidine and 4-(dimethylamino)pyridne. Chem. Phys. 1994, 188, 247–265. [Google Scholar] [CrossRef]
- Gustavsson, T.; Coto, P.B.; Serrano-Andrés, L.; Fujiwara, T.; Lim, E.C. Do fluorescence and transient absorption probe the same intramolecular charge transfer state of 4-(dimethylamino)benzonitrile? J. Chem. Phys. 2009, 131, 031101. [Google Scholar] [CrossRef]
- Fujiwara, T.; Lee, J.-K.; Zgierski, M.Z.; Lim, E.C. Photophysical and spectroscopic manifestations of the low-lying πσ* state of 4-(dimethylamino)benzethyne: Solvent-polarity dependence of fluorescence and excited-state absorptions. Phys. Chem. Chem. Phys. 2009, 11, 2475–2479. [Google Scholar] [CrossRef]
- Coto, P.B.; Serrano-Andrés, L.; Gustavsson, T.; Fujiwara, T.; Lim, E.C. Intramolecular charge transfer and dual fluorescence of 4-(dimethylamino)benzonitrile: Ultrafast branching followed by a two-fold decay mechanism. Phys. Chem. Chem. Phys. 2011, 13, 15182–15188. [Google Scholar] [CrossRef]
- Fujiwara, T.; Zgierski, M.Z.; Lim, E.C. The role of the πσ* state in intramolecular charge transfer of 4-(dimethylamino)benzonitrile. Phys. Chem. Chem. Phys. 2011, 13, 6779–6783. [Google Scholar] [CrossRef]
- Zachariasse, K.A.; Druzhinin, S.I.; Kovalenko, S.A.; Senyushkina, T. Intramolecular charge transfer of 4-(dimethylamino)benzonitrile probed by time-resolved fluorescence and transient absorption: No evidence for two ICT states and a πσ* reaction intermediate. J. Chem. Phys. 2009, 131, 224313. [Google Scholar] [CrossRef] [PubMed]
- Fdez. Galván, I.; Martín, M.E.; Aguilar, M.A. On the absorption properties of the excited states of DMABN. Chem. Phys. Lett. 2010, 499, 100–102. [Google Scholar] [CrossRef]
- Pedersen, S.U.; Bo Christensen, T.; Thomasen, T.; Daasbjerg, K. New methods for the accurate determination of extinction and diffusion coefficients of aromatic and heteroaromatic radical anions in N,N-dimethylformamide. J. Electroanal. Chem. 1998, 454, 123–143. [Google Scholar] [CrossRef]
- Okada, T.; Uesugi, M.; Köhler, G.; Rechthaler, K.; Rotkiewicz, K.; Rettig, W.; Grabner, G. Time-resolved spectroscopy of DMABN and its cage derivatives 6-cyanobenzquinuclidine (CBQ) and benzquinuclidine (BQ). Chem. Phys. 1999, 241, 327–337. [Google Scholar] [CrossRef]
- Chutny, B.; Swallow, A.J. Aromatic anions and free radicals in the pulse radiolysis of aqueous solutions of benzonitrile. Trans. Faraday Soc. 1970, 66, 2847–2854. [Google Scholar] [CrossRef]
- Kwok, W.M.; Ma, C.; Matousek, P.; Parker, A.W.; Phillips, D.; Toner, W.T.; Towrie, M.; Umapathy, S. A determination of the structure of the intramolecular charge transfer state of 4-dimethylaminobenzonitrile (DMABN) by time-resolved resonance Raman spectroscopy. J. Phys. Chem. A 2001, 105, 984–990. [Google Scholar] [CrossRef]
- Rhinehart, J.M.; Challa, J.R.; McCamant, D.W. Multimode charge-transfer dynamics of 4-(dimethylamino)benzonitrile probed with ultraviolet femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 2012, 116, 10522–10534. [Google Scholar] [CrossRef]
- Kwok, W.-M.; George, M.W.; Grills, D.C.; Ma, C.; Matousek, P.; Parker, A.W.; Phillips, D.; Toner, W.T.; Towrie, M. Direct observation of a hydrogen-bonded charge-transfer state of 4-dimethylaminobenzonitrile in methanol by time-resolved IR spectroscopy. Angew. Chem. Int. Ed. 2003, 42, 1826–1830. [Google Scholar] [CrossRef]
- Hashimoto, M.; Hamaguchi, H.-o. Structure of the twisted-intramolecular-charge-transfer excited singlet and triplet states of 4-(dimethylamino)benzonitrile as studied by nanosecond time-resolved infrared spectroscopy. J. Phys. Chem. 1995, 99, 7875–7877. [Google Scholar] [CrossRef]
- Kwok, W.M.; Ma, C.; George, M.W.; Grills, D.C.; Matousek, P.; Parker, A.W.; Phillips, D.; Toner, W.T.; Towrie, M. Solvent effects on the charge transfer excited states of 4-dimethylaminobenzonitrile (DMABN) and 4-dimethylamino-3,5-dimethylbenzonitrile (TMABN) studied by time-resolved infrared spectroscopy: A direct observation of hydrogen bonding interactions. Photochem. Photobiol. Sci. 2007, 6, 987–994. [Google Scholar] [CrossRef]
- Zhao, G.-J.; Han, K.-L. Time-dependent density functional theory study on hydrogen-bonded intramolecular charge-transfer excited state of 4-dimethylamino-benzonitrile in methanol. J. Comput. Chem. 2008, 29, 2010–2017. [Google Scholar] [CrossRef]
- Juchnovski, I.; Tsvetanov, C.; P anayotov, I. IR-spektren der anion-radikale von aromatischen mononitrilen und elektronenübergänge zwischen anion-radikalen und neutralen molekülen. Monatsh. Chem. 1969, 100, 1980–1992. [Google Scholar] [CrossRef]
- Juchnovski, I.N.; Binev, I.G. Relation between the electronic structure and infrared frequencies of the cyano-group in alkali-metal complexes of some substituted benzonitriles. J. Mol. Struct. 1971, 7, 490–494. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Sudholt, W.; Domcke, W. Ab initio investigation of reaction pathways for intramolecular charge transfer in dimethylanilino derivatives. J. Phys. Chem. A 1998, 102, 2716–2722. [Google Scholar] [CrossRef]
- Serrano-Andres, L.; Merchan, M.; Roos, B.O.; Lindh, R. Theoretical study of the internal charge transfer in aminobenzonitriles. J. Am. Chem. Soc. 1995, 117, 3189–3204. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Photoinduced intramolecular charge transfer in 4-(dimethyl)aminobenzonitrile—A theoretical perspective. J. Am. Chem. Soc. 2004, 126, 1277–1284. [Google Scholar] [CrossRef]
- Parusel, A.B.J. A DFT/MRCI study on the excited state charge transfer states of N-pyrrolobenzene, N-pyrrolobenzonitrile and 4-N,N-dimethylaminobenzonitrile. Phys. Chem. Chem. Phys. 2000, 2, 5545–5552. [Google Scholar] [CrossRef]
- Fuß, W.; Pushpa, K.K.; Rettig, W.; Schmid, W.E.; Trushin, S.A. Ultrafast charge transfer via a conical intersection in dimethylaminobenzonitrile. Photochem. Photobiol. Sci. 2002, 1, 255–262. [Google Scholar] [CrossRef]
- Druzhinin, S.I.; Ernsting, N.P.; Kovalenko, S.A.; Lustres, L.P.; Senyushkina, T.A.; Zachariasse, K.A. Dynamics of ultrafast intramolecular charge transfer with 4-(dimethylamino)benzonitrile in acetonitrile. J. Phys. Chem. A 2006, 110, 2955–2969. [Google Scholar] [CrossRef]
- Druzhinin, S.I.; Galievsky, V.A.; Demeter, A.; Kovalenko, S.A.; Senyushkina, T.; Dubbaka, S.R.; Knochel, P.; Mayer, P.; Grosse, C.; Stalke, D.; et al. Two-state intramolecular charge transfer (ICT) with 3,5-dimethyl-4-(dimethylamino)benzonitrile (MMD) and its meta-isomer mMMD. Ground state amino twist not essential for ICT. J. Phys. Chem. A 2015, 119, 11820–11836. [Google Scholar] [CrossRef]
- Dahl, K.; Biswas, R.; Ito, N.; Maroncelli, M. Solvent dependence of the spectra and kinetics of excited-state charge transfer in three (alkylamino)benzonitriles. J. Phys. Chem. B 2005, 109, 1563–1585. [Google Scholar] [CrossRef] [PubMed]
- Gómez, I.; Reguero, M.; Boggio-Pasqua, M.; Robb, M.A. Intramolecular charge transfer in 4-aminobenzonitriles does not necessarily need the twist. J. Am. Chem. Soc. 2005, 127, 7119–7129. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qiao, Q.; Tian, W.; Liu, W.; Chen, J.; Lang, M.J.; Xu, Z. Aziridinyl fluorophores demonstrate bright fluorescence and superior photostability by effectively inhibiting twisted intramolecular charge transfer. J. Am. Chem. Soc. 2016, 138, 6960–6963. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Qiao, Q.; Chi, W.; Chen, J.; Liu, W.; Tan, D.; McKechnie, S.; Lyu, D.; Jiang, X.-F.; Zhou, W.; et al. Quantitative design of bright fluorophores and AIEgens by the accurate prediction of twisted intramolecular charge transfer (TICT). Angew. Chem. Int. Ed. 2020, 59, 10160–10172. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.B.; English, B.P.; Chen, J.; Slaughter, J.P.; Zhang, Z.; Revyakin, A.; Patel, R.; Macklin, J.J.; Normanno, D.; Singer, R.H.; et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods 2015, 12, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yao, L.; Luo, L.; Wang, H.-X.; Yang, Z.; Wang, Z.; Ai, S.-L.; Zhang, Y.; Zou, Q.-C.; Zhang, H.-L. Alkylaminomaleimide fluorophores: Synthesis via air oxidation and emission modulation by twisted intramolecular charge transfer. Org. Chem. Front. 2021, 8, 239–248. [Google Scholar] [CrossRef]
- Hoelzel, C.A.; Hu, H.; Wolstenholme, C.H.; Karim, B.A.; Munson, K.T.; Jung, K.H.; Zhang, H.; Liu, Y.; Yennawar, H.P.; Asbury, J.B.; et al. A general strategy to enhance donor-acceptor molecules using solvent-excluding substituents. Angew. Chem. Int. Ed. 2020, 59, 4785–4792. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, C.; Fang, C. Polarity-dependent twisted intramolecular charge transfer in diethylamino coumarin revealed by ultrafast spectroscopy. Chemosensors 2022, 10, 411. [Google Scholar] [CrossRef]
- Baleeva, N.S.; Zaitseva, S.O.; Gorbachev, D.A.; Smirnov, A.Y.; Zagudaylova, M.B.; Baranov, M.S. The role of N-substituents in radiationless deactivation of aminated derivatives of a locked GFP chromophore. Eur. J. Org. Chem. 2017, 2017, 5219–5224. [Google Scholar] [CrossRef]
- Whitaker, J.E.; Haugland, R.P.; Ryan, D.; Hewitt, P.C.; Haugland, R.P.; Prendergast, F.G. Fluorescent rhodol derivatives: Versatile, photostable labels and tracers. Anal. Biochem. 1992, 207, 267–279. [Google Scholar] [CrossRef]
- Pedone, A. Role of solvent on charge transfer in 7-aminocoumarin dyes: New hints from TD-CAM-B3LYP and state specific PCM calculations. J. Chem. Theory Comput. 2013, 9, 4087–4096. [Google Scholar] [CrossRef] [PubMed]
- Rettig, W. Charge separation in excited states of decoupled systems—TICT compounds and implications regarding the development of new laser dyes and the primary process of vision and photosynthesis. Angew. Chem. Int. Ed. 1986, 25, 971–988. [Google Scholar] [CrossRef]
- Hinckley, D.A.; Seybold, P.G.; Borris, D.P. Solvatochromism and thermochromism of rhodamine solutions. Spectrochim. Acta A 1986, 42, 747–754. [Google Scholar] [CrossRef]
- Rettig, W. Photoinduced charge separation via twisted intramolecular charge transfer states. In Electron Transfer I; Springer: Berlin, Germany, 1994; Volume 169, pp. 253–299. [Google Scholar]
- Farrell, P.G.; Newton, J. Ionization potentials of aromatic amines. J. Phys. Chem. 1965, 69, 3506–3509. [Google Scholar] [CrossRef]
- Aue, D.H.; Webb, H.M.; Bowers, M.T. Quantitative proton affinities, ionization potentials, and hydrogen affinities of alkylamines. J. Am. Chem. Soc. 1976, 98, 311–317. [Google Scholar] [CrossRef]
- Hanaoka, K.; Iwaki, S.; Yagi, K.; Myochin, T.; Ikeno, T.; Ohno, H.; Sasaki, E.; Komatsu, T.; Ueno, T.; Uchigashima, M.; et al. General design strategy to precisely control the emission of fluorophores via a twisted intramolecular charge transfer (TICT) process. J. Am. Chem. Soc. 2022, 144, 19778–19790. [Google Scholar] [CrossRef]
- Bassolino, G.; Nançoz, C.; Thiel, Z.; Bois, E.; Vauthey, E.; Rivera-Fuentes, P. Photolabile coumarins with improved efficiency through azetidinyl substitution. Chem. Sci. 2018, 9, 387–391. [Google Scholar] [CrossRef]
- Lv, X.; Gao, C.; Han, T.; Shi, H.; Guo, W. Improving the quantum yields of fluorophores by inhibiting twisted intramolecular charge transfer using electron-withdrawing group-functionalized piperidine auxochromes. Chem. Commun. 2020, 56, 715–718. [Google Scholar] [CrossRef]
- Lv, X.; Han, T.; Wu, Y.; Zhang, B.; Guo, W. Improving the fluorescence brightness of distyryl Bodipys by inhibiting the twisted intramolecular charge transfer excited state. Chem. Commun. 2021, 57, 9744–9747. [Google Scholar] [CrossRef]
- Grimm, J.B.; Muthusamy, A.K.; Liang, Y.; Brown, T.A.; Lemon, W.C.; Patel, R.; Lu, R.; Macklin, J.J.; Keller, P.J.; Ji, N.; et al. A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 2017, 14, 987–994. [Google Scholar] [CrossRef]
- Rettig, W.; Wermuth, G.; Lippert, E. Photophysical primary processes in solutions of p-substituted dialkylanilines. Ber. Bunsen-Ges. Phys. Chem. 1979, 83, 692–697. [Google Scholar] [CrossRef]
- Abedi, S.A.A.; Chi, W.; Tan, D.; Shen, T.; Wang, C.; Ang, E.C.X.; Tan, C.-H.; Anariba, F.; Liu, X. Restriction of twisted intramolecular charge transfer enables the aggregation-induced emission of 1-(N,N-dialkylamino)-naphthalene derivatives. J. Phys. Chem. A 2021, 125, 8397–8403. [Google Scholar] [CrossRef] [PubMed]
- Haidekker, M.A.; Brady, T.P.; Lichlyter, D.; Theodorakis, E.A. Effects of solvent polarity and solvent viscosity on the fluorescent properties of molecular rotors and related probes. Bioorg. Chem. 2005, 33, 415–425. [Google Scholar] [CrossRef]
- Chen, C.; Tachibana, S.R.; Baleeva, N.S.; Myasnyanko, I.N.; Bogdanov, A.M.; Gavrikov, A.S.; Mishin, A.S.; Malyshevskaya, K.K.; Baranov, M.S.; Fang, C. Developing bright green fluorescent protein (GFP)-like fluorogens for live-cell imaging with nonpolar protein−chromophore interactions. Chem. Eur. J. 2021, 27, 8946–8950. [Google Scholar] [CrossRef] [PubMed]
- Würth, C.; Grabolle, M.; Pauli, J.; Spieles, M.; Resch-Genger, U. Relative and absolute determination of fluorescence quantum yields of transparent samples. Nat. Protoc. 2013, 8, 1535–1550. [Google Scholar] [CrossRef]
- Dahiya, P.; Kumbhakar, M.; Maity, D.K.; Mukherjee, T.; Mittal, J.P.; Tripathi, A.B.R.; Chattopadhyay, N.; Pal, H. Photophysical properties of 2-amino-9,10-anthraquinone: Evidence for structural changes in the molecule with solvent polarity. Photochem. Photobiol. Sci. 2005, 4, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Satpati, A.K.; Kumbhakar, M.; Nath, S.; Pal, H. Photophysical properties of coumarin-7 dye: Role of twisted intramolecular charge transfer state in high polarity protic solvents. Photochem. Photobiol. 2009, 85, 119–129. [Google Scholar] [CrossRef]
- Rettig, W.; Klock, A. Intramolecular fluorescence quenching in aminocoumarines. Identification of an excited state with full charge separation. Can. J. Chem. 1985, 63, 1649–1653. [Google Scholar] [CrossRef]
- Rettig, W.; Bliss, B.; Dirnberger, K. Pseudo-Jahn–Teller and TICT-models: A photophysical comparison of meta- and para-DMABN derivatives. Chem. Phys. Lett. 1999, 305, 8–14. [Google Scholar] [CrossRef]
- Zachariasse, K.A. Comment on “Pseudo-Jahn–Teller and TICT-models: A photophysical comparison of meta- and para-DMABN derivatives” [Chem. Phys. Lett. 305 (1999) 8]: The PICT model for dual fluorescence of aminobenzonitriles. Chem. Phys. Lett. 2000, 320, 8–13. [Google Scholar] [CrossRef]
- Oshima, J.; Shiobara, S.; Naoumi, H.; Kaneko, S.; Yoshihara, T.; Mishra, A.K.; Tobita, S. Extreme fluorescence sensitivity of some aniline derivatives to aqueous and nonaqueous environments: Mechanistic study and its implication as a fluorescent probe. J. Phys. Chem. A 2006, 110, 4629–4637. [Google Scholar] [CrossRef] [PubMed]
- Klymchenko, A.S. Solvatochromic and fluorogenic dyes as environment-sensitive probes: Design and biological applications. Acc. Chem. Res. 2017, 50, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Chatterjee, T.; Das, A.; Mandal, S.; Sen, A.; Ta, M.; Mandal, P.K. Meta-fluors—A unique way to create a 200 Da ultrasmall fluorophore emitting in red with intense Stokes/solvatochromic shift: Imaging subcellular nanopolarity in live stem cells. J. Phys. Chem. C 2019, 123, 24786–24792. [Google Scholar] [CrossRef]
- Chen, C.; Baranov, M.S.; Zhu, L.; Baleeva, N.S.; Smirnov, A.Y.; Zaitseva, S.O.; Yampolsky, I.V.; Solntsev, K.M.; Fang, C. Designing redder and brighter fluorophores by synergistic tuning of ground and excited states. Chem. Commun. 2019, 55, 2537–2540. [Google Scholar] [CrossRef]
- Chen, C.; Fang, C. Devising efficient red-shifting strategies for bioimaging: A generalizable donor-acceptor fluorophore prototype. Chem. Asian J. 2020, 15, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Pal, H.; Nad, S.; Kumbhakar, M. Photophysical properties of Coumarin-120: Unusual behavior in nonpolar solvents. J. Chem. Phys. 2003, 119, 443–452. [Google Scholar] [CrossRef]
- Bozkurt, E.; Onganer, Y. Photophysical features of Coumarin 120 in reverse micelles. J. Mol. Struct. 2018, 1173, 490–497. [Google Scholar] [CrossRef]
- Lopez Arbeloa, T.; Lopez Arbeloa, F.; Tapia, M.J.; Lopez Arbeloa, I. Hydrogen-bonding effect on the photophysical properties of 7-aminocoumarin derivatives. J. Phys. Chem. 1993, 97, 4704–4707. [Google Scholar] [CrossRef]
- Krystkowiak, E.; Dobek, K.; Burdziński, G.; Maciejewski, A. Radiationless deactivation of 6-aminocoumarin from the S1-ICT state in nonspecifically interacting solvents. Photochem. Photobiol. Sci. 2012, 11, 1322–1330. [Google Scholar] [CrossRef]
- Liu, X.; Cole, J.M.; Xu, Z. Substantial intramolecular charge transfer induces long emission wavelengths and mega Stokes shifts in 6-aminocoumarins. J. Phys. Chem. C 2017, 121, 13274–13279. [Google Scholar] [CrossRef]
- Krystkowiak, E.; Dobek, K.; Maciejewski, A. An intermolecular hydrogen-bonding effect on spectral and photophysical properties of 6-aminocoumarin in protic solvents. Photochem. Photobiol. Sci. 2013, 12, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Krystkowiak, E.; Dobek, K.; Maciejewski, A. Deactivation of 6-aminocoumarin intramolecular charge transfer excited state through hydrogen bonding. Int. J. Mol. Sci. 2014, 15, 16628–16648. [Google Scholar] [CrossRef]
- Ayuk, A.A.; Rettig, W.; Lippert, E. Temperature and viscosity effects on an excited state equilibrium as revealed from the dual fluorescence of very dilute solutions of 1-dimethylamino-4-cyanonaphthalene. Ber. Bunsen-Ges. Phys. Chem. 1981, 85, 553–555. [Google Scholar] [CrossRef]
- Davis, B.N.; Abelt, C.J. Synthesis and photophysical properties of models for twisted PRODAN and dimethylaminonaphthonitrile. J. Phys. Chem. A 2005, 109, 1295–1298. [Google Scholar] [CrossRef]
- Chen, T.; Lee, S.W.; Abelt, C.J. 1,5-prodan emits from a planar intramolecular charge-transfer excited state. ACS Omega 2018, 3, 4816–4823. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.S.; Nagirimadugu, N.V.; Abelt, C.J. Fluorescence quenching of carbonyl-twisted 5-acyl-1-dimethylaminonaphthalenes by alcohols. ACS Omega 2019, 4, 14067–14073. [Google Scholar] [CrossRef]
- Lum, K.; Zielinski, S.M.; Abelt, C.J. Dansyl emits from a PICT excited state. J. Phys. Chem. A 2021, 125, 1229–1233. [Google Scholar] [CrossRef] [PubMed]
- Lobo, B.C.; Abelt, C.J. Does PRODAN possess a planar or twisted charge-transfer excited state? Photophysical properties of two PRODAN derivatives. J. Phys. Chem. A 2003, 107, 10938–10943. [Google Scholar] [CrossRef]
- Everett, R.K.; Nguyen, H.A.A.; Abelt, C.J. Does PRODAN possess an O-TICT excited state? Synthesis and properties of two constrained derivatives. J. Phys. Chem. A 2010, 114, 4946–4950. [Google Scholar] [CrossRef]
- Boulanger, S.A.; Chen, C.; Myasnyanko, I.N.; Sokolov, A.I.; Baranov, M.S.; Fang, C. Excited-state dynamics of a meta-dimethylamino locked GFP chromophore as a fluorescence turn-on water sensor. Photochem. Photobiol. 2022, 98, 311–324. [Google Scholar] [CrossRef]
- Chi, W.; Qiao, Q.; Lee, R.; Liu, W.; Teo, Y.S.; Gu, D.; Lang, M.J.; Chang, Y.-T.; Xu, Z.; Liu, X. A photoexcitation-induced twisted intramolecular charge shuttle. Angew. Chem. Int. Ed. 2019, 58, 7073–7077. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.; Chen, J.; Liu, W.; Wang, C.; Qi, Q.; Qiao, Q.; Tan, T.M.; Xiong, K.; Liu, X.; Kang, K.; et al. A general descriptor ΔE enables the quantitative development of luminescent materials based on photoinduced electron transfer. J. Am. Chem. Soc. 2020, 142, 6777–6785. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Bednar, R.M.; Rozanov, N.D.; Hemshorn, M.L.; Mehl, R.A.; Fang, C. Rational design for high bioorthogonal fluorogenicity of tetrazine-encoded green fluorescent proteins. Nat. Sci. 2022, 2, e20220028. [Google Scholar] [CrossRef] [PubMed]
- Daly, B.; Ling, J.; de Silva, A.P. Current developments in fluorescent PET (photoinduced electron transfer) sensors and switches. Chem. Soc. Rev. 2015, 44, 4203–4211. [Google Scholar] [CrossRef]
- Escudero, D. Revising intramolecular photoinduced electron transfer (PET) from first-principles. Acc. Chem. Res. 2016, 49, 1816–1824. [Google Scholar] [CrossRef]
- Sunahara, H.; Urano, Y.; Kojima, H.; Nagano, T. Design and synthesis of a library of BODIPY-based environmental polarity sensors utilizing photoinduced electron-transfer-controlled fluorescence on/off switching. J. Am. Chem. Soc. 2007, 129, 5597–5604. [Google Scholar] [CrossRef]
- de Silva, A.P. Luminescent photoinduced electron transfer (PET) molecules for sensing and logic operations. J. Phys. Chem. Lett. 2011, 2, 2865–2871. [Google Scholar] [CrossRef]
- Filatov, M.A. Heavy-atom-free BODIPY photosensitizers with intersystem crossing mediated by intramolecular photoinduced electron transfer. Org. Biomol. Chem. 2020, 18, 10–27. [Google Scholar] [CrossRef]
- Jiao, G.-S.; Thoresen, L.H.; Burgess, K. Fluorescent, through-bond energy transfer cassettes for labeling multiple biological molecules in one experiment. J. Am. Chem. Soc. 2003, 125, 14668–14669. [Google Scholar] [CrossRef]
- Lee, Y.; Cho, W.; Sung, J.; Kim, E.; Park, S.B. Monochromophoric design strategy for tetrazine-based colorful bioorthogonal probes with a single fluorescent core skeleton. J. Am. Chem. Soc. 2018, 140, 974–983. [Google Scholar] [CrossRef]
- Köhler, G.; Grabner, G.; Rotkiewicz, K. Nonradiative deactivation and triplet states in donor-aryl-acceptor compounds (dialkylaminobenzonitriles). Chem. Phys. 1993, 173, 275–290. [Google Scholar] [CrossRef]
- Zachariasse, K.A.; Grobys, M.; von der Haar, T.; Hebecker, A.; Il’ichev, Y.V.; Jiang, Y.B.; Morawski, O.; Kühnle, W. Intramolecular charge transfer in the excited state. Kinetics and configurational changes. J. Photochem. Photobiol. A 1996, 102, 59–70. [Google Scholar] [CrossRef]
- Suzuki, K.; Demeter, A.; Kühnle, W.; Tauer, E.; Zachariasse, K.A.; Tobita, S.; Shizuka, H. Internal conversion in 4-substituted 1-naphthylamines. Influence of the electron donor/acceptor substituent character. Phys. Chem. Chem. Phys. 2000, 2, 981–991. [Google Scholar] [CrossRef]
- Penfold, T.J.; Gindensperger, E.; Daniel, C.; Marian, C.M. Spin-vibronic mechanism for intersystem crossing. Chem. Rev. 2018, 118, 6975–7025. [Google Scholar] [CrossRef]
- Marian, C.M. Understanding and controlling intersystem crossing in molecules. Annu. Rev. Phys. Chem. 2021, 72, 617–640. [Google Scholar] [CrossRef]
- Das, A.; Ghosh, S.K.; Ramamurthy, V.; Sen, P. Vibration-assisted intersystem crossing in the ultrafast excited-state relaxation dynamics of halocoumarins. J. Phys. Chem. A 2022, 126, 1475–1485. [Google Scholar] [CrossRef]
- Englman, R.; Jortner, J. The energy gap law for radiationless transitions in large molecules. Mol. Phys. 1970, 18, 145–164. [Google Scholar] [CrossRef]
- Robinson, G.W.; Frosch, R.P. Electronic excitation transfer and relaxation. J. Chem. Phys. 1963, 38, 1187–1203. [Google Scholar] [CrossRef]
- Siebrand, W. Radiationless transitions in polyatomic molecules. I. Calculation of Franck–Condon factors. J. Chem. Phys. 1967, 46, 440–447. [Google Scholar] [CrossRef]
- Freed, K.F.; Jortner, J. Multiphonon processes in the nonradiative decay of large molecules. J. Chem. Phys. 1970, 52, 6272–6291. [Google Scholar] [CrossRef]
- Lin, S.H.; Bersohn, R. Effect of partial deuteration and temperature on triplet-state lifetimes. J. Chem. Phys. 1968, 48, 2732–2736. [Google Scholar] [CrossRef]
- Kellmann, A. Intersystem crossing and internal conversion quantum yields of acridine in polar and nonpolar solvents. J. Phys. Chem. 1977, 81, 1195–1198. [Google Scholar] [CrossRef]
- Ermolaev, V.L. The influence of deuteration of complex organic molecules on their fluorescence quantum yield (a review). Opt. Spectrosc. 2016, 121, 567–584. [Google Scholar] [CrossRef]
- Erker, C.; Basché, T. The energy gap law at work: Emission yield and rate fluctuations of single NIR emitters. J. Am. Chem. Soc. 2022, 144, 14053–14056. [Google Scholar] [CrossRef] [PubMed]
- Caspar, J.V.; Kober, E.M.; Sullivan, B.P.; Meyer, T.J. Application of the energy gap law to the decay of charge-transfer excited states. J. Am. Chem. Soc. 1982, 104, 630–632. [Google Scholar] [CrossRef]
- Wang, C.; Otto, S.; Dorn, M.; Kreidt, E.; Lebon, J.; Sršan, L.; Di Martino-Fumo, P.; Gerhards, M.; Resch-Genger, U.; Seitz, M.; et al. Deuterated molecular Ruby with record luminescence quantum yield. Angew. Chem. Int. Ed. 2018, 57, 1112–1116. [Google Scholar] [CrossRef]
- Avouris, P.; Gelbart, W.M.; El-Sayed, M.A. Nonradiative electronic relaxation under collision-free conditions. Chem. Rev. 1977, 77, 793–833. [Google Scholar] [CrossRef]
- Kusinski, M.; Nagesh, J.; Gladkikh, M.; Izmaylov, A.F.; Jockusch, R.A. Deuterium isotope effect in fluorescence of gaseous oxazine dyes. Phys. Chem. Chem. Phys. 2019, 21, 5759–5770. [Google Scholar] [CrossRef]
- Inoue, H.; Hida, M.; Nakashima, N.; Yoshihara, K. Picosecond fluorescence lifetimes of anthraquinone derivatives. Radiationless deactivation via intra- and intermolecular hydrogen bonds. J. Phys. Chem. 1982, 86, 3184–3188. [Google Scholar] [CrossRef]
- Magde, D.; Rojas, G.E.; Seybold, P.G. Solvent dependence of the fluorescence lifetimes of xanthene dyes. Photochem. Photobiol. 1999, 70, 737–744. [Google Scholar] [CrossRef]
- Das, S.; Datta, A.; Bhattacharyya, K. Deuterium isotope effect on 4-aminophthalimide in neat water and reverse micelles. J. Phys. Chem. A 1997, 101, 3299–3304. [Google Scholar] [CrossRef]
- Aharoni, A.; Oron, D.; Banin, U.; Rabani, E.; Jortner, J. Long-range electronic-to-vibrational energy transfer from nanocrystals to their surrounding matrix environment. Phys. Rev. Lett. 2008, 100, 057404. [Google Scholar] [CrossRef] [PubMed]
- Yatsuhashi, T.; Nakajima, Y.; Shimada, T.; Inoue, H. Photophysical properties of intramolecular charge-transfer excited singlet state of aminofluorenone derivatives. J. Phys. Chem. A 1998, 102, 3018–3024. [Google Scholar] [CrossRef]
- Lee, S.F.; Vérolet, Q.; Fürstenberg, A. Improved super-resolution microscopy with oxazine fluorophores in heavy water. Angew. Chem. Int. Ed. 2013, 52, 8948–8951. [Google Scholar] [CrossRef]
- Klehs, K.; Spahn, C.; Endesfelder, U.; Lee, S.F.; Fürstenberg, A.; Heilemann, M. Increasing the brightness of cyanine fluorophores for single-molecule and superresolution imaging. ChemPhysChem 2014, 15, 637–641. [Google Scholar] [CrossRef]
- Maillard, J.; Klehs, K.; Rumble, C.; Vauthey, E.; Heilemann, M.; Fürstenberg, A. Universal quenching of common fluorescent probes by water and alcohols. Chem. Sci. 2021, 12, 1352–1362. [Google Scholar] [CrossRef]
- Braun, C.L.; Smirnov, S.N. Why is water blue? J. Chem. Edu. 1993, 70, 612–614. [Google Scholar] [CrossRef]
- Fita, P.; Fedoseeva, M.; Vauthey, E. Ultrafast excited-state dynamics of Eosin B: A potential probe of the hydrogen-bonding properties of the environment. J. Phys. Chem. A 2011, 115, 2465–2470. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.-J.; Han, K.-L. Hydrogen bonding in the electronic excited state. Acc. Chem. Res. 2012, 45, 404–413. [Google Scholar] [CrossRef]
- Waluk, J. Hydrogen-bonding-induced phenomena in bifunctional heteroazaaromatics. Acc. Chem. Res. 2003, 36, 832–838. [Google Scholar] [CrossRef]
- Oshima, J.; Yoshihara, T.; Tobita, S. Water-induced fluorescence quenching of mono- and dicyanoanilines. Chem. Phys. Lett. 2006, 423, 306–311. [Google Scholar] [CrossRef]
- Biczók, L.; Bérces, T.; Linschitz, H. Quenching processes in hydrogen-bonded pairs: Interactions of excited fluorenone with alcohols and phenols. J. Am. Chem. Soc. 1997, 119, 11071–11077. [Google Scholar] [CrossRef]
- Krystkowiak, E.; Dobek, K.; Maciejewski, A. Origin of the strong effect of protic solvents on the emission spectra, quantum yield of fluorescence and fluorescence lifetime of 4-aminophthalimide: Role of hydrogen bonds in deactivation of S1-4-aminophthalimide. J. Photochem. Photobiol. A 2006, 184, 250–264. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W. Computational studies of the photophysics of hydrogen-bonded molecular systems. J. Phys. Chem. A 2007, 111, 11725–11735. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.-J.; Han, K.-L. Ultrafast hydrogen bond strengthening of the photoexcited fluorenone in alcohols for facilitating the fluorescence quenching. J. Phys. Chem. A 2007, 111, 9218–9223. [Google Scholar] [CrossRef]
- Huang, G.-J.; Ho, J.-H.; Prabhakar, C.; Liu, Y.-H.; Peng, S.-M.; Yang, J.-S. Site-selective hydrogen-bonding-induced fluorescence quenching of highly solvatofluorochromic GFP-like chromophores. Org. Lett. 2012, 14, 5034–5037. [Google Scholar] [CrossRef]
- Yatsuhashi, T.; Inoue, H. Molecular mechanism of radiationless deactivation of aminoanthraquinones through intermolecular hydrogen-bonding interaction with alcohols and hydroperoxides. J. Phys. Chem. A 1997, 101, 8166–8173. [Google Scholar] [CrossRef]
- Dereka, B.; Vauthey, E. Direct local solvent probing by transient infrared spectroscopy reveals the mechanism of hydrogen-bond induced nonradiative deactivation. Chem. Sci. 2017, 8, 5057–5066. [Google Scholar] [CrossRef]
- Zhao, G.-J.; Han, K.-L. Role of intramolecular and intermolecular hydrogen bonding in both singlet and triplet excited states of aminofluorenones on internal conversion, intersystem crossing, and twisted intramolecular charge transfer. J. Phys. Chem. A 2009, 113, 14329–14335. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).