
Antioxidant Determination with the Use of Carbon-Based Electrodes

 , ,
, , 
Abstract
:1. Antioxidants—General Aspects and Main Determination Techniques
1.1. Defining, Classifying and Describing Modes of Action Antioxidants
1.2. Analytical Methods Applied to Antioxidant Determination
1.2.1. General Overview of Methods
1.2.2. Electrochemical Techniques
1.2.3. Biosensor Methods
2. Carbon Electrodes—General Overview
3. Determination of Individual Antioxidants with Carbon-Based Electrodes
4. Determination of Total Antioxidant Activity with Carbon-Based Electrodes
5. Critical Perspectives and Comparative Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A Review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Cimpeanu, C.; Predoi, G. Antioxidant capacity determination in plants and plant-derived products: A review. Oxid. Med. Cell. Longev. 2016, 36, 2016. [Google Scholar] [CrossRef] [Green Version]
- Pisoschi, A.M.; Pop, A.; Iordache, F.; Stanca, L.; Predoi, G.; Serban, A.I. Oxidative stress mitigation by antioxidants-an overview on their chemistry and influences on health status. Eur. J. Med. Chem. 2021, 209, 112891. [Google Scholar] [CrossRef]
- Gulcin, I. Antioxidants and antioxidant methods: An updated overview. Arch. Toxicol. 2020, 94, 651–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, N.; Watanabe, A.; Shi, H. Diverse functions of antioxidants. Free Radic. Res. 2001, 33, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alexandria J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Poljsak, B.; Suput, D.; Milisav, I. Achieving the balance between ROS and antioxidants: When to use the synthetic antioxidants. Oxid. Med. Cell. Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef] [PubMed]
- Santos-Sánchez, N.F.; Salas-Coronado, R.; Villanueva-Cañongo, C.; Hernández-Carlos, B. Chapter 2, Antioxidant compounds and their antioxidant mechanism. In Antioxidants; Shalaby, E., Ed.; InTech Open: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Prior, R.L.; Wu, X.; Schaich, K. Standardized methods for the determination of antioxidant capacity and phenolics in foods and dietary supplements. J. Agric. Food Chem. 2005, 253, 4290–4302. [Google Scholar] [CrossRef]
- Jimenez, A.; Selga, A.; Torres, J.L.; Julia, L. Reducing activity of polyphenols with stable radicals of the TTM series. Electron transfer versus H-abstraction reactions in flavan-3-ols. Org. Lett. 2004, 6, 4583–4586. [Google Scholar] [CrossRef]
- Campesi, I.; Marino, M.; Cipolletti, M.; Romani, A.; Franconi, F. Put “gender glasses” on the effects of phenolic compounds on cardiovascular function and diseases. Eur. J. Nutr. 2018, 57, 2677–2691. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A.; Cimpeanu, C.; Turcus, V.; Predoi, G.; Iordache, F. Nanoencapsulation techniques for compounds and products with antioxidant and antimicrobial activity-a critical view. Eur. J. Med. Chem. 2018, 157, 1326–1345. [Google Scholar] [CrossRef] [PubMed]
- Carocho, M.; Morales, P.; Ferreira, I.C.F.R. Antioxidants: Reviewing the chemistry, food applications, legislation and role as preservatives. Trends Food Sci. Technol. 2018, 71, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Bunaciu, A.A.; Danet, A.F.; Fleschin, S.; Aboul-Enein, H.Y. Recent applications for in vitro antioxidant assay. Crit. Rev. Anal. Chem. 2016, 46, 389–399. [Google Scholar] [CrossRef]
- Apak, R.; Özyürek, M.; Güçlü, K.; Çapanoglu, E. Antioxidant activity/capacity measurement. 1. classification, physicochemical principles, mechanisms, and electron transfer (ET)-based assays. J. Agric. Food Chem. 2016, 64, 997–1027. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Serban, A.; Popa, C.V.; Florescu, M. A nanoparticle-based label-free sensor for screening the relative antioxidant capacity of hydrosoluble plant extracts. Sensors 2019, 19, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brainina, K.; Stozhko, N.; Vidrevich, M. Antioxidants: Terminology, methods, and future considerations. Antioxidants 2019, 8, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisoschi, A.M.; Negulescu, G.P. Methods for total antioxidant activity determination: A review. Biochem. Anal. Biochem. 2012, 1, 106. [Google Scholar] [CrossRef] [Green Version]
- Sadeer, N.B.; Montesano, D.; Albrizio, S.; Zengin, G.; Mahomoodally, M.F. The versatility of antioxidant assays in food science and safety-chemistry, applications, strengths, and limitations. Antioxidants 2020, 9, 709. [Google Scholar] [CrossRef]
- Romanet, R.; Coelho, C.; Liu, Y.; Bahut, F.; Ballester, J.; Nikolantonaki, M.; Gougeon, R.D. The antioxidant potential of white wines relies on the chemistry of sulfur-containing compounds: An optimized DPPH assay. Molecules 2019, 24, 1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Yang, L.; Xue, Q.; Yao, F.; Sun, J.; Yang, F.; Liu, Y. Antioxidant evaluation-guided chemical profling and structureactivity analysis of leaf extracts from five trees in Broussonetia and Morus (Moraceae). Sci. Rep. 2020, 10, 4808. [Google Scholar] [CrossRef] [PubMed]
- Ilyasov, I.R.; Beloborodov, V.L.; Selivanova, I.A.; Terekhov, R.P. ABTS/PP decolorization assay of antioxidant capacity reaction pathways. Int. J. Mol. Sci. 2020, 21, 1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turkiewicz, I.P.; Wojdyło, A.; Tkacz, K.; Nowicka, P.; Golis, T.; Bąbelewski, P. ABTS On-Line antioxidant, α-amylase, α-glucosidase, pancreatic lipase, acetyl-and butyrylcholinesterase inhibition activity of chaenomeles fruits determined by polyphenols and other chemical compounds. Antioxidants 2020, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Payne, A.C.; Mazzer, A.; Clarkson, G.J.J.; Taylor, G. Antioxidant assays-consistent findings from FRAP and ORAC reveal a negative impact of organic cultivation on antioxidant potential in spinach but not watercress or rocket leaves. Food Sci. Nutr. 2013, 1, 439–444. [Google Scholar] [CrossRef]
- Chaves, N.; Santiago, A.; Alías, J.C. Quantification of the antioxidant activity of plant extracts: Analysis of sensitivity and hierarchization based on the method used. Antioxidants 2020, 9, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berker, K.I.; Güçlü, K.; Tor, I.; Demirata, B.; Apak, R. Total antioxidant capacity assay using optimized ferricyanide/prussian blue method. Food Anal. Methods 2010, 3, 154–168. [Google Scholar] [CrossRef]
- Meng, J.; Fang, Y.; Zhang, A.; Chen, S.; Xu, T.; Ren, Z.; Han, G.; Liu, J.; Li, H.; Zhang, Z.; et al. Phenolic content and antioxidant capacity of Chinese raisins produced in Xinjiang Province. Food Res. Int. 2011, 44, 2830–2836. [Google Scholar] [CrossRef]
- Özyürek, M.; Güçlü, K.; Tütem, E.; Başkan, K.S.; Erçağ, E.; Çelik, S.E.; Baki, S.; Yıldız, L.; Karamanc, Ş.; Apak, R. A comprehensive review of CUPRAC methodology. Anal. Methods 2011, 11, 2439–2453. [Google Scholar] [CrossRef]
- Çekiç, S.D.; Demir, A.; Başkan, K.S.; Tütem, E.; Apak, R. Determination of total antioxidant capacity of milk by CUPRAC and ABTS methods with separate characterisation of milk protein fractions. J. Dairy Res. 2015, 82, 177–184. [Google Scholar] [CrossRef]
- Catalán, V.; Frühbeck, G.; Gómez-Ambrosi, J. Inflammatory and oxidative stress markers in skeletal muscle of obese subjects. In Oxidative Stress and Dietary Antioxidants; del Moral, A.M., García, C.M.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 163–189. [Google Scholar]
- De Leon, J.A.D.; Borges, C.R. Evaluation of oxidative stress in biological samples using the thiobarbituric acid reactive substances assay. J. Vis. Exp. 2020, 159, e61122. [Google Scholar] [CrossRef]
- Benbouguerra, N.; Richard, T.; Saucier, C.; Garcia, F. Voltammetric behavior, flavanol and anthocyanin contents, and antioxidant capacity of grape skins and seeds during ripening (Vitis vinifera var. Merlot, Tannat, and Syrah). Antioxidants 2020, 9, 800. [Google Scholar] [CrossRef] [PubMed]
- Mohtar, L.G.; Messina, G.A.; Bertolino, F.A.; Pereira, S.V.; Raba, J.; Nazareno, M.A. Comparative study of different methodologies for the determination the antioxidant activity of Venezuelan propolis. Microchem. J. 2020, 158, 105244. [Google Scholar] [CrossRef]
- Roy, M.K.; Koide, M.; Rao, T.P.; Okubo, T.; Ogasawara, Y.; Juneja, L.R. ORAC and DPPH assay comparison to assess antioxidant capacity of tea infusions: Relationship between total polyphenol and individual catechin content. Int. J. Food Sci. Nutr. 2010, 61, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Litescu, S.C.; Eremia, S.A.V.; Tache, A.; Vasilescu, I.; Radu, G.-L. The use of Oxygen Radical Absorbance Capacity (ORAC) and Trolox Equivalent Antioxidant Capacity (TEAC) assays in the assessment of beverages’ antioxidant properties. In Processing and Impact on Antioxidants in Beverages; Preedy, V., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 245–251. [Google Scholar]
- Romero-Diez, R.; Rodriguez-Rojo, S.; Cocero, M.J.; Duarte, C.M.M.; Matias, A.A.; Bronze, M.R. Phenolic characterization of aging wine lees: Correlation with antioxidant activities. Food Chem. 2018, 259, 188–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denev, P.; Todorova, V.; Ognyanov, M.; Georgiev, Y.; Yanakieva, I.; Tringovska, I.; Grozeva, S.; Kostova, D. Phytochemical composition and antioxidant activity of 63 Balkan pepper (Capsicum annuum L.) accessions. J. Food Meas. Charact. 2019, 13, 2510–2520. [Google Scholar] [CrossRef]
- Gupta, D. Methods for determination of antioxidant capacity: A review. Int. J. Pharm. Sci. Res. 2015, 6, 546–566. [Google Scholar]
- Xiao, F.; Xu, T.; Lu, B.; Liu, R. Guidelines for antioxidant assays for food components. Food Front. 2020, 1, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Tomassetti, M.; Serone, M.; Angeloni, R.; Campanella, L.; Mazzone, E. Amperometric enzyme sensor to check the total antioxidant capacity of several mixed berries. Comparison with two other spectrophotometric and fluorimetric methods. Sensors 2015, 15, 3435–3452. [Google Scholar] [CrossRef] [PubMed]
- Tubaro, F.; Pizzuto, R.; Raimo, G.; Paventi, G. A novel fluorimetric method to evaluate red wine antioxidant activity. Period. Polytech. Chem. Eng. 2019, 63, 57–64. [Google Scholar] [CrossRef]
- Shpigun, L.K.; Arharova, M.A.; Brainina, K.Z.; Ivanova, A.V. Flow injection potentiometric determination of total antioxidant activity of plant extracts. Anal. Chim. Acta 2006, 573–574, 419–426. [Google Scholar] [CrossRef]
- Brainina, K.Z.; Stozhko, N.Y.; Buharinova, M.A.; Khamzina, E.I.; Vidrevich, M.B. Potentiometric method of plant microsuspensions antioxidant activity determination. Food Chem. 2019, 278, 653–658. [Google Scholar] [CrossRef]
- Chevion, S.; Roberts, M.A.; Chevion, M. The use of cyclic voltammetry for the evaluation of antioxidant capacity. Free Radic. Biol. Med. 2000, 28, 860–870. [Google Scholar] [CrossRef]
- Cardenas, A.; Frontana, C. Evaluation of a carbon ink chemically modified electrode incorporating a copper-neocuproine complex for the quantification of antioxidants. Sens. Actuators B Chem. 2020, 313, 128070. [Google Scholar] [CrossRef]
- Lubeckyj, R.A.; Winkler-Moser, J.K.; Fhaner, M.J. Application of differential pulse voltammetry to determine the efficiency of stripping tocopherols from commercial fish oil. J. Am. Oil Chem. Soc. 2017, 94, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Trofin, A.E.; Trinca, L.C.; Ungureanu, E.; Ariton, A.M. CUPRAC voltammetric determination of antioxidant capacity in tea samples by using screen-printed microelectrodes. J. Anal. Methods Chem. 2019, 2019, 8012758. [Google Scholar] [CrossRef] [PubMed]
- Giovagnoli-Vicuña, C.; Pizarro, S.; Briones-Labarca, V.; Delgadillo, A. A square wave voltammetry study on the antioxidant interaction and effect of extraction method for binary fruit mixture extracts. J. Chem. 2019, 3, 1–10. [Google Scholar] [CrossRef]
- Savan, E.K. Square wave voltammetric (SWV) determination of quercetin in tea samples at a single-walled carbon nanotube (SWCNT) modified glassy carbon electrode (GCE). Anal. Lett. 2020, 53, 858–872. [Google Scholar] [CrossRef]
- Gordiienko, A.; Blaheyevskiy, M.; Iurchenko, I. A comparative study of phenolic compound antioxidant activity by the polarography method, using microsomal lipid peroxidation in vitro. Curr. Issues Pharm. Med. Sci. 2018, 31, 186–189. [Google Scholar] [CrossRef]
- Karaman, M.; Tesanovic, K.; Gorjanovic, S.; Pastor, F.T.; Simonovic, M.; Glumac, M.; Pejin, B. Polarography as a technique of choice for the evaluation of total antioxidant activity: The case study of selected Coprinus Comatus extracts and quinic acid, their antidiabetic ingredient. Nat. Prod. Res. 2019, in press. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Cimpeanu, C.; Predoi, G. Electrochemical methods for total antioxidant capacity and its main contributors determination: A review. Open Chem. 2015, 13, 824–856. [Google Scholar] [CrossRef] [Green Version]
- Sazhina, N.N. Determination of antioxidant activity of various bioantioxidants and their mixtures by the amperometric method. Russ. J. Bioorganic Chem. 2017, 43, 771–775. [Google Scholar] [CrossRef]
- Tougas, T.P.; Jannetti, J.M.; Collier, W.G. Theoretical and experimental response of a biamperometric detector for flow injection analysis. Anal. Chem. 1985, 57, 1377–1381. [Google Scholar] [CrossRef]
- Milardovic, S.; Kerekovic, I.; Rumenjak, V. A flow injection biamperometric method for determination of total antioxidant capacity of alcoholic beverages using bienzymatically produced ABTS+. Food Chem. 2007, 105, 1688–1694. [Google Scholar] [CrossRef]
- Moldoveanu, S. The utilization of gas chromatography/mass spectrometry in the profiling of several antioxidants in botanicals. In Advances in Chromatography; Guo, X., Ed.; InTech Open: London, UK, 2014; Chapter 5. [Google Scholar] [CrossRef] [Green Version]
- Viet, T.D.; Xuan, T.D.; Van, T.M.; Andriana, Y.; Rayee, R.; Tran, H.-D. Comprehensive fractionation of antioxidants and GC-MS and ESI-MS fingerprints of celastrus hindsii leaves. Medicines 2019, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Merken, H.M.; Beecher, G.R. Measurement of food flavonoids by High-Performance Liquid Chromatography: A review. J. Agric. Food Chem. 2000, 48, 577–599. [Google Scholar] [CrossRef] [PubMed]
- Ran, J.; Sun, H.; Xu, Y.; Wang, T.; Zhao, R. Comparison of antioxidant activities and high-performance liquid chromatography analysis of polyphenol from different apple varieties. Int. J. Food Prop. 2016, 19, 2396–2407. [Google Scholar] [CrossRef] [Green Version]
- Skorupa, A.; Gierak, A. Detection and visualization methods used in thin-layer chromatography. JPC J. Planar Chromat. 2011, 24, 274–280. [Google Scholar] [CrossRef]
- Gwatidzo, L.; Dzomba, P.; Mangena, M. TLC separation and antioxidant activity of flavonoids from Carissa bispinosa, Ficus sycomorus, and Grewia bicolar fruits. Nutrire 2018, 43, 3. [Google Scholar] [CrossRef]
- Della Pelle, F.; Compagnone, D. Nanomaterial-based sensing and biosensing of phenolic compounds and related antioxidant capacity in food. Sensors 2018, 18, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escarpa, A. Lights and shadows on food microfluidics. Lab Chip. 2014, 14, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Kellner, R.; Mermet, J.M.; Otto, M.; Valcarcel, V.; Widmer, H.M. (Eds.) Analytical Chemistry. A modern Approach to Analytical Science; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Milardovic, S.; Ivekovic, D.; Rumenjak, V.; Grabaric, B.S. Use of DPPH•/DPPH redox couple for biamperometric determination of antioxidant activity. Electroanalysis 2005, 17, 1847–1853. [Google Scholar] [CrossRef]
- Pisoschi, A.M. Biamperometric applications to antioxidant content and total antioxidant capacity assessment: An editorial. Biochem. Anal. Biochem. 2015, 4, 218. [Google Scholar] [CrossRef] [Green Version]
- Pisoschi, A.M.; Cheregi, M.C.; Danet, A.F. Total antioxidant capacity of some commercial fruit juices: Electrochemical and spectrophotometrical approaches. Molecules 2009, 14, 480–493. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Gajaila, I.; Iordache, F.; Dobre, R.; Cazimir, I.; Serban, A.I. Analytical methods applied to the assay of sulfur-containing preserving agents. Microchem. J. 2020, 105, 104681. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Negulescu, G.P.; Pisoschi, A. Ascorbic acid determination by an amperometric ascorbate oxidase-based biosensor. Rev. Chim. 2010, 61, 339–344. [Google Scholar]
- Pisoschi, A.M. Biosensors as bio-based materials in chemical analysis: A Review. J. Biobased Mater. Bioenergy 2013, 7, 19–38. [Google Scholar] [CrossRef]
- Lahcen, A.A.; Rauf, S.; Beduk, T.; Durmus, C.; Aljedaibi, A.; Timur, S.; Alshareef, H.N.; Amine, A.; Wolfbeis, O.S.; Salama, K.N. Electrochemical sensors and biosensors using laser-derived graphene: A comprehensive review. Biosens. Bioelectron. 2020, 168, 112565. [Google Scholar] [CrossRef]
- Kucherenko, I.S.; Soldatkin, O.O.; Dzyadevych, S.V.; Soldatkin, A.P. Electrochemical biosensors based on multienzyme systems: Main groups, advantages and limitations—A review. Anal. Chim. Acta 2020, 1111, 114–131. [Google Scholar] [CrossRef]
- Sochor, J.; Dobes, J.; Krystofova, O.; Ruttkay-Nedecky, B.; Babula, P.; Pohanka, M.; Jurikova, T.; Zitka, O.; Adam, V.; Klejdus, B.; et al. Electrochemistry as a tool for studying antioxidant properties. Int. J. Electrochem. Sci. 2013, 8, 8464–8489. [Google Scholar]
- Peres, A.M.; Sousa, M.E.B.; Veloso, A.C.A.; Estevinho, L.; Dias, L.G. Electrochemical sensors for assessing antioxidant capacity of bee products. In Chemistry, Biology and Potential Applications of Honeybee Plant-Derived Products; Susana, M.C., Artur, M.S.S., Eds.; Bentham Science Publishers: Sharjah, United Arab Emirates, 2016; pp. 196–223. [Google Scholar]
- David, M.; Serban, A.; Radulescu, C.; Danet, A.F.; Florescu, M. Bioelectrochemical evaluation of plant extracts and gold nanozyme-based sensors for total antioxidant capacity determination. Bioelectrochemistry 2019, 129, 124–134. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Florescu, M.; Bala, C. Biosensors for antioxidants detection: Trends and perspectives. Biosensors 2020, 10, 112. [Google Scholar] [CrossRef]
- Ye, Y.; Ji, J.; Sun, Z.; Shen, P.; Sun, X. Recent advances in electrochemical biosensors for antioxidant analysis in foodstuff. Trends Anal. Chem. 2020, 122, 115718. [Google Scholar] [CrossRef]
- Arumugam, R.; Chen, T.-W.; Chen, S.-M.; Rajaji, U.; Chinnapaiyan, S.; Chinnathabmi, S.; Subramanian, B.; Yu, J.; Yu, R. Electrochemical sensors and biosensors for redox analytes implicated in oxidative stress: Review. Int. J. Electrochem. Sci. 2020, 15, 7064–7081. [Google Scholar] [CrossRef]
- Mello, L.D.; Kubota, L.T. Biosensors as a tool for the antioxidant status evaluation. Talanta 2007, 72, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Barroso, M.F.; de-los-Santos-Álvarez, N.; Lobo-Castanón, M.J.; Miranda Ordieres, A.J.; Delerue-Matos, C.; Oliveira, M.B.P.P.; Tunon-Blanco, P. DNA-based biosensor for the electrocatalytic determination of antioxidant capacity in beverages. Biosens. Bioelectron. 2011, 26, 2396–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonanni, A.; Campanella, L.; Gatta, T.; Gregori, E.; Tomassetti, M. Evaluation of the antioxidant and prooxidant properties of several commercial dry spices by different analytical methods. Food Chem. 2007, 102, 751–758. [Google Scholar] [CrossRef]
- Gomes, S.A.; Rebelo, M.J. A new laccase biosensor for polyphenols determination. Sensors 2003, 3, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Böyükbayram, A.; Kıralp, S.; Toppare, L.; Yağci, Y. Preparation of biosensors by immobilization of polyphenol oxidase in conducting copolymers and their use in determination of phenolic compounds in red wine. Bioelectrochemistry 2006, 69, 164–171. [Google Scholar] [CrossRef]
- Gil, D.M.A.; Rebelo, M.J.F. Evaluating the antioxidant capacity of wines: A laccase-based biosensor approach. Eur. Food Res. Technol. 2010, 231, 303–308. [Google Scholar] [CrossRef]
- Raymundo-Pereira, P.A.; Silva, T.A.; Caetano, F.R.; Ribovski, L.; Zapp, E.; Brondani, D.; Bergamini, M.F.; Marcolino, L.H., Jr.; Banks, C.E.; Oliveira, O.N., Jr.; et al. Polyphenol oxidase-based electrochemical biosensors: A review. Anal. Chim. Acta 2020, 1139, 198–221. [Google Scholar] [CrossRef]
- Uslu, B.; Ozkan, S.A. Electroanalytical application of carbon based electrodes to the pharmaceuticals. Anal. Lett. 2007, 40, 817–853. [Google Scholar] [CrossRef]
- Vytras, K.; Svancara, I.; Metelka, R. Carbon paste electrodes in electroanalytical chemistry. J. Serb. Chem. Soc. 2009, 74, 1021–1033. [Google Scholar] [CrossRef]
- Apetrei, C.; Apetrei, I.M.; De Saja, J.A.; Rodriguez-Mendez, M.L. Carbon Paste electrodes made from different carbonaceous materials: Application in the study of antioxidants. Sensors 2011, 11, 1328–1344. [Google Scholar] [CrossRef] [PubMed]
- Svancara, I.; Walcarius, A.; Kalcher, K.; Vytřas, K. Carbon paste electrodes in the new millennium. Cent. Eur. J. Chem. 2009, 7, 598–656. [Google Scholar] [CrossRef]
- Sharma, S. Glassy carbon: A promising material for micro-and nanomanufacturing. Materials 2018, 10, 1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhang, J.; Zhou, Y.; Shuang, S.; Dong, C.; Choi, M.M.F. Electrodeposition of palladium nanoparticles on fullerene modified glassy carbon electrode for methane sensing. Electrochim. Acta. 2012, 76, 288–291. [Google Scholar] [CrossRef]
- Aziz, M.A.; Almadi, R.; Yamani, Z.H. Indium tin oxide nanoparticle-modified glassy carbon electrode for electrochemical sulfide detection in alcoholic medium. Anal. Sci. 2018, 34, 599–604. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Batchelor-McAuley, C.; Compton, R.C. Two-electron, two-proton oxidation of catechol: Kinetics and apparent catalysis. J. Phys. Chem. C. 2015, 119, 1489–1495. [Google Scholar] [CrossRef]
- Thirumalraj, B.; Palanisamy, S.; Chen, S.M.; Kannan, R.S. Alumina polished glassy carbon electrode as a simple electrode for lower potential electrochemical detection of dopamine in its sub-micromolar level. Electroanalysis 2016, 28, 425–430. [Google Scholar] [CrossRef]
- Lima, A.P.; Almeida, P.L.; Sousa, R.M.; Richter, E.M.; Nossol, E.; Munoz, R.A.A. Effect of alumina supported on glassy-carbon electrode on the electrochemical reduction of 2, 4, 6-trinitrotoluene: A simple strategy for its selective detection. J. Electroanal. Chem. 2019, 851, 113385. [Google Scholar] [CrossRef]
- Lima, A.P.; Souza, R.C.; Silva, M.N.T.; Gonçalves, R.F.; Nossol, E.; Richter, E.M.; Lima, R.C.; Munoz, R.A.A. Influence of Al2O3 nanoparticles structure immobilized upon glassy-carbon electrode on the electrocatalytic oxidation of phenolic compounds. Sens. Actuators B Chem. 2018, 262, 646–654. [Google Scholar] [CrossRef]
- Lima, A.P.; dos Santos, W.T.P.; Nossol, E.; Richter, E.M.; Munoz, R.A.A. Critical evaluation of voltammetric techniques for antioxidant capacity and activity: Presence of alumina on glassy-carbon electrodes alters the results. Electrochim. Acta. 2020, 358, 136925. [Google Scholar] [CrossRef]
- Walcarius, A. Analytical applications of silica-modified electrodes-A comprehensive review. Electroanalysis 1998, 10, 1217–1235. [Google Scholar] [CrossRef]
- Kholmanov, I.; Cavaliere, E.; Fanetti, M.; Cepek, C.; Gavioli, L. Growth of curved graphene sheets on graphite by chemical vapor deposition. Phys. Rev. B 2009, 79, 233403–233406. [Google Scholar] [CrossRef]
- Wang, Y.X.; Yang, Z.D.; Gao, L.J. Theoretical study of electronic properties of phenyl covalent functional carbon nanotubes. J. Mol. Sci. 2016, 32, 259–264. [Google Scholar]
- Rao, C.N.R.; Sood, A.K.; Subrahmanyam, K.S.; Govindaraj, A. Graphene: The new two-dimensional nanomaterial. Angew. Chem. Int. Ed. 2009, 48, 7752–7777. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.W.; Li, F.; Liu, M.; Lu, G.Q.; Cheng, H.M. 3D aperiodic hierarchical porous graphitic carbon material for high-rate electrochemical capacitive energy storage. Angew. Chem. Int. Ed. 2008, 47, 373–376. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.; Hyeon, T. Recent progress in the synthesis of porous carbon materials. Adv. Mater. 2006, 18, 2073–2094. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, H.; Huang, Y.; Wang, W.; Wei, S. Hierarchical porous carbon obtained from animal bone and evaluation in electric double-layer capacitors. Carbon 2011, 49, 838–843. [Google Scholar] [CrossRef]
- Hei, Y.; Li, X.; Zhou, X.; Liu, J.; Sun, M.; Sha, T.; Xu, C.; Xue, W.; Bo, X.; Zhou, M. Electrochemical sensing platform based on kelp-derived hierarchical meso-macroporous carbons. Anal. Chim. Acta. 2018, 1003, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Carbon-nanotube based electrochemical biosensors: A review. Electroanalysis 2005, 17, 7–14. [Google Scholar] [CrossRef]
- Sun, N.; Guan, L.; Shi, Z.; Zhu, Z.; Li, N.; Li, M.; Gu, Z. Electrochemistry of fullerene peapod modified electrodes. Electrochem. Commun. 2005, 7, 1148–1152. [Google Scholar] [CrossRef]
- Bhavani, K.S.; Anusha, T.; Brahman, P.K. Fabrication and characterization of gold nanoparticles and fullerene-C60 nanocomposite film at glassy carbon electrode as potential electro-catalyst towards the methanol oxidation. Internat. J. Hydrog. Energy 2019, 44, 25863–25873. [Google Scholar] [CrossRef]
- Wissler, M. Graphite and carbon powders for electrochemical applications. J. Power Sources 2006, 156, 142–150. [Google Scholar] [CrossRef]
- de Oliveira, A.C.; dos Santos, S.X.; Cavalheiro, E.T.G. Graphite–silicone rubber composite electrode: Preparation and possibilities of analytical application. Talanta 2008, 74, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Noked, M.; Soffer, A.; Aurbach, D. The electrochemistry of activated carbonaceous materials: Past, present, and future. J. Solid State Electrochem. 2011, 15, 1563–1578. [Google Scholar] [CrossRef]
- Krivenko, A.G.; Manzhos, R.A.; Komarova, N.S.; Kotkin, A.S.; Kabachov, E.N.; Shulga, Z.M. Comparative study of graphite and the products of its electrochemical exfoliation. Russ. J. Electrochem. 2018, 54, 825–834. [Google Scholar] [CrossRef]
- Karunadasa, K.S.P.; Manoratne, C.H.; Pitawala, H.M.T.G.A.; Rajapakse, R.M.G.A. potential working electrode based on graphite and montmorillonite for electrochemical applications in both aqueous and molten salt electrolytes. Electrochem Commun. 2019, 108, 106562. [Google Scholar] [CrossRef]
- Pleskov, Y.V. Electrochemistry of diamond: A Review. Russ. J. Electrochem. 2002, 38, 1275–1291. [Google Scholar] [CrossRef]
- Einaga, Y. Development of electrochemical applications of boron-doped diamond electrodes. Bull. Chem. Soc. Jpn. 2018, 91, 1752–1762. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.; Rusinek, C.A.; Thompson, C.H.; Setien, M.; Guo, Y.; Rechenberg, R.; Gong, Y.; Weber, A.J.; Becker, M.F.; Purcell, E.; et al. Flexible, diamond-based microelectrodes fabricated using the diamond growth side for neural sensing. Microsyst. Nanoeng. 2020, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Matemadombo, F.; Apetrei, C.; Nyokong, T.; Rodríguez-Méndez, M.L.; de Saja, J.A. Comparison of carbon screen-printed and disk electrodes in the detection of antioxidants using CoPc derivatives. Sens. Actuators B Chem. 2012, 166–167, 457–466. [Google Scholar] [CrossRef]
- Bordonaba, J.G.; Terry, L.A. Electrochemical behaviour of polyphenol rich fruit juices using disposable screen-printed carbon electrodes: Towards a rapid sensor for antioxidant capacity and individual antioxidants. Talanta 2012, 90, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Taleat, Z.; Khoshroo, A.; Mazloum-Ardakan, M. Screen-printed electrodes for biosensing: A review (2008–2013). Microchim. Acta 2014, 181, 865–891. [Google Scholar] [CrossRef]
- Nesakumar, N.; Berchmans, S.; Alwarappan, S. Chemically modified carbon based electrodes for the detection of reduced glutathione. Sens. Actuators B Chem. 2018, 264, 448–466. [Google Scholar] [CrossRef]
- Panizza, M.; Cerisola, G. Electrochemical degradation of gallic acid on a BDD anode. Chemosphere 2009, 77, 1060–1064. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Negulescu, G.P.; Pisoschi, A. Determination of ascorbic acid content of some fruit juices and wine by voltammetry performed at Pt and Carbon Paste electrodes. Molecules 2011, 16, 1349–1365. [Google Scholar] [CrossRef] [PubMed]
- Badea, M.; Chiperea, S.; Bălan, M.; Floroian, L.; Restani, P.; Marty, J.-L.; Iovan, C.; Ţiţ, D.M.; Bungău, S.; Taus, N. New approaches for electrochemical detection of ascorbic acid. Farmacia 2018, 66, 83–87. [Google Scholar]
- Kumar, M.A.; Lakshminarayanan, V.; Ramamurthy, S.S. Platinum nanoparticles-decorated graphene-modified glassy carbon electrode toward the electrochemical determination of ascorbic acid, dopamine, and paracetamol. Comptes Rendus Chim. 2019, 22, 58–72. [Google Scholar] [CrossRef]
- Brainina, Z.K.; Bukharinova, M.A.; Stozhko, N.Y.; Sokolkov, S.V.; Tarasov, A.V.; Vidrevich, M.B. Electrochemical sensor based on a carbon veil modified by phytosynthesized gold nanoparticles for determination of ascorbic acid. Sensors 2020, 20, 1800. [Google Scholar] [CrossRef] [Green Version]
- Karimi-Maleh, H.; Arotiba, O.A. Simultaneous determination of cholesterol, ascorbic acid and uric acid as three essential biological compounds at a carbon paste electrode modified with copper oxide decorated reduced graphene oxide nanocomposite and ionic liquid. J. Colloid. Interface Sci. 2020, 560, 208–212. [Google Scholar] [CrossRef] [PubMed]
- de Lima, A.A.; Sussuchi, E.M.; De Giovani, W.F. Electrochemical and antioxidant properties of anthocyanins and anthocyanidins. Croat. Chem. Acta 2007, 80, 29–34. [Google Scholar]
- Janeiro, P.; Brett, A.M.O. Redox behavior of anthocyanins present in Vitis vinifera L. Electroanalysis 2007, 19, 1779–1786. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, M.J.; Chen, Y.Y.; Isaacs, M.; Matsuhiro, B.; Mendoza, L.; Torres, S. Electrochemical behaviour and antioxidant capacity of anthocyanins from Chilean red wine, grape and raspberry. Food Chem. 2010, 121, 44–48. [Google Scholar] [CrossRef]
- Newair, E.F.; Kilmartin, P.A.; Garcia, F. Square wave voltammetric analysis of polyphenol content and antioxidant capacity of red wines using glassy carbon and disposable carbon nanotubes modified screen-printed electrodes. Eur. Food Res. Technol. 2018, 244, 1225–1237. [Google Scholar] [CrossRef]
- Ziyatdinova, G.; Ziganshina, E.; Budnikov, H. Voltammetric determination of b-carotene in raw vegetables and berries in Triton X100 media. Talanta 2012, 99, 1024–1029. [Google Scholar] [CrossRef]
- Čižmeka, L.; Komorsky-Lovrić, S. Study of electrochemical behaviour of carotenoids in aqueous media. Electroanalysis 2019, 31, 83–89. [Google Scholar] [CrossRef]
- Stefan-van Staden, R.I.; Moscalu-Lungu, A.; van Staden, J.F. Determination of β-carotene in soft drinks using a stochastic sensor based on a graphene–porphyrin composite. Electrochem. Commun. 2019, 109, 106581. [Google Scholar] [CrossRef]
- Čižmeka, L.; Komorsky-Lovrić, S. Electrochemistry as a screening method in determination of carotenoids in crustacean samples used in everyday diet. Food Chem. 2020, 309, 125706. [Google Scholar] [CrossRef]
- Yakovleva, K.E.; Kurzeev, S.A.; Stepanova, E.V.; Fedorova, T.V.; Kuznetsov, B.A.; Koroleva, O.V. Characterization of plant phenolic compounds by cyclic voltammetry. Appl. Biochem. Microbiol. 2007, 43, 661–668. [Google Scholar] [CrossRef]
- Makhotkina, O.; Kilmartin, P.A. The use of cyclic voltammetry for wine analysis: Determination of polyphenols and free sulfur dioxide. Anal. Chim. Acta 2010, 668, 155–165. [Google Scholar] [CrossRef]
- Bisetty, K.; Sabela, M.I.; Khulu, S.; Xhakaza, M.; Ramsarup, L. Multivariate optimization of voltammetric parameters for the determination of total polyphenolic content in wine samples using an immobilized biosensor. Int. J. Electrochem. Sci. 2011, 6, 3631–3643. [Google Scholar]
- Robledo, S.N.; Zachetti, V.G.L.; Zon, M.A.; Fernández, H. Quantitative determination of tocopherols in edible vegetable oils using electrochemical ultra-microsensors combined with chemometric tools. Anal. Chim. Acta 2013, 116, 964–971. [Google Scholar] [CrossRef]
- Malyszko, J.; Karbarz, M. Electrochemical oxidation of trolox and a-tocopherol in acetic acid: A comparative study. J. Electroanal. Chem. 2006, 595, 136–144. [Google Scholar] [CrossRef]
- Kuraya, E.; Nagatomo, S.; Sakata, K.; Kato, D.; Niwa, O.; Nishimi, T.; Kunitake, M. Simultaneous electrochemical analysis of hydrophilic and lipophilic antioxidants in bicontinuous microemulsion. Anal. Chem. 2015, 87, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Sys, M.; Švecová, B.; Švancara, I.; Metelka, R. Determination of vitamin E in margarines and edible oils using square wave anodic stripping voltammetry with a glassy carbon paste electrode. Food. Chem. 2017, 229, 621–627. [Google Scholar] [CrossRef]
- Doblhoff-Dier, O.; Rechnitz, G.A. Amperometric method for the determination of superoxide dismutase activity at physiological pH. Anal. Chim Acta 1989, 222, 247–252. [Google Scholar] [CrossRef]
- Santharaman, P.; Das, M.; Singh, S.K.; Sethy, N.K.; Bhargava, K.; Claussen, J.C.; Karunakaran, C. Label-free electrochemical immunosensor for the rapid and sensitive detection of the oxidative stress marker superoxide dismutase 1 at the point-of-care. Sens. Actuators B Chem. 2016, 236, 546–553. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, Z.; Xie, L.; Liu, M.; Yuan, Z. Determination of the activity of superoxide dismutase using a glassy carbon electrode modified with ferrocene imidazolium salts and hydroxy-functionalized graphene. Microchim. Acta 2017, 184, 289–296. [Google Scholar] [CrossRef]
- Liu, Q.; Bao, J.; Yang, M.; Wang, X.; Lan, S.; Hou, C.; Wang, Y.; Fa, H. A core-shell MWCNT@rGONR heterostructure modified glassy carbon electrode for ultrasensitive electrochemical detection of glutathione. Sens. Actuators B Chem. 2018, 274, 433–440. [Google Scholar] [CrossRef]
- De Almeida Ferraza, N.V.; Vasconcelos, W.S.; Silva, C.S.; Alves Junior, S.; Amorim, C.G.; da Conceição Branco, S.M.; Montenegro, M.; Cunha Areias, M.C. Gold-copper metal-organic framework nanocomposite as a glassy carbon electrode modifier for the voltammetric detection of glutathione in commercial dietary supplements. Sens. Actuators B Chem. 2020, 307, 127636. [Google Scholar] [CrossRef]
- Stojanović, Z.S.; Đurović, A.D.; Ashrafi, A.M.; Koudelková, Z.; Zítka, O.; Richtera, L. Highly sensitive simultaneous electrochemical determination of reduced and oxidized glutathione in urine samples using antimony trioxide modified carbon paste electrode. Sens. Actuators B Chem. 2020, 318, 128141. [Google Scholar] [CrossRef]
- Das, S.C.; Pandey, R.R.; Devkota, T.; Chusuei, C.C. Raman spectroscopy as an assay to disentangle zinc oxide carbon nanotube composites for optimized uric acid detection. Chemosensors 2018, 6, 65. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Yue, H.; Zhang, J.; Gao, X.; Zhang, H.; Lin, X.; Wang, B.; Bychanok, D. Electrochemical determination of uric acid in the presence of ascorbic acid by hybrid of ZnO nanorods and graphene nanosheets. Ionics 2018, 24, 2499–2507. [Google Scholar] [CrossRef]
- Fukuda, T.; Muguruma, H.; Iwasa, H.; Tanaka, T.; Hiratsuka, A.; Shimizu, T.; Tsuji, K.; Kishimoto, T. Electrochemical determination of uric acid in urine and serum with uricase/carbon nanotube /carboxymethylcellulose electrode. Anal. Biochem. 2020, 590, 113533. [Google Scholar] [CrossRef] [PubMed]
- Thangamuthu, M.; Gabriel, W.E.; Santschi, C.; Martin, O.J.F. Electrochemical sensor for bilirubin detection using screen printed electrodes functionalized with carbon nanotubes and graphene. Sensors 2018, 18, 800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raveendran, J.; Stanley, J.; Satheesh Babu, T.G. Voltammetric determination of bilirubin on disposable screen printed carbon electrode. J. Electroanal. Chem. 2018, 818, 124–130. [Google Scholar] [CrossRef]
- Akhoundian, M.; Alizadeh, T.; Pan, G. Investigation of electrochemical behavior of bilirubin at unmodified carbon paste electrode. Anal. Bioanal. Electrochem. 2019, 9, 1166–1175. [Google Scholar]
- Apetrei, I.M.; Apetrei, C. Voltammetric determination of melatonin using a graphene-based sensor in pharmaceutical products. Int. J. Nanomed. 2016, 11, 1859–1866. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Goyal, R.N. Electrochemical behavior of melatonin and its sensing in pharmaceutical formulations and in human urine. Curr. Pharm. Anal. 2017, 13, 85–90. [Google Scholar] [CrossRef]
- Castagnola, E.; Woeppel, K.; Golabchi, A.; McGuier, M.; Chodapaneedi, N.; Metro, J.; Taylor, I.M.; Tracy Cui, X. Electrochemical detection of exogenously administered melatonin in the brain. Analyst 2020, 145, 2612–2620. [Google Scholar] [CrossRef] [PubMed]
- Michalkiewicz, S. Voltammetric determination of coenzyme Q10 in pharmaceutical dosage forms. Bioelectrochemistry 2008, 73, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Petrova, E.V.; Korotkova, E.I.; Kratochvil, B.; Voronova, O.A.; Dorozhko, E.V.; Bulycheva, E.V. Investigation of Coenzyme Q10 by voltammetry. Procedia Chem. 2014, 10, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Charoenkitamorn, K.; Chaiyo, S.; Chailapakul, O.; Siangproh, W. Low-cost and disposable sensors for the simultaneous determination of coenzyme Q10 and α-lipoic acid using manganese (IV) oxide-modified screen-printed graphene electrodes. Anal. Chim. Acta 2018, 1004, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Cincotto, F.H.; Canevari, T.C.; Machado, S.A.S. Highly sensitive electrochemical sensor for determination of Vitamin D in mixtures of water-ethanol. Electroanalysis 2014, 26, 2783–2788. [Google Scholar] [CrossRef]
- Men, K.; Chen, Y.; Liu, J.B.; Wei, D.J. Electrochemical detection of Vitamin D2 and D3 based on a Au-Pd modified glassy carbon electrode. Int. J. Electrochem. Sci. 2017, 12, 9555–9564. [Google Scholar] [CrossRef]
- Durovic, A.; Stojanovic, Z.; Kravic, S.; Kos, J.; Richtera, L. Electrochemical determination of Vitamin D3 in pharmaceutical products by using boron doped diamond electrode. Electroanalysis 2020, 32, 741–748. [Google Scholar] [CrossRef]
- Ferreira, A.P.M.; Dos Santos Pereira, L.N.; Santos da Silva, I.; Tanaka, S.M.C.N.; Tanaka, A.A.; Angnes, L. Determination of alpha-lipoic acid on a pyrolytic graphite electrode modified with cobalt phthalocyanine. Electroanalysis 2014, 26, 2138–2144. [Google Scholar] [CrossRef]
- Dos Santos Pereira, L.N.; da Silva, I.S.; Araújo, T.P.; Tanaka, A.A.; Angnes, L. Fast quantification of α-lipoic acid in biological samples and dietary supplements using batch injection analysis with amperometric detection. Talanta 2016, 154, 249–254. [Google Scholar] [CrossRef]
- Ziyatdinova, G.; Antonova, T.; Vorobev, V.; Osin, Y.; Budnikov, H. Selective voltammetric determination of α-lipoic acid on the electrode modified with SnO2 nanoparticles and cetyltriphenylphosphonium bromide. Monatsh. Chem. 2019, 150, 401–410. [Google Scholar] [CrossRef]
- Nunes Angelis, P.; de Cássia Mendonça, J.; Rianne da Rocha, L.; Boareto Capelari, T.; Carolyne Prete, M.; Gava Segatelli, M.; Borsato, D.; Ricardo Teixeira Tarley, C. Feasibility of a nano-carbon black paste electrode for simultaneous voltammetric determination of antioxidants in food samples and biodiesel in the presence of surfactant. Electroanalysis 2020, 32, 1198–1207. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. Comparative sulfite assay by voltammetry using Pt electrodes, photometry and titrimetry: Application to cider, vinegar and sugar analysis. Open Chem. 2018, 16, 1248–1256. [Google Scholar] [CrossRef]
- Dar, R.A.; Brahman, P.K.; Khurana, N.; Wagay, J.A.; Lone, Z.A.; Ganaie, M.A.; Pitre, K.S. Evaluation of antioxidant activity of crocin, podophyllotoxin and kaempferol by chemical, biochemical and electrochemical assays. Arab. J. Chem. 2017, 10, S1119–S1128. [Google Scholar] [CrossRef] [Green Version]
- Korotkova, E.I.; Lipskikh, O.I.; Kiseleva, M.A.; Ivanov, V.V. Voltammetric study of the antioxidant properties of catalase and superoxide dismutase. Pharm. Chem. J. 2008, 42, 485–487. [Google Scholar] [CrossRef]
- Wei, Y.; Zhang, S. Study on the electroreduction process of oxygen to superoxide ion by using acetylene black powder microelectrode. Russ. J. Electrochem. 2008, 44, 967–971. [Google Scholar] [CrossRef]
- Blasco, A.J.; Rogerio, M.C.; Gonzalez, M.C.; Escarpa, A. “Electrochemical Index” as a screening method to determine “total polyphenolics” in foods: A proposal. Anal. Chim. Acta 2005, 539, 237–244. [Google Scholar] [CrossRef]
- Abdel-Hamid, R.; Newair, E.F. Voltammetric determination of polyphenolic content in pomegranate juice using a poly(gallic acid)/multiwalled carbon nanotube modified electrode. Beilstein J. Nanotechnol. 2016, 7, 1104–1112. [Google Scholar] [CrossRef] [Green Version]
- Raymundo-Pereira, P.A.; Campos, A.M.; Prado, T.M.; Furini, L.N.; Boas, N.V.; Calegaro, M.L.; Machado, S.A.S. Synergy between Printex nano-carbons and silver nanoparticles for sensitive estimation of antioxidant activity. Anal. Chim Acta 2016, 926, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Eguílaz, M.; Gutierrez, A.; Gutierrez, F.; Gonzalez-Domínguez, J.M.; Anson-Casaos, A.; Hernandez-Ferrer, J.; Ferreyra, N.F.; Martínez, M.T.; Rivas, G. Covalent functionalization of single-walled carbon nanotubes with polytyrosine: Characterization and analytical applications for the sensitive quantification of polyphenols. Anal. Chim. Acta 2016, 909, 51–59. [Google Scholar] [CrossRef]
- Yuan, Y.; Bao, Z.H.; Li, S.M.; Zhao, K. Electrochemical evaluation of antioxidant capacity in pharmaceutical antioxidant excipient of drugs on guanine-based modified electrode. J. Electroanal. Chem. 2016, 772, 58–65. [Google Scholar]
- Oliveira-Neto, J.R.; Garcia Rezende, S.; de Fátima Reis, C.; Rathinaraj Benjamin, S.; Lavorenti Rocha, M.; de Souza Gil, E. Electrochemical behavior and determination of major phenolic antioxidants in selected coffee samples. Food Chem. 2016, 190, 506–512. [Google Scholar] [CrossRef]
- Tirawattanakoson, R.; Rattanarat, P.; Ngamrojanavanich, N.; Rodthongkum, N.; Chailapakul, O. Free radical scavenger screening of total antioxidant capacity in herb and beverage using graphene/PEDOT: PSS-modified electrochemical sensor. J. Electroanal. Chem. 2016, 767, 68–75. [Google Scholar] [CrossRef]
- Ziyatdinova, G.; Kozlova, E.; Budnikov, H. Chronocoulometry of wine on multi-walled carbon nanotube modified electrode: Antioxidant capacity assay. Food. Chem. 2016, 196, 405–410. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Neto, J.R.; Rezende, S.G.; Lobón, G.S.; Garcia, T.A.; Macedo, I.Y.L.; Garcia, L.F.; Alves, V.F.; Sapateiro Torres, I.M.; Santiago, M.F.; Schmidt, F.; et al. Electroanalysis and laccase-based biosensor on the determination of phenolic content and antioxidant power of honey samples. Food Chem. 2017, 237, 1118–1123. [Google Scholar] [CrossRef]
- Della Pelle, F.; Di Battista, R.; Vázquez, L.; Palomares, F.J.; Del Carlo, M.; Sergi, M.; Compagnone, D.; Escarpa, A. Press-transferred carbon black nanoparticles for class-selective antioxidant electrochemical detection. Appl. Mater. Today 2017, 9, 29–36. [Google Scholar] [CrossRef] [Green Version]
- de Menezes Peixoto, C.R.; Fraga, S.; Justim, J.D.; Gomes, M.S.; Carvalho, D.G.; Jarenkow, J.A.; de Moura, N.F. Voltammetric determination of total antioxidant capacity of Bunchosia glandulifera tree extracts. J. Electroanal. Chem. 2017, 799, 519–524. [Google Scholar] [CrossRef]
- Ziyatdinova, G.K.; Kozlova, E.V.; Budnikov, H.C. Chronoamperometric evaluation of the antioxidant capacity of tea on a polyquercetin-modified electrode. J. Anal. Chem. 2017, 72, 382–389. [Google Scholar] [CrossRef]
- Jara-Palacios, M.J.; Escudero-Gilete, M.L.; Hernández-Hierro, J.M.; Heredia, F.J.; Hernanz, D. Cyclic voltammetry to evaluate the antioxidant potential in winemaking by-products. Talanta 2017, 165, 211–215. [Google Scholar] [CrossRef]
- Samoticha, J.; Jara-Palacios, M.J.; Hernández-Hierro, J.M.; Heredia, F.J.; Wojdyło, A. Phenolic compounds and antioxidant activity of twelve grape cultivars measured by chemical and electrochemical methods. Eur. Food Res. Technol. 2018, 244, 1933–1943. [Google Scholar] [CrossRef]
- De Siqueira Leite, K.C.; Garcia, L.F.; Lobón, G.S.; Thomaz, D.V.; Goncalves Moreno, E.K.; de Carvalho, M.F.; Rocha, M.L.; dos Santos, W.T.P.; de Souza Gil, E. Antioxidant activity evaluation of dried herbal extracts: An electroanalytical approach. Rev. Bras. Farmacogn. 2018, 28, 325–332. [Google Scholar] [CrossRef]
- Lugonja, N.M.; Stanković, D.M.; Miličić, B.; Spasić, S.D.; Marinković, V.; Vrvić, M.M. Electrochemical monitoring of the breast milk quality. Food Chem. 2018, 240, 567–572. [Google Scholar] [CrossRef]
- Fernández, E.; Vidal, L.; Canals, A. Rapid determination of hydrophilic phenols in olive oil by vortex-assisted reversed-phase dispersive liquid-liquid microextraction and screen-printed carbon electrodes. Talanta 2018, 181, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, I.G.; Litescu, S.C.; Popa, D.E.; Buleandra, M.; Iordache, L.; Albu, C.; Alecu, A.; Penu, R.L. Voltammetric analysis of naringenin at a disposable pencil graphite electrode-application to polyphenol content determination in citrus juice. Anal. Methods 2018, 10, 5763–5772. [Google Scholar] [CrossRef]
- Arantes, I.V.S.; Stefano, J.S.; Sousa, R.M.F.; Richter, E.M.; Foster, C.W.; Banks, C.E.; Munoz, R.A.A. Fast determination of antioxidant capacity of food samples using continuous amperometric detection on polyester screen-printed graphitic electrodes. Electroanalysis 2018, 30, 1192–1197. [Google Scholar] [CrossRef]
- Wagay, J.A.; Nayik, G.A.; Wani, S.A.; Mir, R.A.; Ahmad, M.A.; Rahman, Q.I.; Vyas, D. Phenolic profiling and antioxidant capacity of Morchella esculenta L. by chemical and electrochemical methods at multiwall carbon nanotube paste electrode. J. Food Meas. Charact. 2019, 13, 1805–1819. [Google Scholar] [CrossRef]
- Djitieu Deutchoua, A.D.; Ngueumaleu, Y.; Dedzo, G.K.; Tonle, I.K.; Ngamen, E. Electrochemical study of DPPH incorporated in carbon paste electrode as potential tool for antioxidant properties determination. Electroanalysis 2019, 31, 335–342. [Google Scholar] [CrossRef]
- Muhammad, H.; Tahiri, I.A.; Qasim, M.; Versiani, M.A.; Hanif, M.; Gul, B.; Ali, S.T.; Ahmed, S. Electrochemical determination of antioxidant activity and HPLC profling of some dry fruits. Monatsh. Chem. 2019, 150, 1195–1203. [Google Scholar] [CrossRef]
- Nikolic, M.D.; Pavlovic, A.N.; Mitic, S.S.; Tosic, S.B.; Mitic, M.N.; Kalicanin, B.M.; Manojlovic, D.D.; Stankovic, D.M. Use of cyclic voltammetry to determine the antioxidant capacity of berry fruits: Correlation with spectrophotometric assays. Eur. J. Hortic. Sci. 2019, 84, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Ricci, A.; Teslic, N.; Petropolus, V.-I.; Parpinello, G.P.; Versari, A. Fast analysis of total polyphenol content and antioxidant activity in wines and oenological tannins using a flow injection system with tandem diode array and electrochemical detections. Food Anal. Methods 2019, 12, 347–354. [Google Scholar] [CrossRef]
- Pilaquinga, F.; Amaguaña, D.; Morey, J.; Moncada-Basualto, M.; Pozo-Martínez, J.; Olea-Azar, C.; Fernández, L.; Espinoza-Montero, P.; Jara-Negrete, E.; Meneses, L.; et al. Synthesis of silver nanoparticles using aqueous leaf extract of Mimosa albida (Mimosoideae): Characterization and antioxidant activity. Materials 2020, 13, 503. [Google Scholar] [CrossRef] [Green Version]
- Aravena-Sanhueza, F.; Pérez-Rivera, M.; Castillo-Felices, R.; Mundaca-Uribe, R.; Aranda Bustos, M.; Peña Farfal, C. Determination of antioxidant capacity (orac) of Greigia sphacelata and correlation with voltammetric methods. J. Chil. Chem. Soc. 2020, 65, 4925–4928. [Google Scholar] [CrossRef]
- Banica, F.; Bungau, S.; Tit, D.M.; Behl, T.; Otrisal, P.; Nechifor, A.C.; Gitea, D.; Pavel, F.-M.; Nemeth, S. Determination of the total polyphenols content and antioxidant activity of Echinacea Purpurea extracts using newly manufactured glassy carbon electrodes modified with carbon nanotubes. Proceses 2020, 8, 833. [Google Scholar] [CrossRef]
- Schilder, W.H.; Tanumihardja, E.; Leferink, A.M.; van den Berg, A.; Olthuis, W. Determining the antioxidant properties of various beverages using staircase voltammetry. Heliyon 2020, 6, e04210. [Google Scholar] [CrossRef]
- Stevanovic, M.; Stevanovic, S.; Mihailovic, M.; Kiprovski, B.; Bekavac, G.; Mikulic-Petkovsek, M.; Lovic, J. Antioxidant capacity of dark red corn-biochemical properties coupled with electrochemical evaluation. Rev. Chim. 2020, 71, 31–41. [Google Scholar] [CrossRef]
- Gevaerd, A.; da Silva, B.M.; de Oliveira, P.R.; Marcolino, L.H.; Bergamini, M.F. A carbon fiber ultramicroelectrode as a simple tool to direct antioxidant estimation based on caffeic acid oxidation. Anal. Methods 2020, 12, 3608–3616. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; You, Z.; Xiao, A.; Liu, L.; Zhou, W. Electrochemical evaluation of the antioxidant capacity of natural compounds on glassy carbon electrode modified with guanine-, polythionine-, and nitrogen-doped graphene. Open Chem. 2020, 18, 1054–1063. [Google Scholar] [CrossRef]
- Demir, E.; Senocak, A.; Tassembedo-Koubangoye, M.F.; Demirbas, E.; Aboul-Enein, H.Y. Electrochemical evaluation of the total antioxidant capacity of Yam food samples on a polyglycine-glassy carbon modified electrode. Curr. Anal. Chem. 2020, 16, 176–183. [Google Scholar] [CrossRef]
- Zrinski, I.; Pungjunun, K.; Martinez, S.; Zavagnik, J.; Stankovic, D.; Kalcher, K.; Mehmeti, E. Evaluation of phenolic antioxidant capacity in beverages based on laccase immobilized on screen-printed carbon electrode modified with graphene nanoplatelets and gold nanoparticles. Microchem. J. 2020, 152, 104282. [Google Scholar] [CrossRef]
- Oualcadi, Y.; Aityoub, A.; Berrekhis, F. Investigation of different antioxidant capacity measurements suitable for bioactive compounds applied to medicinal plants. J. Food Meas. Charact. 2021, 15, 71–83. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Serban, A.I.; Fafaneata, C. Electrochemical methods for ascorbic acid determination. Electrochim. Acta 2014, 121, 443–460. [Google Scholar] [CrossRef]
- Rueda, M.; Aldaz, A.; Sanchez-Burgos, F. Oxidation of L-ascorbic acid on a gold electrode. Electrochim. Acta 1978, 23, 419–424. [Google Scholar] [CrossRef]
- Koh, S.N.; Tan, W.T.; Zainal, Z.; Zawawi, R.M.; Zidan, M. Detection of ascorbic acid at glassy carbon electrode modified by single-walled carbon nanotube/zinc oxide. Int. J. Electrochem. Sci. 2013, 8, 10557–10567. [Google Scholar]
- Olana, B.N.; Kitte, S.A.; Soreta, T.R. Electrochemical determination of ascorbic acid at p-phenylenediamine film–holes modified glassy carbon electrodes. J. Serb. Chem. Soc. 2015, 80, 1161–1175. [Google Scholar] [CrossRef]
- Janeiro, P.; Brett, A.M.O. Catechin electrochemical oxidation mechanisms. Anal. Chim. Acta 2004, 518, 109–115. [Google Scholar] [CrossRef] [Green Version]
- McCreery, R.L. Advanced carbon electrode materials for molecular electrochemistry. Chem. Rev. 2008, 108, 2646–2687. [Google Scholar] [CrossRef]
- Rocheleau, M.-J.; Purdy, W.C. The application of quaternary ammonium ionic polymers to electroanalysis: Part 2. Voltammetric studies with quaternary ammonium functionalized polymer film-coated electrodes. Electroanalysis 1991, 3, 935–939. [Google Scholar] [CrossRef]
- Dalmasso, P.R.; Pedano, M.L.; Rivas, G.A. Electrochemical determination of ascorbic acid and paracetamol in pharmaceutical formulations using a glassy carbon electrode modified with multi-wall carbon nanotubes dispersed in polyhistidine. Sens. Actuators B Chem. 2012, 173, 732–736. [Google Scholar] [CrossRef]
- Rychagov, A.Y.; Urisson, N.A.; Volfkovich, Y.M. Electrochemical characteristics and properties of the surface of activated carbon electrodes in a double-layer capacitor. Russ. J. Electrochem. 2001, 37, 1172–1179. [Google Scholar] [CrossRef]
- Hassanpour, S.; Behnam, B.; Baradaran, B.; Hashemzaei, M.; Oroojalian, F.; Mokhtarzadeh, A.; de la Guardia, M. Carbon based nanomaterials for the detection of narrow therapeutic index pharmaceuticals. Talanta 2021, 221, 121610. [Google Scholar] [CrossRef]
- Yang, C.; Denno, M.E.; Pyakurel, P.; Venton, B.J. Recent trends in carbon nanomaterial-based electrochemical sensors for biomolecules: A review. Anal. Chim. Acta 2015, 887, 17–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jariwala, D.; Sangwan, V.K.; Lauhon, L.J.; Marks, T.J.; Hersam, M.C. Carbon nanomaterials for electronics, optoelectronics, photovoltaics, and sensing. Chem. Soc. Rev. 2013, 42, 2824–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Ratinac, K.R.; Ringer, S.P.; Thordarson, P.; Gooding, J.J.; Braet, F. Carbon nanomaterials in biosensors: Should you use nanotubes or graphene? Angew. Chem. Int. Ed. 2010, 49, 2114–2138. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, A.; Omidi, M.; Choolaei, M.; Nazarzadeh, M.; Yadegari, A.; Haghierosadat, F.; Oroojalian, F.; Azhdari, M. Electromechanical properties of vertically aligned carbon nanotube. In Advanced Materials Research; Trans Tech Publ: Stafa-Zurich, Switzerland, 2013; Volume 705, pp. 332–336. [Google Scholar] [CrossRef]
- Eguílaz, M.; Dalmasso, P.R.; Rubianes, M.D.; Gutierrez, F.; Rodríguez, M.C.; Gallay, P.A.; Mujica, M.E.L.; Ramírez, M.L.; Tettamanti, C.S.; Montemerlo, A.E. Recent advances in the development of electrochemical hydrogen peroxide carbon nanotube-based (bio) sensors. Curr. Opin. Electrochem. 2019, 14, 157–165. [Google Scholar] [CrossRef]
- Scida, K.; Stege, P.W.; Haby, G.; Messina, G.A.; García, C.D. Recent applications of carbon-based nanomaterials in analytical chemistry: Critical review. Anal. Chim. Acta 2011, 691, 6–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Wu, P.F.; Yue, H.Y.; Gao, X.; Song, S.S.; Guo, X.R.; Chen, H.T. ZnO nanosheet arrays/graphene foam: Voltammetric determination of dopamine in the presence of ascorbic acid and uric acid. J. Mater. Sci. Mater. Electron. 2019, 30, 16510–16517. [Google Scholar] [CrossRef]
- Svancara, I.; Vytras, K.; Barek, J.; Zima, J. Carbon paste electrodes in modern electroanalysis. Crit. Rev. Anal. Chem. 2001, 31, 311–345. [Google Scholar] [CrossRef]
- Wang, J. Analytical Electrochemistry, 2nd ed.; Wiley-VCH: New York, NY, USA, 2000. [Google Scholar]
- Ambaye, A.D.; Kefeni, K.K.; Mishra, S.B.; Nxumalo, E.N.; Ntsendwana, B. Recent developments in nanotechnology-based printing electrode systems for electrochemical sensors. Talanta 2021, 225, 121951. [Google Scholar] [CrossRef]

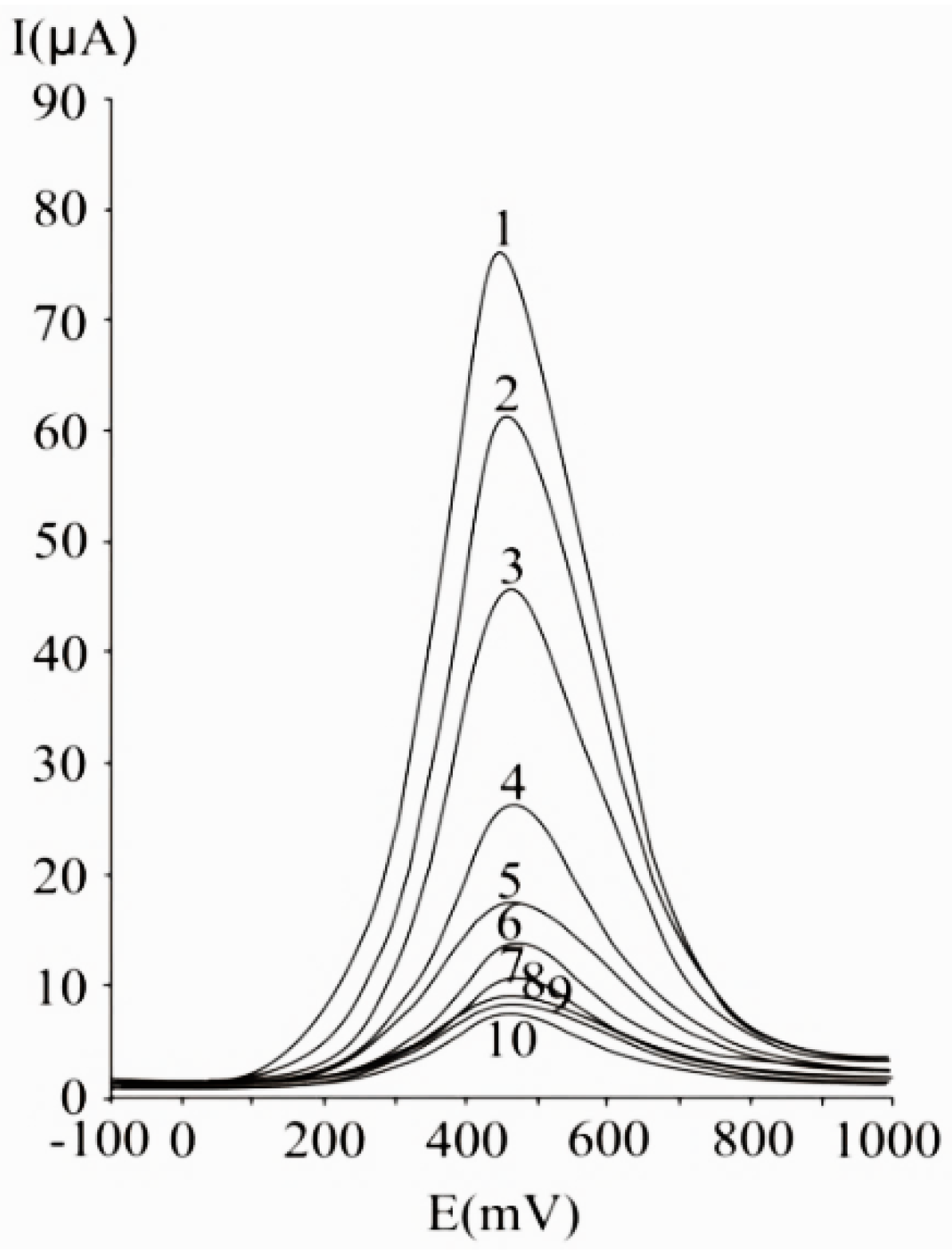

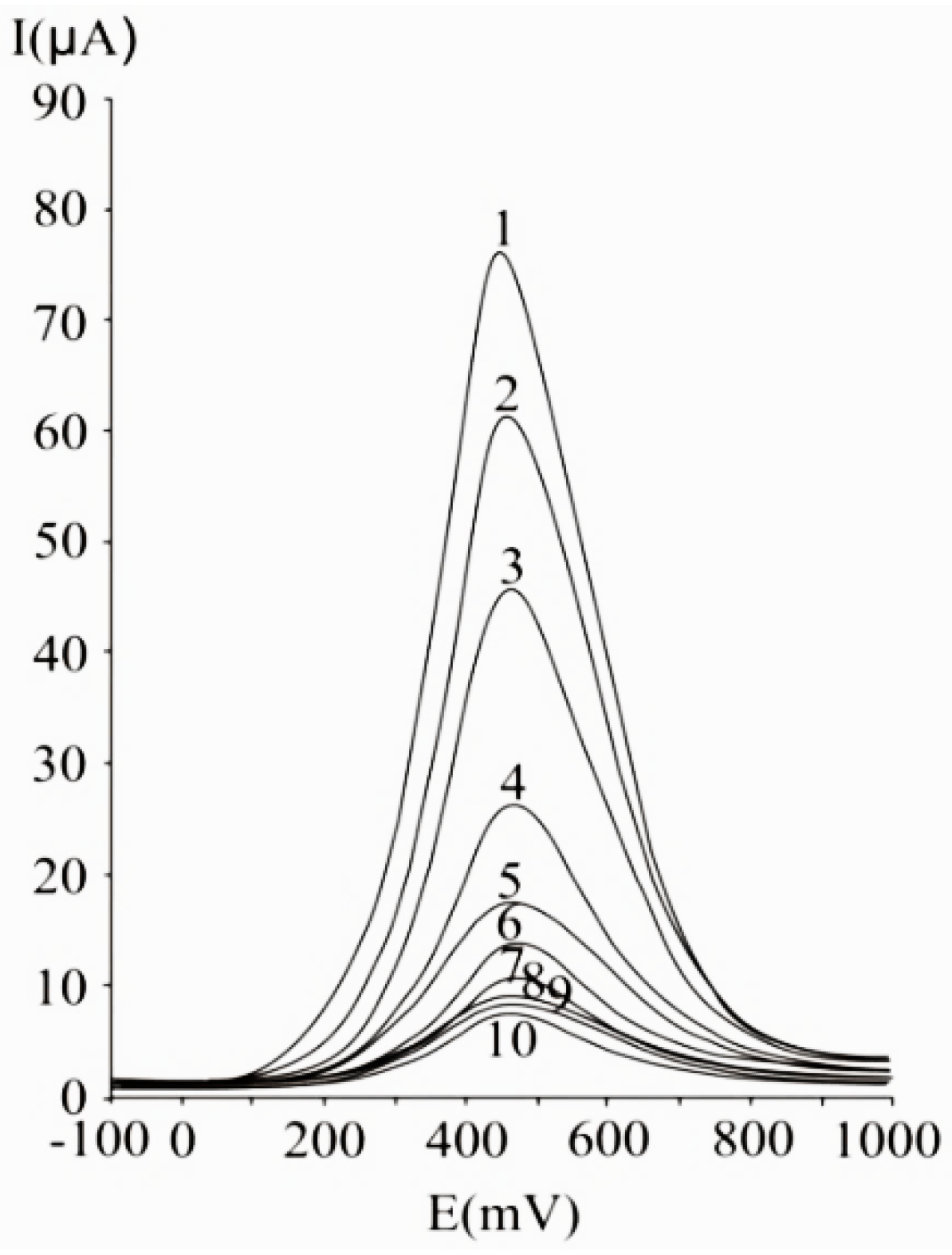
{kind=link}
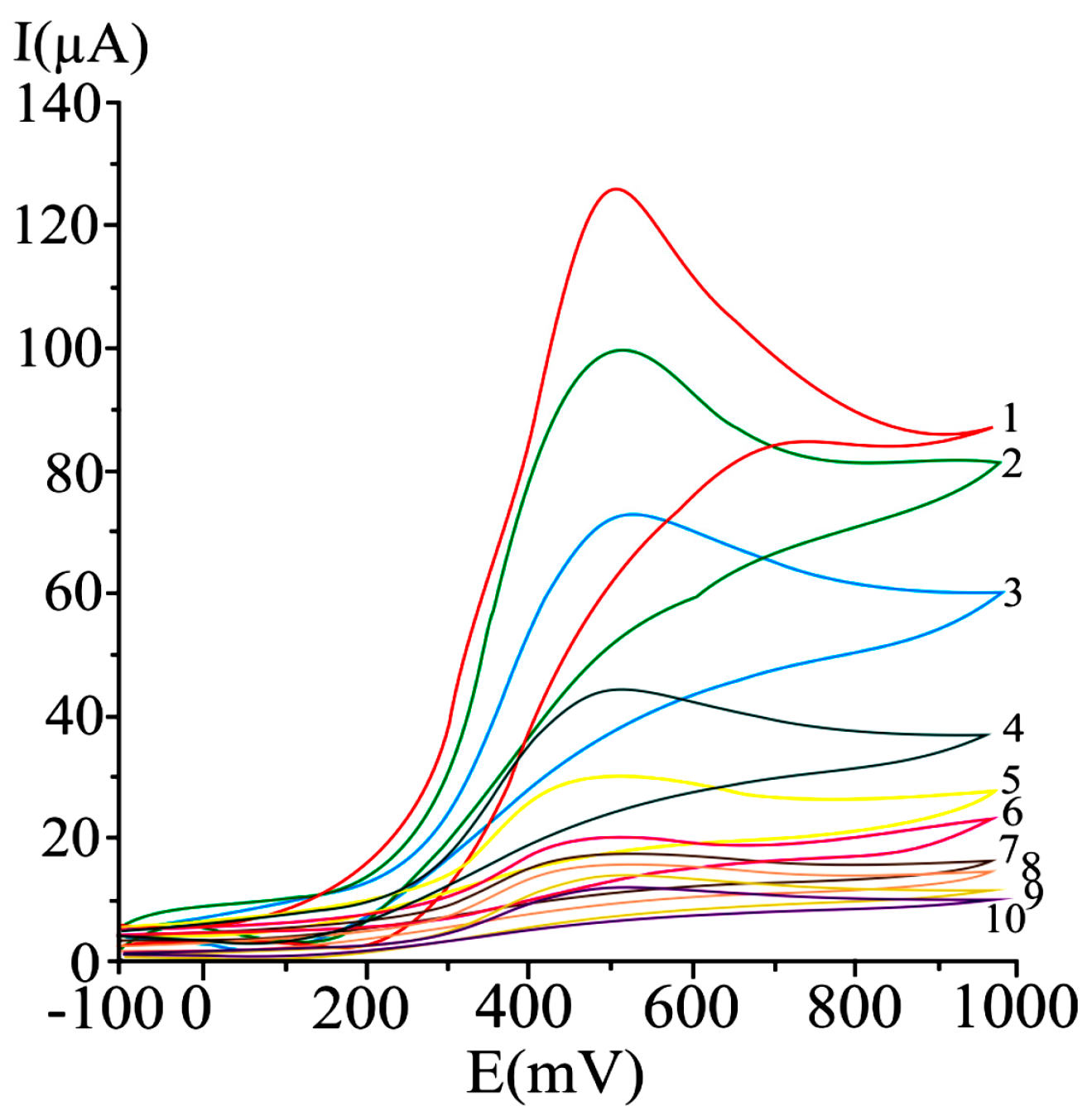
{kind=link}
| Total Antioxidant Capacity or Its Main Contributors’ Assay | Method’s Principle | Detection of the End-Product | Ref. |
|---|---|---|---|
| Spectrometry | |||
| DPPH | Antioxidants react with an organic radical | Colorimetric | [21,22] |
| ABTS | Antioxidants react with an organic cation radical | Colorimetric | [23,24] |
| FRAP | Antioxidants react with ferric-tripyridyltriazine complex | Colorimetric | [25,26] |
| PFRAP | Potassium ferricyanide is reduced by antioxidants to potassium ferrocyanide, that reacts with Fe3+ yielding ferric ferrocyanide | Colorimetric | [27,28] |
| CUPRAC | Antioxidants reduce Cu (II) complex to a Cu (I) complex | Colorimetric | [29,30] |
| Thiobarbituric Acid Reactive Species (TBARS) Assay | The generation of malonyl dialdehyde can be detected after its reaction with thiobarbituric acid, yielding a pink chromogen | Colorimetric assay of malonyl dialdehyde-thiobarbituric acid adduct | [31,32] |
| Folin-Ciocalteu | Phenolics react with a mixture of phosphomolybdate/phosphotungstate in the presence of sodium carbonate 20%. | Absorbance of the blue molybdenum-tungsten complex resulted is measured, versus gallic acid as reference antioxidant | [33,34] |
| ORAC | Peroxyl radicals, induced by AAPH (2,2′-azobis-2-amidino-propane) decomposition are reduced by antioxidants | Loss of fluorescence indicated by fluorescein | [35,36] |
| HORAC | Co(II)-based Fenton systems result in OH radicals generation, followed by quenching by antioxidants | Loss of fluorescence indicated by fluorescein | [37,38] |
| TRAP | Luminol-derived radicals, formed by AAPH decomposition, are scavenged by antioxidants | Quenching of chemiluminescence | [39,40] |
| Fluorimetry | Emission of electromagnetic radiation (generally in the visible range) that follows an absorbtion process (generally in the UV domain) | Recording of excitation/emission spectra of fluorescent reagent | [41,42] |
| Electrochemical Techniques | |||
| Potentiometry | Antioxidants interact with the oxidized form of a redox couple, changing the ratio between the oxidized form and the reduced form concentration | The analytical signal recorded is the potential shift of the mediator system, resulting from interaction with antioxidants | [43,44] |
| Cyclic voltammetry (CV) | Linear variation of the potential of a working electrode following a triangular waveform, and recording of current intensity | The intensity value corresponding the cathodic/anodic peak is measured | [45,46] |
| Differential pulse voltammetry (DPV) | Voltage pulses are superimposed on the potential scan, that is varied linearly or stairstep-wise | First intensity value is sampled before applying the pulse, and the second towards the end of the pulse | [47,48] |
| Square-wave voltammetry (SWV) | A square wave is superimposed on a potential staircase sweep variation | Current intensity recorded at the end of each potential change | [49,50] |
| Polarography | Determination of the antioxidant potential of radical scavengers relied on the anodic oxidation of dropping mercury electrode | Diminution of the anodic limiting current of the hydroxoperhydroxo-mercury(II) complex, [Hg(O2H) (OH)], generated in H2O2 solution at alkaline pH, at the potential of Hg oxidation | [51,52] |
| Amperometry | Measurement of the current intensity at a fixed potential value of the working electrode, with respect to a reference one | The intensity of the current, occurring as result of oxidation/reduction of the analyte at constant potential, is measured | [53,54] |
| Biamperometry | Reaction of the antioxidant with the oxidized form of a reversible indicator redox couple | The current flowing between two identical working electrodes is measured, at a small potential difference; the measuring solution contains the antioxidant(s) in the presence of a reversible redox couple | [55,56] |
| Chromatography | |||
| Gas chromatography (GC) | The compounds to be separated and quantified are differentially distributed between a liquid stationary phase and a gaseous mobile phase | Detection based on thermal conductivity or flame ionisation | [57,58] |
| High performance liquid chromatography (HPLC) | The compounds to be separated suffer different repartition between a solid stationary phase and a liquid mobile phase with various polarities, at high values of pressure of the mobile phase and flow rate | Diode array (UV-VIS), mass spectrometry, fluorescence, or electrochemical detection | [59,60] |
| Thin layer chromatography (TLC) | Compound separation relies on the repartition between a solid stationary phase (silica gel, alumina) and a liquid mobile phase (methyl acetate/formic acid, ethanol/hexane, or methanol/chloroform/ hexane) | UV-VIS vizualization, fluorescence or phosphorescence detection | [61,62] |
| No | Antioxidant | Analytical Method | Carbon-Based Working Electrode | Analytical Characteristics | Ref. |
|---|---|---|---|---|---|
| 1. | Ascorbic acid (AA) | Cyclic voltammetry; Differential pulse voltammetry; | -carbon paste electrode; | -linear range 0.07–20 mM -supporting electrolyte: KCl 0.1 M; -RSD 2.35% in DPV and 2.29% in CV; -LOD 0.018 mM (CV) and 0.02 mM (DPV), calculated as 3× square mean error (for 10 determinations of the blank)/the slope of the calibration graph; -LOQ 0.062 mM (CV) and 0.068 mM (DPV), calculated as 10× square mean error (for 10 determinations of the blank)/the slope of the calibration graph; -analysis of commercial and home-made fruit juices; -oxidation peaks at 470 mV in DPV and at 510 mV in CV (vs SCE); | [123] |
| 2. | Ascorbic acid | Differential pulse voltammetry; | -screen-printed carbon electrode; | -determinations performed in phosphate buffer solutions (pH: 5.8, 7.0 and 7.4); -oxidation peak potentials increasing with concentration, noticed between −0.02 V and 0.11 V (vs. Ag/AgCl); -linear range of 1 to 4 µM (pH 5.8) and 2–10 µM (pH 7.0), as present in calibration curve; -assay of injectable vitamin C solutions; | [124] |
| 3. | Ascorbic acid Dopamine Paracetamol | Cyclic voltammetry; Differential pulse voltammery; Chronoamperometry; | -platinum nanoparticles-decorated graphene nanocomposite electrode, compared with graphene-modified glassy carbon electrode and bare glassy carbon electrode; | -supporting electrolyte: KCl 0.1 M; -linear range 300 μM to 20.89 mM (for AA in CV); -LOD of 300 μM (for AA in CV); -AA exhibited two linear DPV ranges, 300 μM to 7.36 mM and 8.12 to 39.87 mM; -LOD 5 μM (AA in DPV); -linear range from 420 μM to 29.26 mM for ascorbic acid in chronoamperometry, at 0.0 V vs. Ag/AgCl used as reference; | [125] |
| 4. | Ascorbic acid | Cyclic voltammetry; Chrono-amperometry; Linear sweep voltammetry (LSV); | -carbon veil electrode modified with phytosynthesized gold nanoparticles; | -LSVs recorded from 0.0 V to +0.8 V, vs. Ag/AgCl, at a scan rate of 0.05 Vs−1; -supporting electrolyte: Phosphate buffer pH 5.0 to 8.0; -modification with gold nanoparticles shifted the cyclic voltammetric potential of AA oxidation with more than 0.4 V towards less positive values; -linear response to AA 1 µM–5.75 mM in anodic voltammetry; -LOD 0.05 µM and LOQ 0.15 µM (CV); -most increased oxidation current of AA obtained in pH 6.0 phosphate buffer solution; -analysis of fruit juices; | [126] |
| 5. | Ascorbic acid Uric acid Cholesterol | Cyclic voltammetry; Square-wave voltammetry; | -carbon paste electrode modified with copper oxide-decorated reduced graphene; | -supporting electrolyte: Phosphate buffer pH 7.4; -scan rates (CV) of 10, 25, 50, 100, 150, 200, 250, 300, 500 mV s−1, for 500.0 µM cholesterol, using Ag/AgCl as reference; -linear response to AA 0.04–240.0 µM, with a LOD of 9 nM (SWV); -linear response to uric acid 0.04–400 µM, with a LOD of 8 nM (SWV); -linear response to cholesterol 0.03–300 µM, with a LOD of 9 nM (SWV); -differences between peak potentials (SWV) as follows: 430 mV (between cholesterol and ascorbic acid), 270 mV (between ascorbic acid and uric acid) and 700 mV (between cholesterol and uric acid); | [127] |
| 6. | Delphinidin, Cyanidin, Pelargonidin, Kuromanin, Callistephin | Cyclic voltammetry; Differential pulse voltammetry; | -glassy carbon electrode; | -supporting electrolyte: Methanol containing 0.1 mol L−1 lithium perchlorate or 0.1 mmol L−1 Britton-Robinson buffer; -pulse amplitude of 50 mV, pulse width of 70 ms, and scan rate of 10 mV s−1 (DPV); -cyclic voltammetric scan rates ranging from 25 mV s−1 to 500 mV s−1; -oxidation peak potentials comprised between 519 and 1115 mV vs Ag/AgCl (CV); -the larger the number of hydroxyl groups in the B ring, the lower the oxidation potential; -sugar moieties result in displacement of peak potentials to more positive values; | [128] |
| 7. | Oenin chloride; Malvin chloride; Kuromanine chloride; Cyanin chloride; Myrtillin chloride; Petunidin chloride; | Cyclic voltammetry; Differential pulse voltammetry; Square-wave voltammetry; | -glassy carbon electrode; | -supporting electrolytes: Acetic acid/acetate buffer pH 3.5 and 4.5, as well as phosphate buffer pH = 7.0; -voltammetric scans in the potential range of 0 to + 1.4 V vs. Ag/AgCl; -differential pulse voltammetric pulse amplitude 50 mV, pulse width 70 ms and scan rate 5 m Vs.−1; -square-wave voltammetric frequency 13, 25 and 50 Hz; amplitude 50 mV and potential increment 2 mV; -first cyclic voltammetric oxidation peak appears at 0.3 V for kuromanine chloride as well as for cyanin chloride, with a corresponding cathodic peak at 0.23 V in phosphate buffer pH = 7.0; -kuromanine chloride showed a DPV oxidation peak potential at 0.49 V and peonidin-3-O-glucoside at 0.39 V, in 0.2 M acetate buffer, pH 3.5; | [129] |
| 8. | Delphinidin-3-O-glucoside; Malvidin-3-O-glucoside-catechin; Peonidin-3-O-glucoside-4-vinylphenol, etc.; | Cyclic voltammetry; Differential pulse voltammetry; | -glassy carbon electrode; | -supporting electrolytes: Acetate-acetic acid buffer pH 3.6, and acetate-acetic acid buffer pH 3.6, containing 12% ethanol; -cyclic voltammograms obtained in the range of 0 to +0.8 mV, at a scan rate of 100 mV/s; -differential pulse voltammetric measurements performed with a pulse amplitude of 50 mV and a pulse width of 50 ms; -20-fold diluted wine presented DPV peak potentials of 443 mV and 666 mV vs Ag/AgCl, similar to those of wine extract, and to those of malvidin-3-O-glucoside (53.6% of the total anthocyanin content in grape extract); -20-fold diluted wine presented one oxidation peak, at 491 mV in CV; -ascorbic acid (0.05–0.1 mg/mL) used as reference, presented an oxidation peak at 270 mV in DPV; | [130] |
| 9. | Malvidin-3-glucoside, Catechin, Epicatechin, Gallic acid, Hydroxycinnamic acids, etc. | Square-wave voltammetry; | -glassy carbon electrode; | -disposable unmodified screen-printed carbon electrodes; -screen-printed carbon electrodes modified with single- and multi-walled carbon nanotubes; -determinations performed in a model wine solution: 12% (v/v) ethanol, containing 33 mM l-tartaric acid, at pH 3.6; -Ag/AgCl as reference electrode; -characterization of red wine polyphenols; -at the single-walled carbon nanotubes-modified screen-printed carbon electrode, first peak obtained between 138 mV (gallic acid) and 340 mV (malvidin-3-O-glucoside); -at the multi-walled carbon nanotubes-modified screen-printed carbon electrode, first peak obtained between 120 mV (gallic acid) and 370 mV (malvidin-3-O-glucoside); | [131] |
| 10. | β-carotene | Cyclic voltammetry; | -glassy carbon electrode; | -supporting electrolyte: 0.1 M LiClO4 in ethanol containing 10% CH2Cl2; -potential scan rate of 100 mV s1; -potential range from 0 to 1500 mV; -beta-carotene irreversibly oxidized at 500 and 920 mV vs Ag/AgCl reference; -linear analytical range of 10 to 380 mM; -LOD 2.5 mM; -LOQ 8.3 mM; | [132] |
| 11. | β-caro tene | Square-wave voltammetry; | -paraffin impregnated graphite electrode; | -supporting electrolyte: 0.1 M HClO4; -pulse amplitude 50 mV; -step potential 2 mV; -SWV showed oxidation peaks at 0.88 V and 1.09 V versus Ag/AgCl for β-carotene and astaxanthin; -analysis of raw vegetables and fruits; | [133] |
| 12. | β-carotene | Chrono-amperometry; | -stochastic sensor based on a graphene–porphyrin composite; | -supporting electrolyte: Acetate buffer pH = 3.0 -working potential of 125 mV versus Ag/AgCl; -linear response in the range between 1.0 × 10−15 mol L−1 and 1.0 × 10−3 mol L−1; -LOQ 1.0 × 10−15 mol L−1; -sensitivity 8.66 × 1010 s−1/mol L−1; -analysis of soft drinks; | [134] |
| 13. | Astaxanthin | Square-wave voltammetry; | -paraffin-impregnated graphite rod electrode | -two electrolyte solutions: 0.1 mol L−1 HClO4 and 0.1 mol L−1 KNO3; -frequency of 100 Hz, pulse amplitude 50 mV and step potential 2 mV. -LOD 15.77 µmol L−1 and LOQ 47.80 µmol L−1; -first reversible oxidation at −0.276 V vs. Ag/AgCl; second, not well defined, oxidation peak at −0.032 V; third reversible voltammetric response at 0.335 V; | [135] |
| 14 | Quercetin, dihydroquercetin, ferulic acid, synapic acid, gallic acid, caffeic acid etc. | Cyclic voltammetry; | -pyrographite electrode; | -determinations performed in: 0.2 M potassium phosphate buffer pH 6.0; 0.05 M potassium citrate buffer pH 5.0, and 0.1 M citrate-phosphate buffer pH 3.5; for increasing conductivity, 0.1 M KCl was added as an auxiliary electrolyte; -scanning speed 25 mV/s; -analyzed phenolics showed oxidation peaks in the range 235–834 mV, vs. Ag/AgCl; | [136] |
| 15. | Catechin, caffeic acid, coumaric acid, syringic acid, quercetin, mailvidin trans-resveratrol; estimation of total polyphenols levels; | Cyclic voltammetry; | -glassy carbon electrode | -determinations performed in model wine solution, consisting of 12% (v/v) ethanol, 33 mM l-tartaric acid, pH = 3.0, with Ag/AgCl as reference; -the anodic peak area in the range −100 to 1200 mV accounted for about 70% of total phenolics that absorbed at 280 nm; -catechol and galloyl containing polyphenols present in wine were quantitated relying on the size of the first anodic peak at around 450 mV after treatment with acetaldehyde; -flavonols were quantitated on the basis of the anodic peak current at 1120 mV; -good correlation of total flavanols with HPLC; | [137] |
| 16. | Catechin; estimation of total polyphenols levels; | Differential pulse voltammetry; | -glassy carbon electrode modified with green apple-sourced polyphenol oxidase (biosensor); | -supporting electrolyte phosphate buffer pH 7.65; -the anodic peak for reversible catechin oxidation, noticed at 0.219 V, with a cathodic peak at 0.128 V vs Ag/AgCl reference; -LOD 1.76 μg L−1; -LOQ 5.86 μg L−1; -RSD 2.5%; -detection of polyphenols in wine; | [138] |
| 17 | α-tocopherol; | Square-wave voltammetry; | -carbon fiber disk ultramicroelectrode | -determinations performed in benzene/ethanol and 0.1 mol L−1 H2SO4; -square wave amplitude 50 mV; -staircase step height 0.005 V; -frequency 25 Hz; -peak potential between 0.6 and 0.7 V versus SCE; | [139] |
| 18. | α-tocopherol; | Cyclic voltammetry; | -glassy carbon electrode | -supporting electrolyte: Glacial acetic acid and acetonitrile, containing 0.4 M sodium perchlorate; -100 mV s−1 scan rate; -external silver chloride reference; -peak potential 548 mV at first electron loss (that leads to phenoxyl radical) and 517 mV for second electron loss (that leads to phenoxonium cation radical); | [140] |
| 19. | α-tocopherol; Gallic acid; ascorbic acid; | Cyclic voltammetry; | -hydrophilic indium tin oxide electrode, lipophilic fluorinated nanocarbon film electrode and glassy carbon electrode; | -supporting electrolyte: Phosphate buffer saline (pH = 7.0), sodium dodecyl sulfate surfactant, 2-butanol cosurfactant, and toluene; -antioxidants analyzed in bicontinuous microemulsion, in which water and oil phases coexisted at microscopic scale; -using the indium tin oxide electrode, hydrosoluble gallic acid, ascorbic acid, and amphiphilic trolox exhibited irreversible anodic oxidation peaks at 0.61, 0.41, and 0.72 V, respectively, vs saturated calomel; -using the lipophilic fluorinated nanocarbon film electrode, amphiphilic trolox and lipophilic α-tocopherol, gave irreversible oxidations at 0.90 and 0.69 V, respectively, vs saturated calomel; | [141] |
| 20. | α-tocopherol; | Square wave anodic stripping voltammetry; | -glassy carbon paste electrode, | -supporting electrolyte: 0.1 M HNO3; -linear ranges of 5 × 10−7− 4 × 10−5 and 5 × 10−8− 1×10−5 mol L−1; -LOD of 1 × 10−7 mol L−1; -anodic peak potential at 520 mV vs. Ag/AgCl reference, -analyte extracted into glassy carbon paste electrode with 10% silicone oil, from 60% aqueous-acetonic mixture; -analysis of margarine and edible oils; | [142] |
| 21. | Superoxide dismutase; | Amperometry; | -glassy carbon electrode; | -detection performed at 0.0 V vs saturated calomel reference -LOD 8 × l0−11 M at pH 7.0, and 2 × 10−12 M at pH 9.0; -assay of buttermilk-sourced superoxide dismutase solution, in 0.1 M phosphate buffer, pH 8.0, containing 1 × 10−4 M EDTA; | [143] |
| 22. | Superoxide dismutase; | Cyclic voltammetry; | -screen-printed carbon electrode modified with self-assembled monolayers of gold nanoparticles in electropolymerized polypyrrole, and biofunctionalized with monoclonal anti-SOD1 antibody (immunosensor); | -supporting electrolyte: 0.1 M phosphate buffer solution containing 100 μM nitrite; -scan rate of 50 m Vs−1, using Ag/AgCl reference; -peak current recorded at the potential 0.8 V for UV-A treated cells was significantly higher than for control cells; -linear working range 0.5 nM to 5 μM -LOD 0.5 nM; -analysis of cultured human epidermal keratinocytes; | [144] |
| 23. | Superoxide dismutase; | Cyclic voltammetry; Electrochemiluminescence: Based on the signal (originating from the superoxide anion radical) emitted by methyl-cypridinalucifrin analogue at the electrode; | -glassy carbon electrode modified with a composite consisting of ferrocene imidazolium salts and hydroxy-functionalized graphene, in a Nafion matrix; | -supporting electrolyte: Phosphate buffer pH 5.3; -peak accounting for superoxide dismutase activity present between 0.6 and 0.7 V in electrochemiluminescence, and between 0.7 and 0.8 in cyclic voltammetry; -calibration plot linear in the 0.5 to 6.5 U·mL−1 SOD activity range, with LOD = 0.2 U·mL−1 in electrochemiluminescence; light emission lowered as SOD activity increased, correlated with oxygen generation from superoxide. -RSD (n = 11) 2.3% for 2.0 U·mL−1 SOD in electrochemiluminiscence; -scan range between 1.0 and −1.2 V.vs. Ag/AgCl in cyclic voltammetry and electrochemiluminescence -scan rate: 0.1 V·s−1 | [145] |
| 24. | Glutathione; | Cyclic voltammetry; Amperometry; Differential pulse voltammetry; | -multiwalled carbon nanotubes@reduced graphene oxide nanoribbons core-shell heterostructure-modified glassy carbon electrode; | -supporting electrolyte: 0.01 M phosphate buffered saline, pH 7.0; -CV measurements carried out from−0.2 V to +0.6 V, or from +0.3 V to +0.8 V at a scan rate of 50 mV s−1, showing enhanced electrocatalytical activity for the developed electrode; -DPV measurements carried out by scanning from +0.2 V to +0.7 V at a pulse amplitude of 50 mV; -highest voltammetric response obtained at 0.55 V vs. Ag/AgCl; -amperometric measurements carried out in 0.01 M phosphate buffer pH 7.0 at + 0.55 V vs. Ag/AgCl; -LOD 0.039 μM (amperometry); -two linear ranges: 0.05–266.3 μM and 266.3–766.3 μM (amperometry); -RSD 3.53% (amperometry); -analysis of real human serum samples; | [146] |
| 25 | Glutathione; | Cyclic voltammetry; Square wave voltammetry; | -gold-copper metal-organic framework immobilized on the surface of a glassy carbon electrode; | -supporting electrolyte: 0.1 mol L−1 phosphate buffer, pH 3.0; -anodic oxidation peak appeared at around +0.30 V vs. Ag/AgCl in CV and +0.25 V vs. Ag/AgCl in SWV; -linear dynamic range 1–10 μmol L−1 in SWV; -LOD 0.30 μmol L−1 in SWV; -sensitivity 0.89 ± 0.02 μA μmol L−1 in SWV; -repeatability 2.14% in SWV; -analysis of commercial tablets with more than 98% recovery; | [147] |
| 26. | Glutathione; Glutathione disulphide; | Cyclic voltammetry; Square-wave voltammetry; | -antimony trioxide–modified-carbon paste electrode; | -supporting electrolyte: 0.2 mol/L Britton-Robinson buffer; -oxidation potentials of +1.08 V for glutathione and +1.36 V for oxidized glutathione, vs. Ag/AgCl reference in CV and SWV; -SWV responses linear in the concentration range of 2 to 300 μmol/L glutathione; -LOD of 0.34 μmol/L glutathione and 0.1 μmol/L for oxidized glutathione in SWV; -determination of glutathione and glutathione disulphide in urine samples; -ascorbic acid, cysteine, glucose, glutamic acid and uric acid gave no significant interferences; | [148] |
| 27. | Uric acid; | Cyclic voltammetry; Chrono-amperometry; | -glassy carbon electrode, modified with a ZnO/carboxylic acid/multiwalled nanotube composite; | -supporting electrolyte: Phosphate buffer solution, pH = 7.0; -cyclic voltammetric peak at 0.5 V vs. Ag/AgCl; -chronoamperometric measurements performed at +0.577 V vs. Ag/AgCl; -rapid current response time (<5 s); -selective measurement of uric acid at clinically relevant concentrations (100–900 µM) by chronoamperometry; | [149] |
| 28. | Uric acid; | Cyclic voltammetry; Differential pulsevoltammetry; | -ZnO nanorods and graphene nanosheets hybrid electrode sprayed on indium tin oxide (ITO) glass; | -supporting electrolyte: Phosphate buffer saline, 0.01 M, pH = 7.4; -potential range: −0.2 to 0.6 V, vs. Ag/AgCl reference electrode (CV and DPV); -CVs were recorded at a scan rate of 50 mV s−1; -cyclic voltammetric oxidation peak potentials of uric acid and ascorbic acid, were 0.36 V and 0.28 V, respectively, -DPV analytical responses recorded with: A pulse height of 50 mV, a step height of 4 mV, a pulse width of 0.2 s, and a step time of 0.5 s; -sensitivity for uric acid 0.3 μA μM−1 cm−2 (DPV); -peak current intensities linearly related to the uric acid concentration in the range of 5–80 μM (DPV); -LOD 5 μM (DPV); -potentially applicable to clinical determination of uric acid; | [150] |
| 29. | Uric acid; | Cyclic voltammetry; | uricase/carboxymethylcellulose dispersed carbon nanotube/gold thin film biosensor; | -supporting electrolyte: 0.05 M phosphate buffer solution (pH 7.4); -cyclic voltammetric sweep rate: 50 mV s−1; -sensitivity of 233 μA mM−1 cm−2 at +0.35 V vs. Ag/AgCl reference; -linear range 0.02–2.7 mM; -detection limit of 2.8 μM; -detection of uric acid in serum and urine; -negligible interferents effect from urea and ascorbic acid at physiological amounts; | [151] |
| 30. | Bilirubin; | Cyclic voltammetry; Amperometry; | -carbon electrode modified with multiwalled carbon nanotubes or electrochemically reduced graphene oxide; | -supporting electrolyte: 0.1 M phosphate buffer solution (pH 7.2); -scan rate of 50 mV s−1; -two cyclic voltammetric oxidation peaks: At +0.25 V, corresponding to the oxidation of bilirubin to biliverdin and another at +0.48 V, corresponding to the oxidation of biliverdin to purpurine; -graphene type electrode: Amperometric linear range 0.1–600 µM and LOD 0.1 ± 0.018 nM; sensitivity 30 nA µM−1 cm−2, at 0.48 V vs. Ag/AgCl reference; -multiwalled carbon nanotube type: Amprometric linear range 0.5–500 μM; LOD 0.3 ± 0.022 μM; sensitivity 15 nA µM−1 cm−2, at 0.48 V vs Ag/AgCl reference; -no interferences from glucose, ascorbic acid, uric acid, and glutathione; -analysis of blood serum samples; | [152] |
| 31 | Bilirubin; | Linear sweep voltammetry; Differential pulse voltammetry; | -disposable screen-printed carbon electrodes obtained using graphite carbon ink printed on a PET substrate; | -supporting electrolyte: 0.05 M Trizma buffer, pH 8.5; -LSVs were recorded at a potential window of 0 to 0.6 V at a scan rate of 0.1 V/s; -DPVs were obtained at a potential window of 0 to 0.6 V with pulse amplitude of 0.05 V and pulse width of 0.05 s; -two anodic voltammetric peaks noticed on DPVs at around 0.25 V and 0.35 V (vs Ag/AgCl reference) corresponding to the oxidation of bilirubin to biliverdin, and of biliverdin to purpurin; -linear range 5–600 µM (LSV); -sensitivity 95 μA μM−1 cm−2 (LSV); -good selectivity in the presence of glucose, creatinine and ethanol; -application to serum samples; | [153] |
| 32. | Bilirubin; | Cyclic voltammetry; Differential pulsevoltammetry; | -carbon paste electrode; | -supporting electrolyte: 0.05 M phosphate buffer solution (pH 8.0); -two step oxidation process at around 300 mV and around 500 mV (CV); -DPV parameters: Pulse time 10 ms, potential step 5 mV and 150 ms optimized pulse amplitude; -linear range 3.5–25 µmol L−1 in DPV, considering the signal of irreversible anodic oxidation at 320 mV vs. Ag/AgCl; -LOD 1.2 µmol L−1 in DPV; | [154] |
| 33. | Melatonin; | Cyclic voltammetry; Fixed-potential amperometry; | -screen-printed carbon electrode modified with graphene; | -supporting electrolyte: 0.1 M phosphate buffer (pH 7.0); -oxidation CV peaks at 0.22 V and 0.80 V; reduction peaks at 0.12 V and 0.75 V vs pseudosilver/silver chloride reference electrode, -linear range of 1–300 μM in amperometry, at 0.8 V; -LOD 0.87 × 10−6 M and LOQ 2.91 × 10−6 M in amperometry; -RSD = 1.24% at the assay of Bien Dormir tablets (CV); | [155] |
| 34. | Melatonin; | Square wave voltammetry; | -glassy carbon electrode; | -linear concentration range of 5–200 µM; -LOD of 0.3432 µM; -analytical peak present at about 650 mV; -determination in pharmaceutical formulations and in human urine; | [156] |
| 35. | Melatonin; | Square wave voltammetry; | -carbon fiber microelectrode; | -reliably quantified melatonin concentrations in the visual cortex of anesthetized mice after intraperitoneal injections of different melatonin doses; -SWV enabled sensitive detection of oxidation peak at about 0.7 V vs. Ag/AgCl, discriminating melatonin from most common interferents; | [157] |
| 36. | Coenzyme Q10; | Differential pulse voltammetry; | -glassy carbon electrode; | -supporting electrolyte: Acetic acid containing 20% acetonitrile and 0.5 M CH3COONa; -DPV pulse amplitude of 20 mV, scan rate of 20 mV s−1 and pulse width of 80 ms allowed both fast recording and good resolution; -well-configured DPV cathodic peak attributed to reduction of CoQ10 at −20 mV vs. silver chloride external reference -LOD 0.014 mM (12 mg L−1); -LOQ 0.046 mM (40 mg L−1); -linearity up to 1 mM, with excellent corelation (r = 0.9989); -determination in commercial capsules; | [158] |
| 37. | conzyme Q10; | Direct current voltammetry; | -glassy carbon electrode; | -supporting electrolyte: Phosphate buffer solution (pH 6.86); -reversible oxidation peak at +0.4 V, corresponding to oxidation of hydroquinone group; reduction peak at −0.6 V vs. silver chloride reference; consistent to the previously confirmed reduction of ubiquinone to ubiquinol; -linear range 2.0 × 10−5–2.0 × 10−4 M; -assay of coenzyme Q10 in pharmaceuticals; | [159] |
| 38. | Coenzyme Q10; α-lipoic acid; | Cyclic voltammetry; Square wave anodic stripping voltammetry; | -MnO2-modified screen-printed graphene electrodes; | -determinations performed in 20:80 (v/v) ratio of ethanol/acetate buffer 0.1 M at pH 4.0; -the anodic peak of alpha lipoic acid present at a potential of 0.64 V, and that of coenzyme Q10 at 0.22 V, vs Ag/AgCl paste reference electrode (CV); -optimized square wave parameters: 5 mV step potential, 20 mV amplitude, and 25 Hz frequency; - linear range 2.0–75.0 μg mL−1 for coenzyme Q10, and 0.3–25 μg mL-1 for α-lipoic acid in square wave anodic stripping voltammetry; -LOD 0.56 μg mL−1 for coenzyme Q10 and 0.088 μg mL−1 for α-lipoic acid in square wave anodic stripping voltammetry; -determination in dietary supplements with good specificity in the presence of other vitamins and ionic species; | [160] |
| 39. | Vitamin D2 and D3; | Cyclic voltammetry; Differential pulsevoltammetry; | -glassy carbon electrode; | -supporting electrolyte: 40% ethanol/60% water containing LiClO4; -potential range of 0.0 to +1.5 V and scan rate of 50 mV s−1 (CV); -three well-configured, separate DPV peaks: Vitamin D around 0.594 V, vitamin E around 0.334 V, and vitamin A around 0.841 V vs. Ag/AgCl reference; -LOD 1.3 × 10−7 (vitamin D2) and 1.18 × 10−7 mol/L (vitamin D3) in DPV; -determination in vitamin D3 tablets; | [161] |
| 40. | Vitamin D2 and D3; | Cyclic voltammetry; Differential pulse voltammetry; | -glassy carbon electrode modified with AuPd; | -supporting electrolyte: Ethanol/water (40%/60%: v/v) containing lithium perchlorate; -CV scan rate of 50 mV/s, in the domain 0.0–1.5 V; -DPV scan rate: 10 mV/s, sampling time: 20 ms, pulse interval: 100 ms. -detection potential of +0.4 V vs. Ag/AgCl enabled diminution of interferences and good separation from vitamins A and E (DPV); -linear ranges 1–10 μM vitamin D2, 5–50 μM vitamin D3 in DPV; -LOD 0.15 μM vitamin D2 and 0.18 μM vitamin D3 (DPV); -detection of vitamin D3 in drug specimen; | [162] |
| 41. | Vitamin D3; | Square wave voltammetry; | -boron-doped diamond electrode; | -supporting electrolyte: 0.02 mol L−1 Britton-Robinson buffer pH 5.0 prepared in 50% ethanol; -well-defined voltammetric peak at around +1.00 V vs. Ag/AgCl; -linear range 2 to 200 mol L−1; -LOD 0.17 μmol L−1; -LOQ 0.51 μmol L−1; -determination in pharmaceutical products; | [163] |
| 42. | Alpha lipoic acid; | Cyclic voltammetry; Chronoamperometry; Differential pulse voltammetry; | -pyrolytic graphite electrode modified with cobalt phthalocyanine; | -supporting electrolyte: Phosphate buffer solution (pH = 7.0); -scan rate (CV) 25 m Vs.−1; -oxidation peak present at 0.84 V vs. SCE (CV), with highest response resulted from modification with cobalt phthalocyanine; -LOD 2.5 × 10−7 mol L−1 and LOQ 8.3 × 10−7 mol L−1 (CV); -LOD 9.8 × 10−8 mol L−1 and LOQ 3.2 × 10−7 mol L−1 (Chronoamperometry); -LOD 3.4 × 10−9 mol L−1 and LOQ 1.2 ×1 0−8 mol L−1 (DPV); -determination in pharmaceutical dietary supplement samples; | [164] |
| 43. | Alpha lipoic acid; | Amperometry; | -cobalt phthalocyanine–modified pyrolytic graphite electrode, integrated in a batch injection analysis set-up; | -supporting electrolyte: 0.1 mol L−1 phosphate buffer, pH 7.0; -applied potential of 0.9 V vs. Ag/AgCl; -linear response in the range 1.0 × 10−5–1.3 × 10−4 mol L−1; -LOD 1.5 × 10−8 mol L−1; -quantification in dietary supplements and in synthetic urine; | [165] |
| 44. | Alpha lipoic acid; | Cyclic voltammetry; Differential pulse voltammetry; | -SnO2 nanoparticles-modified glassy carbon electrode; | -supporting electrolyte: Britton-Robinson buffer pH 4.5; -well-defined DPV oxidation peak at 0.843 V vs. Ag/AgCl; -two linear dynamic ranges of 0.50–50 and 50–400 μmol L−1 (DPV); -LOD 0.13 μmol L−1 (DPV); -LOQ 0.43 μmol L−1 (DPV); -analysis of pharmaceutical dosage forms, with RSD between 0.45 and 6.2%; | [166] |
| 45. | Tert-butylhydroquinone and butylated hydroxyanisole | Cyclic voltammetry; Square-wave voltammetry; | -carbon black paste electrode; | -optimum conditions of electrolyte: 0.2 mol L−1 phosphate buffer (pH 7.0), 600.0 μmol L−1 surfactant cetylpyridinium bromide; -scan rate 50.0 mVs−1; -anodic cyclic voltammetric peak at cca 0 V vs. Ag/AgCl for tert-butylhydroquinone and at 0.4 V for butylated hydroxyanisole; -LOQ for tert-butylhydroquinone 0.23 μmol L−1 (SWV) and 0.27 μmol L−1 (DPV); -LOQ for butylated hydroxyanisole 0.26 μmol L−1 (SWV) and 0.23 μmol L−1 (DPV); -determination in mayonnaise, margarine, biodiesel; | [167] |
| No. | Analytical Method | Carbon-Based Working Electrode | Analytical Characteristics | Ref. |
|---|---|---|---|---|
| 1. | Cyclic voltammetry; Chronoamperometry; Square-wave voltammetry; | -poly(gallic acid)/multiwalled carbon nanotube modified glassy carbon electrode; | -supporting electrolyte 0.2 M H3PO4; -cyclic voltammetric scan rate 50 mV/s; -catalytic rate constant of 2.75 × 104 mol L−1 s−1, in chronoamperometry; -voltammetric oxidation peak for gallic acid at 0.53 V vs Ag/AgCl, in CV and SWV; -linear range of 4.975 × 10−6 to 3.381 × 10−5 M (SWV); -LOD 3.22 × 10−6 M gallic acid (SWV); -the SWVs of a fresh pomegranate juice sample shows three anodic peaks at 0.60, 0.70 and 1.0 V; signals can be attributed to the oxidation of different polyphenolic compounds, including gallic acid and catechin; -determination of total phenolic content in pomegranate juice, as gallic acid equivalent; -lack of interference of ascorbic acid, fructose, potassium nitrate and barbituric acid; | [173] |
| 2. | Cyclic voltammetry; Differential pulse voltammetry; | -nanocarbon-nanosilver hybrid electrode; | -supporting electrolyte: Phosphate buffer solution, pH 7.0; -CV studies confirmed that silver nanoparticles were efficiently immobilized on the Printex carbon surface; anodic and cathodic peak potentials noticed, were assigned to the redox pair Ag0/Ag+, whose presence was confirmed in the nanocomposite’s structure; -DPV peak of gallic acid at 0.091 V vs. Ag/AgCl; -sensitivity 0.254 μA/mol L−1 in DPV; -LOD 0.0663 μM in DPV; -linear range 5.0 × 10−7–8.5 × 10−6 in DPV; -estimation of antioxidant activity in wine; | [174] |
| 3. | Cyclic voltammetry; Amperometry; | -single-walled carbon nanotubes electrode, covalently functionalized with polytyrosine; | -supporting electrolyte 0.050 M phosphate buffer solution, pH 7.40; -CVs recorded between −0.200 V and 0.800 V (vs. Ag/AgCl) at a scan rate of 0.100 V s−1; -cyclic voltammetric oxidation peak potential for gallic acid at 0.2 V; -amperometric working potential 0.200 V; -amperometric sensitivity 163.2 mA/mol L−1; -amperometric LOD 8.8 × 10−9 M; -quantification of polyphenols in tea extracts: Green-Patagonia, red-Patagonia, classic-Green Hill and herbal (Taragüí); | [175] |
| 4. | Differential pulse Voltammetry; | -TiO2 nanoparticles/ multiwalled carbon nanotubes-modified glassy carbon electrode; -guanine biosensor based on TiO2 nanoparticles and multiwalled carbon nanotubes, immobilized on glassy carbon electrode; | -supporting electrolyte: phosphate buffer solution, pH 7.4; -oxidation DPV peak at 0.80 V (vs. SCE) corresponding to the electro-oxidation of guanine at the developed biosensor; -the peak intensity value of guanine oxidation increased linearly with increasing metabisulfite (employed as OH radical scavenger) concentration from 1 to 30 mmol L−1; -LOD 0.54 mmol L−1 for the guanine biosensor; -quantification of the antioxidant capacity in drug samples (adrenaline hydrochloride injection); | [176] |
| 5. | Cyclic voltammetry; Differential pulse Voltammetry; | -carbon paste electrode; | -supporting electrolyte: 0.1 M phosphate buffer, pH 5.0; -a cyclic voltammetric anodic peak at 0.33 V, with a corresponding cathodic peak at 0.28 V, vs Ag/AgCl, for 1% coffee sample in 0.1 M phosphate buffer pH 5.0; -two further anodic peaks at 0.55 V and 0.78 V were observed in DPVs of 0.5% coffee sample, in the same electrolyte; -good correlation with DPPH photometry and HPLC; -evaluation of the antioxidant activity of roasted coffee samples; -determination of electrochemical index of roasted coffee samples on the basis of the sum of the ratios of anodic peak currents to anodic peak potentials noticed on DPVs; | [177] |
| 6. | Cyclic voltammetry; Square-wave voltammetry; Chronoamperometry | -nanocomposite-graphene/poly (3,4-ethylenedioxythiophene): Poly (styrenesulfonate) modified screen-printed carbon electrode; | -supporting electrolyte: Ethanolic phosphate buffer solution based on 60% ethanol and 0.1 M phosphate buffer saline, pH 7.0; -method relied on DPPH reduction by antioxidants; -the presence of Trolox yielded a well–countoured anodic peak at around 0.9 V and a small cathodic peak at 0.3 V. -cathodic cyclic voltammetric peak potentials of catechin and caffeic acid were present at −0.03 V and−0.025 V vs. Ag/AgCl; -square voltammetric peak of DPPH at 0.25 V; -chronoamperometric DPPH detection at 0.2 V vs. Ag/AgCl; the linear calibration between the difference of cathodic DPPH currents (in the presence and absence of standard Trolox solution) and Trolox concentration in a range of 5–30 μM; -LOD 0.59 μM and LOQ 1.97 μM (chronoamperometry); -RSD of reproducibility is 2.13% (chronoamperometry); -RSD of repeatability 2.78% (chronoamperometry); -evaluation of the antioxidant activity in Thai herb and herbal beverage, expressed as mg of Trolox/g of sample; | [178] |
| 7. | Differential pulse voltammetry; | -multi-walled carbon nanotubes-modified glassy carbon electrode; | -supporting electrolyte: 0.1 M phosphate buffer (pH 4.0–7.0); -three DPV oxidation peaks observed at 0.39, 0.61 and 0.83 V for red dry wine and at 0.39, 0.80 and 1.18 V vs. Ag/AgCl for white dry wine, in phosphate buffer pH 4.0; -RSD% (as gallic acid equivalents) comprised between 1.0 and 6.9, as function of the wine sample; -evaluation of red and white dry wine antioxidant capacity, as gallic acid equivalents per 1 L of wine; | [179] |
| 8. | Differential pulse voltammetry; | -carbon paste electrode; -laccase-based modified carbon paste biosensor for the determination of phenolic content; | -supporting electrolyte: 0.1 mol L−1 phosphate buffer, pH 6.0; -biosensor was characterized by enhanced activity in mild acid medium and the response time (corresponding to the time required for enzyme oxidation of phenolic compounds), was lower than 30 s, but gradually increased up to 240 s, when a plateau was reached; -honey samples presented 2 to 3 anodic DPV peaks, the first at about 0.2 V, the second at about 0.5 V and the third nearby 0.8 V vs. Ag/AgCl; -electrochemical index determination, based on the sum of ratios of peak currents to peak potentials; -determination of phenolic content in honey samples; | [180] |
| 9. | Cyclic voltammetry; Differential pulse voltammetry; | -carbon black nanoparticles press imprinted films; | -supporting electrolyte: Phosphate buffer pH 7.40; -scan rate of 50 mV s−1 in the potential range of -0.20 V to +1.0 V vs. Ag/AgCl (CV); -pulse amplitude 50 mV/s, scan rate 10 m Vs−1 (DPV); -anodic peaks of o-diphenols and m-phenols present in olive oil extract, noticed in the range 0.120–0.160 V and 0.590–0.610 V (vs. Ag/AgCl), respectively; consistency with results obtained for the standards (DPV); -good repeatability for o-phenols; -RSD < 6% (o-phenols), RSD < 15% (m-phenols) in CV; -stable and reproducible voltammetric response of carbon black nanoparticles-based electrode; -determination of phenolic content and electrochemical indexes in olive oil extracts, using hydroxytyrosol and tyrosol as standards; | [181] |
| 10. | Cyclic voltammetry; Differential pulse voltammetry; | -glassy carbon electrode; | -supporting electrolyte: Dimethylsulfoxide, with tetrabutylammonium hexafluorophosphate 0.1 mol L−1; -the CVs were obtained at a scan rate of 100 mV s−1; -DPV pulse width = 5 mV, pulse amplitude = 60 mV and scan rate = 20 V s−1; -oxidation of ascorbic acid at 0.90 V in cyclic voltammetry and around 0.75 V vs. Ag/AgCl in differential pulse voltammetry; -in the CVs of the bark extract, a very well contoured peak was observed at 1.3 V, corresponding to meta-diphenols and isolated phenols; -in the CVs of the root and leaf extracts, an additional peak at 0.9 V indicates the presence of phenolics with ortho- or para-diphenol groups, in low amounts; -determination of the antioxidant capacity of Bunchosia glandulifera (Jacq.) Kunth (Malpighiaceae) extracts, using ascorbic acid as standard; | [182] |
| 11. | Chrono-amperometry; Differential pulse voltammetry; | -glassy carbon electrode modified with multi-walled carbon nanotubes; -glassy carbon polyquercetin-modified electrode; | -supporting electrolyte: Phosphate buffer pH 7.0; -antioxidant capacity using gallic acid as reference; -DPVs recorded from 0 to 0.8 V (pulse amplitude 50 mV, pulse time 50 ms and potential scan rate 10 mV/s); -DPVs of tea on the polyquercetin-modified electrode exhibited oxidation peaks at 0.080 and 0.19 V depending on the type of tea and a less configured oxidation step between 0.55 and 0.62 V vs Ag/AgCl; -chronoamperograms recorded at a constant potential of 0.2 V, potential corresponding to oxidation of tea antioxidants; -RSD = 0.5–20%, as function of the tea type (chronoamperometry); -determination of the antioxidant capacity of tea, highest content for Green Sencha; | [183] |
| 12. | Cyclic voltammetry; | -glassy carbon electrode; | -supporting electrolyte: Phosphate buffer pH = 7.0; -cyclic voltammetric scans performed between 0.0 and 0.5 V vs Ag/AgCl at a scanning rate of 5 mV/s; -anodic peak at 244 mV for pomace and its parts (skins and stems), and 252 mV for seeds; -analyse of winemaking by-products (pomace, skins, seeds and stems separated from pomace); | [184] |
| 13. | Cyclic voltammetry; | -glassy carbon electrode; | -supporting electrolyte: 0.1 M sodium acetate–acetic acid buffer at pH 3.6; -all the grapes revealed peak I at 0.26–0.31 V, peak II between 0.42 and 0.55 V, and peak III at approximately 0.66 V vs Ag/AgCl; -correlations of anodic peak area with phenolic content and antioxidant activity were assessed; -determination of phenolic contents and antioxidant capacity in 12 grape cultivars; | [185] |
| 14. | Cyclic voltammetry; Square-wave voltammetry; Differential pulse voltammetry | -glassy carbon electrode; -laccase-modified carbon paste electrode; | -supporting electrolyte: 0.1 M phosphate buffer solution, pH 6.0, using Ag/AgCl reference; -CV: Scan rate of 100 mV s−1 within the range 0–1.4 V; -SWV: Pulse amplitude 50 mV, frequency 50 Hz and a potential increment of 2 mV, scan rate of 100 mV s−1; -first peak present between 100 and 400 mV, second between 0.55 and 0.7, and third at around 1 V, in CV/SWV; -solutions of the extracts yielded highest DPV peaks at 0.2 V, alongside peaks present at 0.6 and 0.9 V; -electrochemical indexes were calculated based on the sum of ratios of peak currents to peak potentials in DPV; -antioxidant activity evaluation of dried herbal extracts; -highest electrochemical indexes obtained for Gingko biloba and Hypericum perforatum, consistent with the results obtained by spectrophotometry; | [186] |
| 15. | Cyclic voltammetry; Differential pulse voltammetry; | -glassy carbon electrode; | -supporting electrolyte 0.1 M KCl; -CV scan from 0 to +1000 mV at a scan rate of 100 mV s−1; -DPV scan from 0 to +1000 mV at a scan rate of 100 mV s−1; -the first peak of mature-phase milk occurred at around 400 mV; colostrum, had oxidation peaks at very high potential, around 800 mV (DPV) vs Ag/AgCl; -mature-phase milk yielded a peak at around 400 mV; pasteurized milk had a peak at around 500 mV (CV); -areas below oxidation peaks proportional to the amount of antioxidant compounds; -free radical scavenging activity was highest for fresh breast milk and lowest for pasteurized breast milk, confirming the results obtained in DPPH assay; -correlation between DPV and CV (r = 0.602, p < 0.001); correlation between DPV and DPPH method (r = 0.339, p = 0.003); correlation between CV and DPPH method (r = 0.468 p < 0.000); | [187] |
| 16. | Cyclic voltammetry; Differential pulse voltammetry; | -screen-printed carbon electrodes; | -supporting electrolyte 0.1 M HCl; -in CV, the potential recorded between 0.0 V and +1.2 V, at 100 mV s−1 scan rate, using silver pseudo-reference electrode; -optimum DPV parameters: 100 mV modulation amplitude, 10 mV step potential, 0.05 s modulation time and 0.5 s interval time; -ortho-diphenols (oleuropein, hydroxytyrosol and caffeic acid) show one anodic peak between 0.5 and 0.6 V, and one cathodic peak between 0.4 and 0.6 V (CV); same compounds present an anodic peak at +0.5 V, in standard and real sample (DPV); -ferulic acid gave an oxidation peak at higher potential (0.7 V), in the standard solution, whereas this signal was almost negligible in the real sample (DPV); -tyrosol is oxidized at +0.93 V, alongside other mono-phenols (such as vanillic acid) that suffer oxidation around this potential value, in standard and real sample (DPV); -LOD of 0.022 mg L−1 for caffeic acid and tyrosol, in DPV; -determination of hydrophilic phenols in olive oil; | [188] |
| 17. | Cyclic voltammetry; Differential pulse voltammetry; Square-wave voltammetry; | -electroactivated pencil graphite electrode; | -supporting electrolyte: 0.05 mol L−1 potassium hydrogen phthalate; -naringenin is irreversibly oxidized, giving rise to two pH-dependent peaks due to mixed (diffusion- and adsorption-controlled) electrode processes involving two electrons and one proton; -LOD = 3.06 × 10−8 mol L−1, and LOQ = 1.02 × 10−7 mol L−1 for DPV, expressed as naringenin; -LOD = 4.40 × 10−8 mol L−1, and LOQ = 1.11 × 10−7 mol L−1 for SWV, expressed as naringenin; -application to determination of polyphenol content in citrus juice; | [189] |
| 18. | Amperometry; Cyclic voltammetry; | -disposable polyester screen-printed graphitic macroelectrodes; | -supporting electrolyte: 1:1 (v/v) methanol: Ethanol mixture containing 0.05 mol/L−1 LiCl; -CV scans between −0.3 and +1.0 V, scan rate 50 mV s−1, for DPPH 1 mmol/L−1 (in 50 mmol L−1 LiCl prepared in methanol:ethanol) and for DPPH in the presence of antioxidants; -chlorogenic acid, caffeic acid, catechin and quercetin were oxidized between +0.7 and +0.9 V (CV); -oxidation processes of tocopherol and BHT occurred at more positive potentials, around +1.0 V (CV); -amperometric detection of DPPH remaining after reaction with antioxidants, at +0.1 V (vs. pseudo AgCl). -analysis of edible oils; | [190] |
| 19. | Cyclic voltammetry; Differential pulse voltammetry; Linear sweep voltammetry; | -multi-walled carbon nanotube paste electrode; | -supporting electrolyte: 0.02 M acetate-acetic acid buffer/4% methanol (pH 4.5); -CV scan between 0 and 1.5 V, at 100 mV s−1; -DPV pulse amplitude 50 mV and scan rate 100 mV s−1 -CV oxidation potential at 1.12 V; -DPV oxidation potential at 1.19 V; -the average Tafel slopes of mushroom extract was found to be 1.258 mV per decade, in LSV; -assay of Morchella esculenta L. as ethnomedicinal food; -obtained net electrochemical antioxidant power as 2.7 ± 0.12 mg per gram, using ascorbic acid as reference; | [191] |
| 20. | Cyclic voltammetry; | -carbon paste electrode incorporating 2,2-diphenyl-1-picrylhydrazyl; | -supporting electrolyte: Phosphate buffer solution 0.1 M, pH 7.0; -potential ranges investigated: 0.00 V to −1.00 V; 0.60 V to −0.20 V and 0.45 V to 1.10 V, vs. Ag/AgCl; -a peak potential of −833 mV, due to the irreversible reduction of the nitro functions on the phenyl group, present in the structure of DPPH; -for tea extract analyzed, signals recorded in the potential window 0.4 V–1.1 V; -tea extracts presented an anodic peak at about 0.8 V and a cathodic one at around 0.75 V; | [192] |
| 21. | Cyclic voltammetry; Amperometry; | -single walled carbon nanotubes-, graphene- and gold nanoparticles-based screen-printed electrodes; | -supporting electrolyte: Sodium phosphate buffer solution 0.1 M, pH 7.0; -assessment of the quenching capacity of plant extracts (Hippophae fructus and Lavandula Flowers) in the presence of H2O2 (chosen as model reactive oxygenated species); -cyclic voltammograms reveal anodic peaks below 0.45 V vs Ag/AgCl, in the presence of extract; -a marked cyclic voltammetric anodic peak at 0.09 V, and a small cathodic peak at 0.35 V noticed for lavender extracts; -amperometric assay based on sensor’s sensitivity to H2O2 in the absence / presence of the extract; -best sensitivity obtained at the gold nanoparticles-modified sensor: 6.43 ± 0.2 µA cm−2 mM−1; -amperometric determinations at constant potential of 0.55 V, with linearity of 2 to 30 mM hydrogen peroxide: -antioxidant capacity determination of hydrosoluble plant extracts; | [17] |
| 22. | Cyclic voltammetry; | -glassy carbon electrode; | -supporting electrolyte: Tetrabutylammonium perchlorate, in dimethyl sulfoxide 99%; -scan rate 25 mV/s in CV; -the voltammograms of the figs and almond extracts presented redox signals at positive potentials, the anodic oxidation being noticed at 1.175 V and 1.218 V, respectively, vs saturated calomel reference; -determination of antioxidant activity of dry fruits (almond, apricot, cashew, figs, peanut, pistachio, raisins, and walnut); | [193] |
| 23. | Cyclic voltammetry; | -glassy carbon electrode; | -supporting electrolyte: Sodium acetate-acetic acid buffer (0.1 mol L−1, pH = 4.5) in acidified 80% methanol; -analytical cyclic voltammetric signals for all target phenolic compounds present between 0 mV and 800 mV, at a scan rate of 100 mV s−1; -cyclic voltamogramms of berry fruits presented anodic peaks between 310 mV (quercetin) and 0.756 mV (coumaric acid) vs. Ag/AgCl -antioxidant capacity quantification relied on the area underneath the anodic peak, corresponding to the charge up to a potential value of 500 mV (Q500); -evaluation of antioxidant activity of 15 berry samples (strawberries, blackberries, blueberries and red raspberries); | [194] |
| 24. | Amperometry; | -glassy carbon electrode integrated in a flow injection system with sequential diode array and amperometric detection; | -supporting electrolyte: Ethanol 12% v/v and tartaric acid 2 g/L, pH 3.6; -amperometric determinations at 800 mV vs. Ag/AgCl; -calibration curve over the range 0–0.19 mM gallic acid equivalents; -determination of total polyphenol content and antioxidant activity of white, red wines and oenological tannins; -total wine phenolic content between 1.08 and 15.4 mM gallic acid; -concentration range 0.07–0.34 mM gallic acid obtained for tannin solutions; | [195] |
| 25. | Cyclic voltammetry; Differential pulse voltammetry; | -glassy carbon electrode; | -supporting electrolyte: Sodium acetate 0.1 mol L−1; -two CV oxidation waves at potentials of 0.45 V and 0.84 V vs Ag/AgCl, pointing towards the presence in the extract of minimum two kinds of reducing species, or a single reducing species that can be oxidized by two stable intermediates; -extracts showed no voltammetric waves in the range of reduction potentials, suggesting that the reducing species in the extract of Mimosa albida leaves can exhibit antioxidant potential; -two oxidation waves noticed on DPVs, indicating the existence of two antioxidant compounds: One species with greater antioxidant capacity with oxidation potential at 0.34 V, and the other one with lower antioxidant power, at 0.79 V; -oxidation signal for Mimosa albida-modified silver nanoparticles at +0.3792 V (CV); -analysis of aqueous leaf extract of Mimosa albida and assay of antioxidant capacity of Mimosa albida-modified silver nanoparticles; | [196] |
| 26. | Differential pulse voltammetry; | -glassy carbon electrode; | -supporting electrolyte: Sodium phosphate buffer solution 0.1 M, pH 7.4; -scan rate 50 mV/s; pulse period 35 ms; potential step 10 mV; -at increasing amounts of added extract, DPV oxidation peaks were noticed, at approximately 0.270 V, 0.430V, and 0.880 V vs. Ag/AgCl; -determination of the antioxidant capacity of the Greigia Sphacelata fruit; | [197] |
| 27. | Cyclic voltammetry; Differential pulse voltammetry; | -glassy carbon electrodes modified with carbon nanotubes and chitosan; | -supporting electrolyte: Britton-Robinson electrolyte buffer, at pH 3.0; -chicoric acid anodic peak at 0.610 ± 0.060 V and cathodic peak at 435 ± 0.055 V vs Ag/AgCl, at 300 mV s−1 scan rate; -the intensities of oxidation and reduction currents linearly vary with the square root of the scanning speed, in cyclic voltammetry; -DPVs showed oxidation peaks for caftaric acid at 0.505 ± 0.002 V, and for chicoric acid at 0.515 ± 0.001 V vs Ag/AgCl, which are consistent with the results obtained at the assay of pharmaceutical forms; -determination of total polyphenol content and antioxidant activity of Echinacea purpurea extracts in 3 different pharmaceutical forms (capsules, tablets and tincture); | [198] |
| 28. | Staircase voltammetry; | glassy carbon electrode; | -supporting electrolyte-100 mM KNO3; -staircase voltammograms recorded successively for 4 cycles between +1.0 and −1.2 V vs Ag/AgCl; -scan rate 50 mV/s, starting and ending in +1.0 V; -the half-wave potential (E1/2), or the potential corresponding to half the anodic peak current (Ipa) was considered; lower E1/2 values are correlated to higher antioxidant potential; -the peak intensity or, more accurately the surface area under the oxidation peak, provided quantitative informations: Antioxidant concentration/antioxidant capacity; -analytical peaks present between −0.6 and 0 V, and around 0.5 V; -evaluation of antioxidant activity for teas, wines and (superfood) juices; -antioxidant index calculated relying on the maximum charge of oxidation (Qmax), the standard potential of the oxygen evolution reaction (vs. Ag/AgCl) and the standard potential of hydrogen evolution reaction (vs. Ag/AgCl); | [199] |
| 29. | Cyclic voltammetry; | -glassy carbon electrode; | -supporting electrolyte −0.1 M H2SO4 solution; -scan rate investigated in the range of 20–160 mV s−1; -dependence of charge under the anodic peak, on the concentration of tested red corn pigments, quantified in the region −0.2–1.2 V; -CV for dark red corn seeds extract (1 mg mL−1) presents two anodic peaks at about 0.4 V and 0.65 V; a cathodic peak at the reverse scan, at about 0.2 V vs saturated calomel reference; -evaluation of total phenolic and flavonoid contents in red corn; | [200] |
| 30. | Voltammetry; | -carbon fiber ultramicroelectrodes; | -linear relationship between anodic peak current and caffeic acid (reference antioxidant) concentration from 3.0 to 500 μmol L−1; -repeatability illustrated by a RSD of 2.7%; -sensitivity 12 μA L mol−1; -Ag/AgCl electrode used as reference; -LOD 0.41 μmol L−1; -LOQ 1.26 μmol L−1; -estimation of antioxidant capacity in three different wines, and in green and red grape samples; | [201] |
| 31 | Cyclic voltammetry; Square-wave voltammetry; | -carbon electrode modified with guanine-, polythionine-, and nitrogen-doped graphene; | -determinations performed in PBS pH = 1.5; -1.0 mg/mL−1 guanine solution as optimum for modification of the electrode; -a pair of redox peaks found between 0 and 0.3 V in CV; peak currents increased with increasing scan times; a thin blue membrane formed on the surface of electrode, showed that thionine was successfully polymerized; -oxidation peak for ascorbic acid at about 1.1 V (SWV); -linear range for ascorbic acid (standard antioxidant) analytical response ranged from 0.5 to 3.0 mg L−1 in SWV; -LOD 0.21 mg L−1 (SWV); -RSD 3.1% (SWV); -determination of antioxidant capacity of fruit juices (grape juice, guava juice, and orange juice) and jute leaves extract, ramie leaves extract, and hemp leaves extract; | [202] |
| 32 | Differential pulse stripping voltammetry (DPSV); Cyclic voltammetry; | -glassy carbon electrode modified with polyglycine; | -supporting electrolyte: Britton-Robinson electrolyte buffer, pH 3.0; -well-configured oxidation peak of quercetin (model antioxidant) occurs at around +460 mV, a corresponding cathodic peak being visible at 420 mV vs Ag/AgCl; -peaks shifted towards less positive potentials when the scan rates increased from 20 to 400 mV/s in CV; -oxidation peak current assigned to phenolic compounds of yam, at 430 mV, consistent to the peak potential of quercetin, on differential pulse stripping voltammograms; -DPSV-pulse amplitude of 50 mV, pulse width of 500 ms; -LOD 0.39 µg L−1 (DPSV); -LOQ 1.39 µg L−1 (DPSV); -electrode modification resulted in 3.15-fold increase of sensitivity, when compared to the bare glassy carbon; -total antioxidant capacity of 0.1 kg of yam, obtained as 96.15 +/− 0.85 µg/L of equivalents quercetin at 95% confidence level; -relative standard deviation of 0.88%; | [203] |
| 33 | Cyclic voltammetry; Chronoamperometry; | -biosensor based on laccase immobilized onto a gold nanoparticles/graphene nanoplatelets-modified screen-printed carbon electrode; | -supporting electrolyte: Sodium phosphate buffer solution 0.1M, pH 7.0; -potential range from −0.6 V to 1.2 V with a scan rate of 0.05 V/s and a step potential of 2.0 mV (CV); -anodic and cathodic CV peaks of hydroquinone at 0.2 V vs Ag/AgCl and 0 V, respectively, at a scan rate of 0.05 V s−1; -excellent electrocatalytic activity towards oxidation of hydroquinone at a potential of −0.05 V in hydrodynamic amperometry; -linear range 4–130 µM (chronoamperometry); -LOD 1.5 µM chronoamperometry) -LOQ 5 µM (chronoamperometry); -determination of phenolic antioxidant capacity in wine and blueberry syrup; | [204] |
| 34. | Cyclic voltammetry; | -glassy carbon electrode; | -supporting electrolyte: 0.1 M sodium acetate/acetic acid bufer solution, pH 3.6; -potential scans performed from −0.4 V to 1 V vs Ag/AgCl at a scan rate of 25 mV s−1; -total antioxidant capacity expressed as ascorbic acid equivalents; -crude medicinal plant extracts exhibited an oxidation peak around 750 mV on cyclic voltammograms; -medicinal plant extracts have less than 36 times smaller total antioxidant capacity, when compared to ascorbic acid; -it was concluded that cyclic voltammetry and FRAP are recommended for flavonoid quantitation; | [205] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisoschi, A.M.; Pop, A.; Iordache, F.; Stanca, L.; Bilteanu, L.; Serban, A.I. Antioxidant Determination with the Use of Carbon-Based Electrodes. Chemosensors 2021, 9, 72. https://doi.org/10.3390/chemosensors9040072
Pisoschi AM, Pop A, Iordache F, Stanca L, Bilteanu L, Serban AI. Antioxidant Determination with the Use of Carbon-Based Electrodes. Chemosensors. 2021; 9(4):72. https://doi.org/10.3390/chemosensors9040072
Chicago/Turabian StylePisoschi, Aurelia Magdalena, Aneta Pop, Florin Iordache, Loredana Stanca, Liviu Bilteanu, and Andreea Iren Serban. 2021. "Antioxidant Determination with the Use of Carbon-Based Electrodes" Chemosensors 9, no. 4: 72. https://doi.org/10.3390/chemosensors9040072
APA StylePisoschi, A. M., Pop, A., Iordache, F., Stanca, L., Bilteanu, L., & Serban, A. I. (2021). Antioxidant Determination with the Use of Carbon-Based Electrodes. Chemosensors, 9(4), 72. https://doi.org/10.3390/chemosensors9040072