
Transfer RNA-Derived Fragments and isomiRs Are Novel Components of Chronic TBI-Induced Neuropathology


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
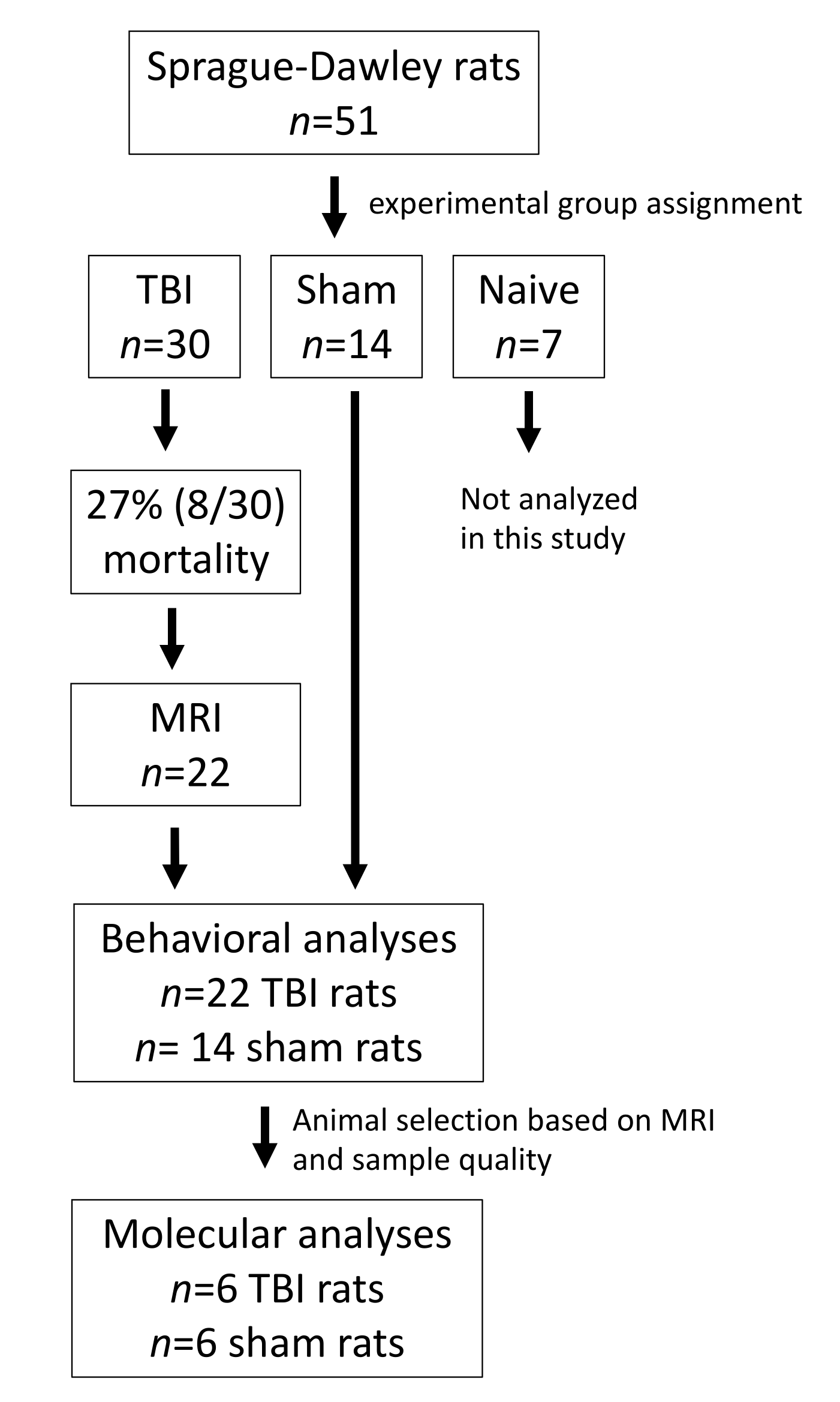
2.1. Animals
2.2. Induction of TBI with Lateral Fluid-Percussion
2.3. Composite Neuroscore
2.4. Magnetic Resonance Imaging
2.5. Morris Water Maze
2.6. Sampling of Brain Tissue
2.7. Histologic Analysis
2.8. Isolation of Total RNA from Brain Tissue
2.9. Small RNA and RNA Sequencing from Brain Tissue
2.9.1. Small RNA Sequencing
2.9.2. Messenger RNA Sequencing
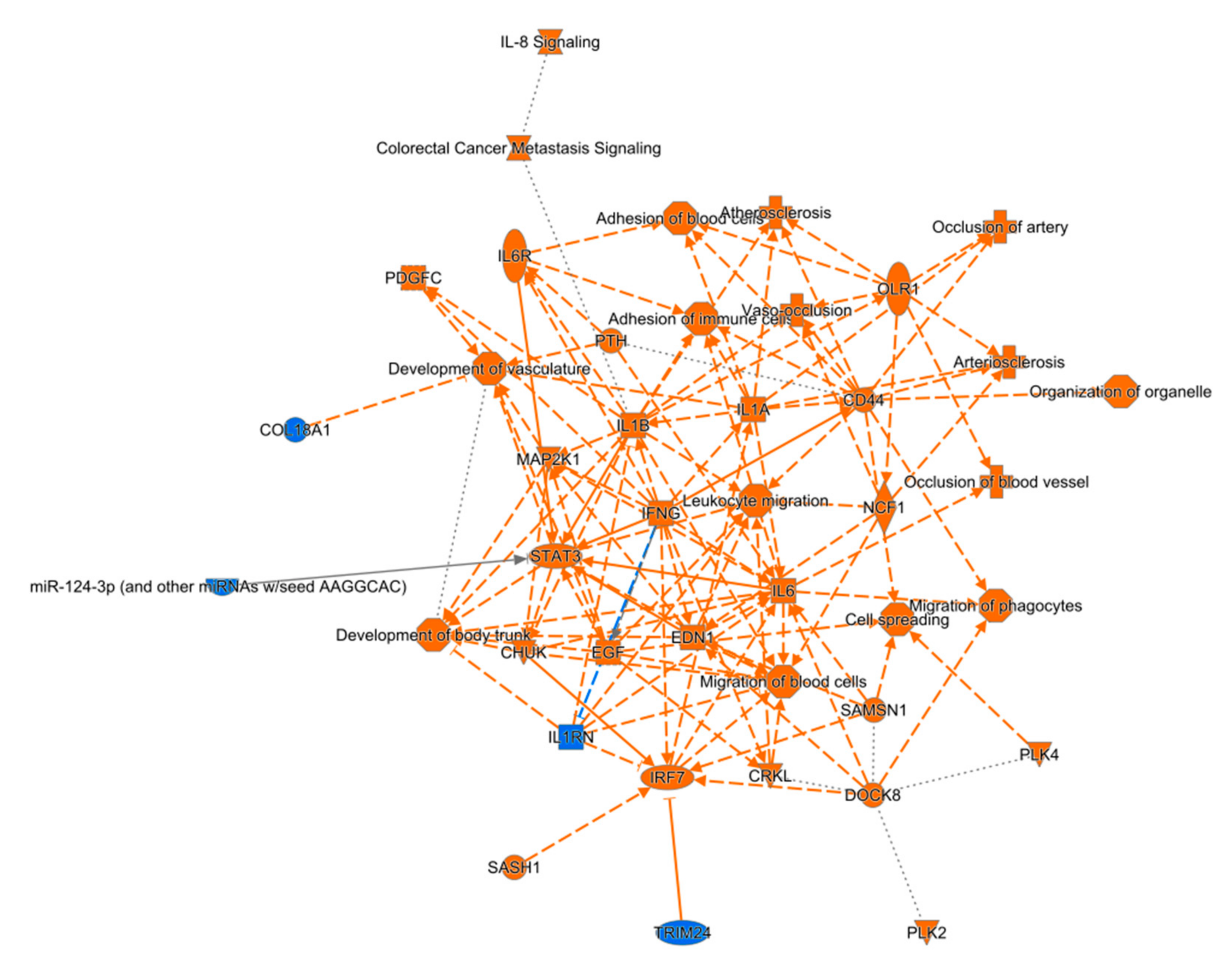
2.10. Pathway Analysis of RNA-Seq Data
2.11. Validation of the Small RNA-Seq Data with Droplet Digital PCR and Quantitative PCR
2.11.1. microRNA
2.11.2. Transfer RNA Derived Fragments
2.12. In-Silico Prediction and qPCR Validation of mRNA Targets for the Validated miRNAs
2.13. In-Silico Prediction and qPCR Vvalidation of mRNA Targets for the Validated tRFs
2.14. qPCR Analysis of tRF Cleaving Enzyme Angiogenin
2.15. Data Analysis and Statistics
3. Results
3.1. Mortality after Lateral FPI
3.2. Anatomic Analysis and Assessment of Chronic Neuroinflammation
3.3. Differentially Expressed miRNAs Identified from sncRNA-Seq
3.4. mRNA Targets Predicted for the Validated miRNA Candidates
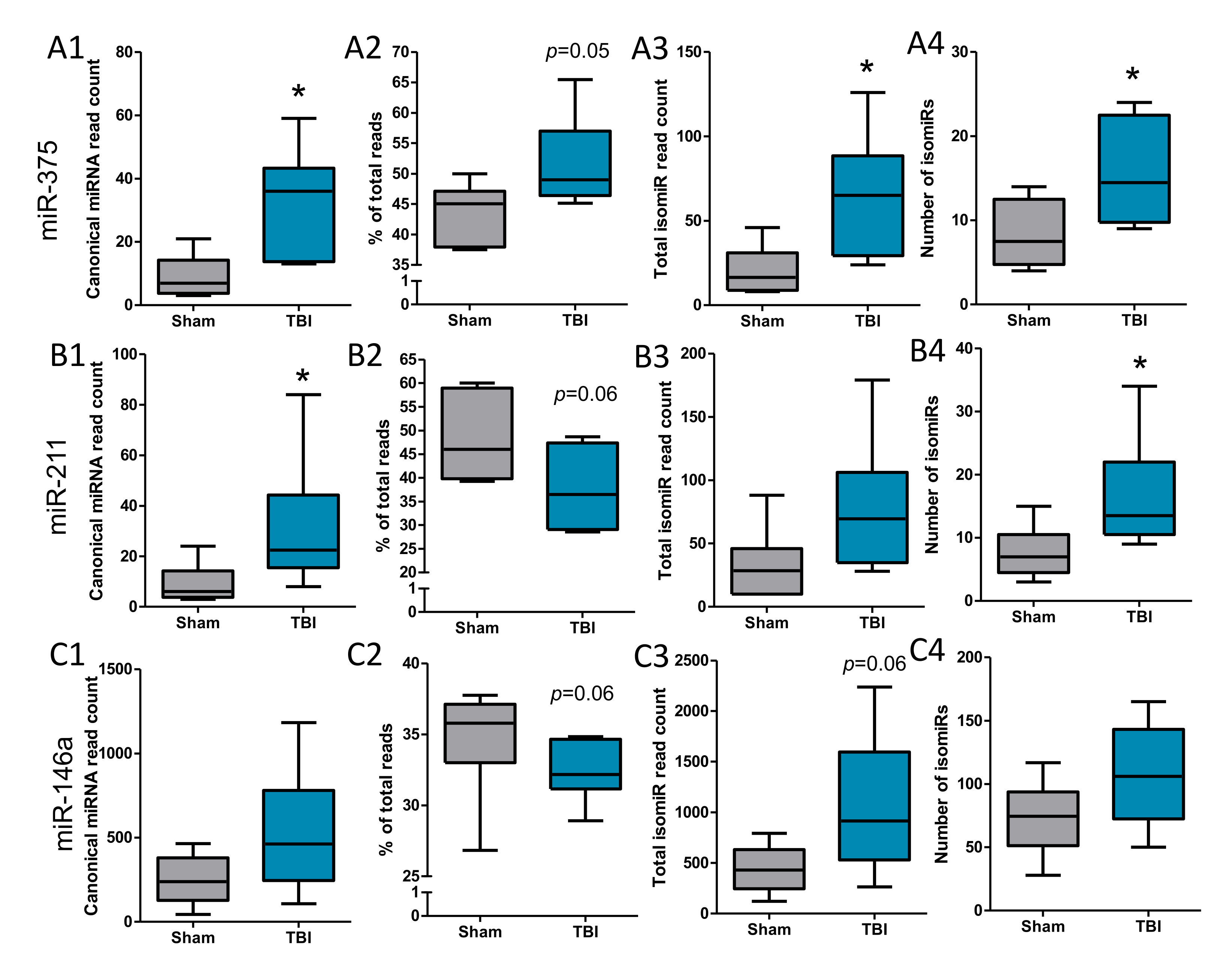
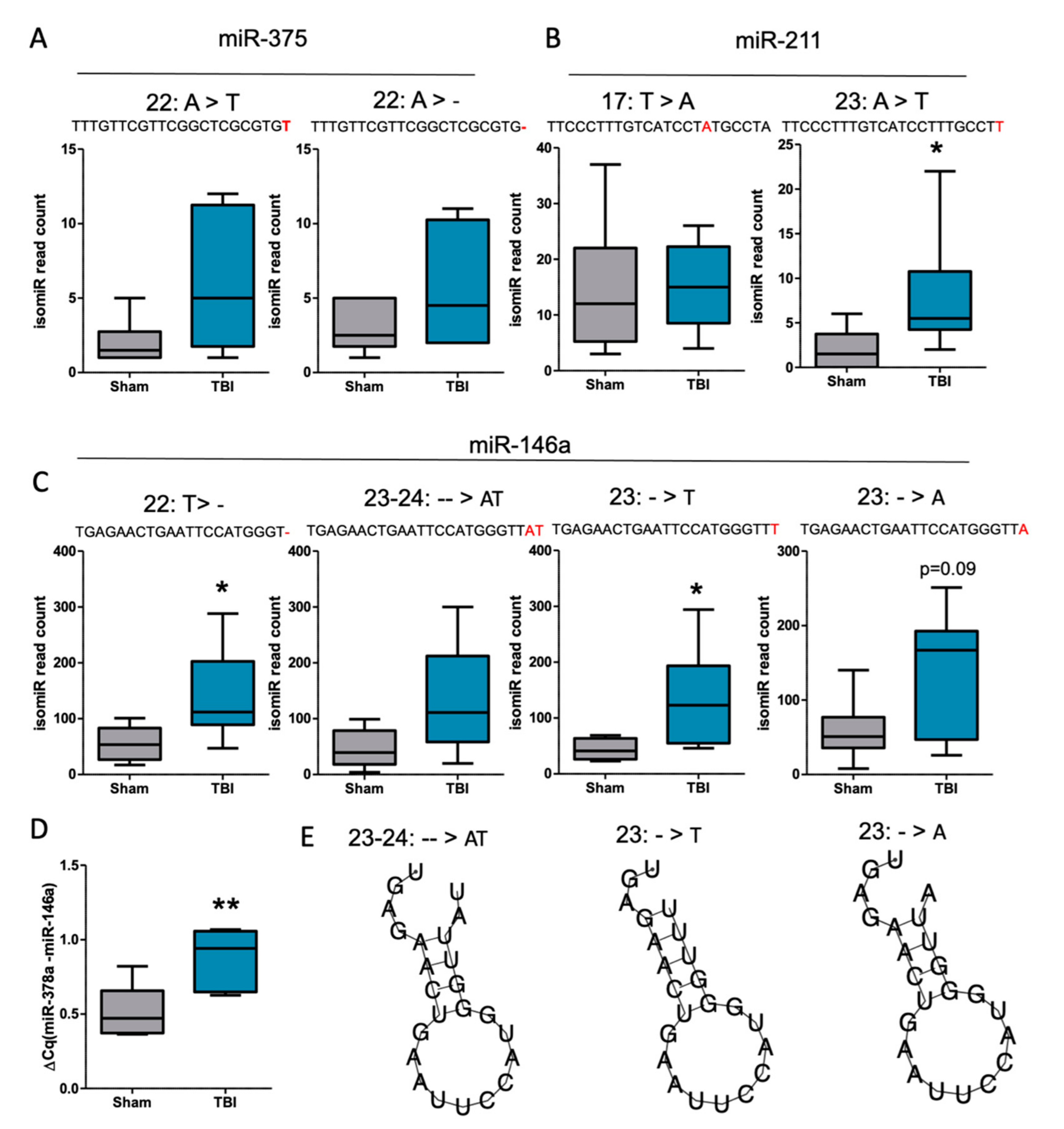
3.5. IsomiRs Identified for the Validated Differentially Expressed miRNAs
3.6. Elevation of Transfer RNA-Derived Fragments after TBI
3.7. mRNA Targets Predicted for the Validated tRF Candidates
3.8. No Clear Upregulation of tRF-Cleaving Enzyme Angiogenin after TBI
3.9. Elevated 3′tRF and miR-146a Levels Relate to Worse Behavioral Outcome after TBI
4. Discussion
4.1. Chronic Neuroinflammation after TBI
4.2. Differentially Expressed miRNAs after TBI
4.3. Are 3′isomiRs a Specific Feature of TBI?
4.4. Variable Region Cleaved 3′tRFs Are Upregulated after TBI
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carroll, L.J.; Cassidy, J.D.; Holm, L.; Kraus, J.; Coronado, V.G. Methodological issues and research recommendations for mild traumatic brain injury: The WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. J. Rehabil. Med. 2004, 113–125. [Google Scholar] [CrossRef] [Green Version]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet. Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.-H.; Cao, X.-Y.; Li, Y.-Y.; Zhou, C.-C.; Li, L.; Wang, K.; Li, H.; Yu, P.; Jin, Y.; Gao, L. Gene expression profile of the hippocampus of rats subjected to traumatic brain injury. J. Cell. Biochem. 2019, 120, 15776–15789. [Google Scholar] [CrossRef]
- Lipponen, A.; Paananen, J.; Puhakka, N.; Pitkänen, A. Analysis of Post-Traumatic Brain Injury Gene Expression Signature Reveals Tubulins, Nfe2l2, Nfkb, Cd44 and S100a4 as Treatment Targets. Sci. Rep. 2016, 6, 31570. [Google Scholar] [CrossRef] [Green Version]
- Johnson, V.E.; Stewart, J.E.; Begbie, F.D.; Trojanowski, J.Q.; Smith, D.H.; Stewart, W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013, 136, 28–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thelin, E.P.; Tajsic, T.; Zeiler, F.A.; Menon, D.K.; Hutchinson, P.J.A.; Carpenter, K.L.H.; Morganti-Kossmann, M.C.; Helmy, A. Monitoring the Neuroinflammatory Response Following Acute Brain Injury. Front. Neurol. 2017, 8, 351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleminger, S.; Oliver, D.L.; Lovestone, S.; Rabe-Hesketh, S.; Giora, A. Head injury as a risk factor for Alzheimer’s disease: The evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 2003, 74, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Risacher, S.L.; McAllister, T.W.; Saykin, A.J. Traumatic brain injury and age at onset of cognitive impairment in older adults. J. Neurol. 2016, 263, 1280–1285. [Google Scholar] [CrossRef] [Green Version]
- Walsh, S.; Donnan, J.; Fortin, Y.; Sikora, L.; Morrissey, A.; Collins, K.; MacDonald, D. A systematic review of the risks factors associated with the onset and natural progression of epilepsy. Neurotoxicology 2017, 61, 64–77. [Google Scholar] [CrossRef]
- Li, Q.; Hu, B.; Hu, G.W.; Chen, C.Y.; Niu, X.; Liu, J.; Zhou, S.M.; Zhang, C.Q.; Wang, Y.; Deng, Z.F. TRNA-Derived Small Non-Coding RNAs in Response to Ischemia Inhibit Angiogenesis. Sci. Rep. 2016, 6, 20850. [Google Scholar] [CrossRef] [Green Version]
- Bratkovič, T.; Modic, M.; Camargo Ortega, G.; Drukker, M.; Rogelj, B. Neuronal differentiation induces SNORD115 expression and is accompanied by post-transcriptional changes of serotonin receptor 2c mRNA. Sci. Rep. 2018, 8, 5101. [Google Scholar] [CrossRef] [Green Version]
- Peschansky, V.J.; Wahlestedt, C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics 2014, 9, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKiernan, R.C.; Jimenez-Mateos, E.M.; Bray, I.; Engel, T.; Brennan, G.P.; Sano, T.; Michalak, Z.; Moran, C.; Delanty, N.; Farrell, M.; et al. Reduced mature microRNA levels in association with dicer loss in human temporal lobe epilepsy with hippocampal sclerosis. PLoS ONE 2012, 7, e35921. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Redell, J.B.; Liu, Y.; Dash, P.K. Traumatic brain injury alters expression of hippocampal microRNAs: Potential regulators of multiple pathophysiological processes. J. Neurosci. Res. 2009, 87, 1435–1448. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, A.M.; Ando, Y.; de Hoon, M.J.L.; Tomaru, Y.; Nishibu, T.; Ukekawa, R.; Funakoshi, T.; Kurokawa, T.; Suzuki, H.; Hayashizaki, Y.; et al. A comprehensive survey of 3’ animal miRNA modification events and a possible role for 3’ adenylation in modulating miRNA targeting effectiveness. Genome Res. 2010, 20, 1398–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burroughs, A.M.; Ando, Y. Identifying and characterizing functional 3’ nucleotide addition in the miRNA pathway. Methods 2019, 152, 23–30. [Google Scholar] [CrossRef]
- Raina, M.; Ibba, M. tRNAs as regulators of biological processes. Front. Genet. 2014, 5, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, D.M.; Parker, R. Stressing out over tRNA cleavage. Cell 2009, 138, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Xu, Z.; Sheng, J. tRNA-Derived Small RNA: A Novel Regulatory Small Non-Coding RNA. Genes 2018, 9, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberbauer, V.; Schaefer, M.R. tRNA-Derived Small RNAs: Biogenesis, Modification, Function and Potential Impact on Human Disease Development. Genes 2018, 9, 607. [Google Scholar] [CrossRef] [Green Version]
- Thompson, D.M.; Lu, C.; Green, P.J.; Parker, R. tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA 2008, 14, 2095–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haussecker, D.; Huang, Y.; Lau, A.; Parameswaran, P.; Fire, A.Z.; Kay, M.A. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA 2010, 16, 673–695. [Google Scholar] [CrossRef] [Green Version]
- Krishna, S.; Raghavan, S.; DasGupta, R.; Palakodeti, D. tRNA-derived fragments (tRFs): Establishing their turf in post-transcriptional gene regulation. Cell. Mol. Life Sci. 2021, 78, 2607–2619. [Google Scholar] [CrossRef] [PubMed]
- Hogg, M.C.; Raoof, R.; El Naggar, H.; Monsefi, N.; Delanty, N.; O’Brien, D.F.; Bauer, S.; Rosenow, F.; Henshall, D.C.; Prehn, J.H. Elevation in plasma tRNA fragments precede seizures in human epilepsy. J. Clin. Investig. 2019, 129, 2946–2951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapur, M.; Ganguly, A.; Nagy, G.; Adamson, S.I.; Chuang, J.H.; Frankel, W.N.; Ackerman, S.L. Expression of the Neuronal tRNA n-Tr20 Regulates Synaptic Transmission and Seizure Susceptibility. Neuron 2020, 108, 193–208. [Google Scholar] [CrossRef]
- Qin, C.; Xu, P.-P.; Zhang, X.; Zhang, C.; Liu, C.-B.; Yang, D.-G.; Gao, F.; Yang, M.-L.; Du, L.-J.; Li, J.-J. Pathological significance of tRNA-derived small RNAs in neurological disorders. Neural Regen. Res. 2020, 15, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Vuokila, N.; Das Gupta, S.; Huusko, R.; Tohka, J.; Puhakka, N.; Pitkänen, A. Elevated Acute Plasma miR-124-3p Level Relates to Evolution of Larger Cortical Lesion Area after Traumatic Brain Injury. Neuroscience 2020, 433, 21–35. [Google Scholar] [CrossRef]
- Kharatishvili, I.; Nissinen, J.P.; McIntosh, T.K.; Pitkänen, A. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience 2006, 140, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, J.; Andrade, P.; Natunen, T.; Hiltunen, M.; Malm, T.; Kanninen, K.; Soares, J.I.; Shatillo, O.; Sallinen, J.; Ndode-Ekane, X.E.; et al. Disease-modifying effect of atipamezole in a model of post-traumatic epilepsy. Epilepsy Res. 2017, 136, 18–34. [Google Scholar] [CrossRef] [Green Version]
- Halonen, T.; Nissinen, J.; Jansen, J.A.; Pitkänen, A. Tiagabine prevents seizures, neuronal damage and memory impairment in experimental status epilepticus. Eur. J. Pharmacol. 1996, 299, 69–81. [Google Scholar] [CrossRef]
- Huusko, N.; Römer, C.; Ndode-Ekane, X.E.; Lukasiuk, K.; Pitkänen, A. Loss of hippocampal interneurons and epileptogenesis: A comparison of two animal models of acquired epilepsy. Brain Struct. Funct. 2015, 220, 153–191. [Google Scholar] [CrossRef] [PubMed]
- Peña-Llopis, S.; Brugarolas, J. Simultaneous isolation of high-quality DNA, RNA, miRNA and proteins from tissues for genomic applications. Nat. Protoc. 2013, 8, 2240–2255. [Google Scholar] [CrossRef] [Green Version]
- Lipponen, A.; El-Osta, A.; Kaspi, A.; Ziemann, M.; Khurana, I.; KN, H.; Navarro-Ferrandis, V.; Puhakka, N.; Paananen, J.; Pitkänen, A. Transcription factors Tp73, Cebpd, Pax6, and Spi1 rather than DNA methylation regulate chronic transcriptomics changes after experimental traumatic brain injury. Acta Neuropathol. Commun. 2018, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC, a Quality Control Tool for High throughput Sequence Data, Version 3; Babraham Bioinformatics: Cambridge, UK, 2010.
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Nielsen, C.B.; Shomron, N.; Sandberg, R.; Hornstein, E.; Kitzman, J.; Burge, C.B. Determinants of targeting by endogenous and exogenous microRNAs and siRNAs. RNA 2007, 13, 1894–1910. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.P.; Lowe, T.M. GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016, 44, D184–D189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, P.; Liu, D.Z.; Jickling, G.C.; Sharp, F.R.; Yin, K.-J. MicroRNA-based therapeutics in central nervous system injuries. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2018, 38, 1125–1148. [Google Scholar] [CrossRef]
- Klein, P.; Dingledine, R.; Aronica, E.; Bernard, C.; Blümcke, I.; Boison, D.; Brodie, M.J.; Brooks-Kayal, A.R.; Engel, J.J.; Forcelli, P.A.; et al. Commonalities in epileptogenic processes from different acute brain insults: Do they translate? Epilepsia 2018, 59, 37–66. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, H.; Zhong, J.; Yang, J.; Darwazeh, R.; Tian, X.; Huang, Z.; Jiang, L.; Cheng, C.; Wu, Y.; et al. Significant changes in circular RNA in the mouse cerebral cortex around an injury site after traumatic brain injury. Exp. Neurol. 2019, 313, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.A.; Moss, L.D.; Lee, J.-Y.; Tajiri, N.; Acosta, S.; Hudson, C.; Parag, S.; Cooper, D.R.; Borlongan, C.V.; Bickford, P.C. Long noncoding RNA MALAT1 in exosomes drives regenerative function and modulates inflammation-linked networks following traumatic brain injury. J. Neuroinflammation 2018, 15, 204. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; He, J.; Tian, X.; Luo, Y.; Zhong, J.; Zhang, H.; Li, H.; Cen, B.; Jiang, T.; Sun, X. microRNA-9-5p alleviates blood-brain barrier damage and neuroinflammation after traumatic brain injury. J. Neurochem. 2020, 153, 710–726. [Google Scholar] [CrossRef]
- Henry, R.J.; Doran, S.J.; Barrett, J.P.; Meadows, V.E.; Sabirzhanov, B.; Stoica, B.A.; Loane, D.J.; Faden, A.I. Inhibition of miR-155 Limits Neuroinflammation and Improves Functional Recovery After Experimental Traumatic Brain Injury in Mice. Neurother. J. Am. Soc. Exp. Neurother. 2019, 16, 216–230. [Google Scholar] [CrossRef] [Green Version]
- Corps, K.N.; Roth, T.L.; McGavern, D.B. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015, 72, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Ramlackhansingh, A.F.; Brooks, D.J.; Greenwood, R.J.; Bose, S.K.; Turkheimer, F.E.; Kinnunen, K.M.; Gentleman, S.; Heckemann, R.A.; Gunanayagam, K.; Gelosa, G.; et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann. Neurol. 2011, 70, 374–383. [Google Scholar] [CrossRef]
- Webster, K.M.; Sun, M.; Crack, P.; O’Brien, T.J.; Shultz, S.R.; Semple, B.D. Inflammation in epileptogenesis after traumatic brain injury. J. Neuroinflammation 2017, 14, 10. [Google Scholar] [CrossRef] [Green Version]
- Albert, V.; Arulselvi, S.; Agrawal, D.; Pati, H.P.; Pandey, R.M. Early posttraumatic changes in coagulation and fibrinolysis systems in isolated severe traumatic brain injury patients and its influence on immediate outcome. Hematol. Oncol. Stem Cell Ther. 2019, 12, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, R.; Liu, L.; Watkins, T.; Zhang, F.; Dong, J. Traumatic brain injury-associated coagulopathy. J. Neurotrauma 2012, 29, 2597–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, S.C.; Smith, D.H. Coagulopathy in traumatic brain injury. Neurocrit. Care 2004, 1, 479–488. [Google Scholar] [CrossRef]
- Ou, J.; Kou, L.; Liang, L.; Tang, C. MiR-375 attenuates injury of cerebral ischemia/reperfusion via targetting Ctgf. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-P.; Bi, Y.-J.; Liu, D.-M.; Wang, L.-Y. Hsa-miR-375 promotes the progression of inflammatory bowel disease by upregulating TLR4. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7543–7549. [Google Scholar] [CrossRef]
- Bekenstein, U.; Mishra, N.; Milikovsky, D.Z.; Hanin, G.; Zelig, D.; Sheintuch, L.; Berson, A.; Greenberg, D.S.; Friedman, A.; Soreq, H. Dynamic changes in murine forebrain miR-211 expression associate with cholinergic imbalances and epileptiform activity. Proc. Natl. Acad. Sci. USA 2017, 114, E4996–E5005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korotkov, A.; Puhakka, N.; Das Gupta, S.; Vuokila, N.; Broekaart, D.W.M.; Anink, J.J.; Heiskanen, M.; Karttunen, J.; van Scheppingen, J.; Huitinga, I.; et al. Increased expression of miR142 and miR155 in glial and immune cells after traumatic brain injury may contribute to neuroinflammation via astrocyte activation. Brain Pathol. 2020, 30, 897–912. [Google Scholar] [CrossRef]
- van Scheppingen, J.; Iyer, A.M.; Prabowo, A.S.; Mühlebner, A.; Anink, J.J.; Scholl, T.; Feucht, M.; Jansen, F.E.; Spliet, W.G.; Krsek, P.; et al. Expression of microRNAs miR21, miR146a, and miR155 in tuberous sclerosis complex cortical tubers and their regulation in human astrocytes and SEGA-derived cell cultures. Glia 2016, 64, 1066–1082. [Google Scholar] [CrossRef]
- Wang, X.; Yin, F.; Li, L.; Kong, H.; You, B.; Zhang, W.; Chen, S.; Peng, J. Intracerebroventricular injection of miR-146a relieves seizures in an immature rat model of lithium-pilocarpine induced status epilepticus. Epilepsy Res. 2018, 139, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Cui, G.; Tang, H.; Kong, L.; Wang, X.; Cui, C.; Xiao, Q.; Ji, H. Silencing of microRNA-146a alleviates the neural damage in temporal lobe epilepsy by down-regulating Notch-1. Mol. Brain 2019, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Puhakka, N.; Bot, A.M.; Vuokila, N.; Debski, K.J.; Lukasiuk, K.; Pitkänen, A. Chronically dysregulated NOTCH1 interactome in the dentate gyrus after traumatic brain injury. PLoS ONE 2017, 12, e0172521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, C.; Huang, C.; Xu, X.; Teng, J. miR-155 Knockdown Protects against Cerebral Ischemia and Reperfusion Injury by Targeting MafB. Biomed Res. Int. 2020, 2020, 6458204. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: MicroRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, N.; Wani, S.; Xu, Q.; Gu, J.; Lea, K.; Heater, S.; Barbacioru, C.; Steptoe, A.L.; Martin, H.C.; Nourbakhsh, E.; et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol. 2011, 12, R126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muljo, S.A.; Kanellopoulou, C.; Aravind, L. MicroRNA targeting in mammalian genomes: Genes and mechanisms. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Avcilar-Kucukgoze, I.; Kashina, A. Hijacking tRNAs From Translation: Regulatory Functions of tRNAs in Mammalian Cell Physiology. Front. Mol. Biosci. 2020, 7, 610617. [Google Scholar] [CrossRef]
- Kuscu, C.; Kumar, P.; Kiran, M.; Su, Z.; Malik, A.; Dutta, A. tRNA fragments (tRFs) guide Ago to regulate gene expression post-transcriptionally in a Dicer-independent manner. RNA 2018, 24, 1093–1105. [Google Scholar] [CrossRef] [Green Version]
- Kümmel, D.; Krishnakumar, S.S.; Radoff, D.T.; Li, F.; Giraudo, C.G.; Pincet, F.; Rothman, J.E.; Reinisch, K.M. Complexin cross-links prefusion SNAREs into a zigzag array. Nat. Struct. Mol. Biol. 2011, 18, 927–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.-H.; Hoover, R.; McIntosh, T.K.; Hazell, A.S. Early, transient increase in complexin I and complexin II in the cerebral cortex following traumatic brain injury is attenuated by N-acetylcysteine. J. Neurotrauma 2006, 23, 86–96. [Google Scholar] [CrossRef]
- Xie, Y.; Yao, L.; Yu, X.; Ruan, Y.; Li, Z.; Guo, J. Action mechanisms and research methods of tRNA-derived small RNAs. Signal Transduct. Target. Ther. 2020, 5, 109. [Google Scholar] [CrossRef]
- Olson, K.A.; Verselis, S.J.; Fett, J.W. Angiogenin is regulated in vivo as an acute phase protein. Biochem. Biophys. Res. Commun. 1998, 242, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, P.; O’Day, E.; Emara, M.M.; Wagner, G.; Lieberman, J.; Anderson, P. G-quadruplex structures contribute to the neuroprotective effects of angiogenin-induced tRNA fragments. Proc. Natl. Acad. Sci. USA 2014, 111, 18201–18206. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, S.; Ivanov, P.; Hu, G.-F.; Anderson, P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J. Cell Biol. 2009, 185, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastià, J.; Kieran, D.; Breen, B.; King, M.A.; Netteland, D.F.; Joyce, D.; Fitzpatrick, S.F.; Taylor, C.T.; Prehn, J.H.M. Angiogenin protects motoneurons against hypoxic injury. Cell Death Differ. 2009, 16, 1238–1247. [Google Scholar] [CrossRef] [Green Version]
- Skorupa, A.; King, M.A.; Aparicio, I.M.; Dussmann, H.; Coughlan, K.; Breen, B.; Kieran, D.; Concannon, C.G.; Marin, P.; Prehn, J.H.M. Motoneurons secrete angiogenin to induce RNA cleavage in astroglia. J. Neurosci. 2012, 32, 5024–5038. [Google Scholar] [CrossRef] [PubMed]
- Megel, C.; Hummel, G.; Lalande, S.; Ubrig, E.; Cognat, V.; Morelle, G.; Salinas-Giegé, T.; Duchêne, A.-M.; Maréchal-Drouard, L. Plant RNases T2, but not Dicer-like proteins, are major players of tRNA-derived fragments biogenesis. Nucleic Acids Res. 2019, 47, 941–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, A.; Zurolo, E.; Prabowo, A.; Fluiter, K.; Spliet, W.G.M.; van Rijen, P.C.; Gorter, J.A.; Aronica, E. MicroRNA-146a: A key regulator of astrocyte-mediated inflammatory response. PLoS ONE 2012, 7, e44789. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puhakka, N.; Das Gupta, S.; Vuokila, N.; Pitkänen, A. Transfer RNA-Derived Fragments and isomiRs Are Novel Components of Chronic TBI-Induced Neuropathology. Biomedicines 2022, 10, 136. https://doi.org/10.3390/biomedicines10010136
Puhakka N, Das Gupta S, Vuokila N, Pitkänen A. Transfer RNA-Derived Fragments and isomiRs Are Novel Components of Chronic TBI-Induced Neuropathology. Biomedicines. 2022; 10(1):136. https://doi.org/10.3390/biomedicines10010136
Chicago/Turabian StylePuhakka, Noora, Shalini Das Gupta, Niina Vuokila, and Asla Pitkänen. 2022. "Transfer RNA-Derived Fragments and isomiRs Are Novel Components of Chronic TBI-Induced Neuropathology" Biomedicines 10, no. 1: 136. https://doi.org/10.3390/biomedicines10010136
APA StylePuhakka, N., Das Gupta, S., Vuokila, N., & Pitkänen, A. (2022). Transfer RNA-Derived Fragments and isomiRs Are Novel Components of Chronic TBI-Induced Neuropathology. Biomedicines, 10(1), 136. https://doi.org/10.3390/biomedicines10010136