
From NAFLD to MAFLD: Aligning Translational In Vitro Research to Clinical Insights

 , , , ,
, , , ,
Abstract
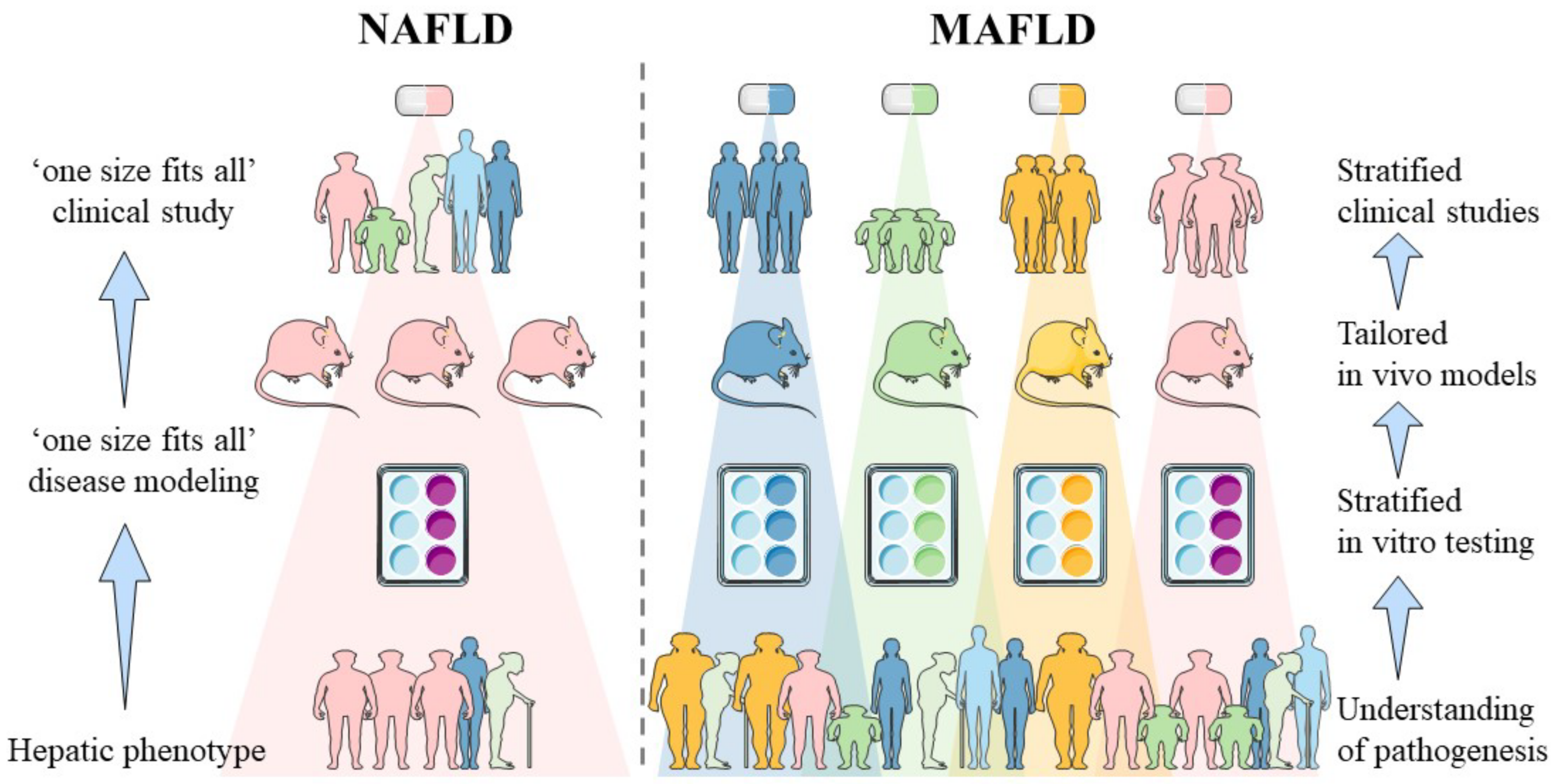
:1. Introduction
2. Sources of Heterogeneity in MAFLD and Their In Vitro Implementation
2.1. Sex and Hormonal Status
2.2. Pediatric/Juvenile MAFLD
2.3. Fructose Consumption
2.4. Genetic Predisposition and Ethnicity
2.5. Epigenetics
2.6. Obesity and Body Fat Distribution
2.7. Lean MAFLD
2.8. Microbiota
2.9. Zonation
2.10. Disease Progression and Regression
3. Dual-Etiology in MAFLD: Fatty Liver with Multiple Faces
3.1. Drug Intake
3.2. Ethanol Consumption
3.3. Viral Hepatitis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cotter, T.G.; Rinella, M. Nonalcoholic fatty liver disease 2020: The state of the disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Human-based systems: Mechanistic NASH modelling just around the corner? Pharmacol. Res. 2018, 134, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Brunt, E.M.; Kleiner, D.E.; Kowdley, K.V.; Chalasani, N.; Lavine, J.E.; Ratziu, V.; Mccullough, A. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology 2011, 54, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Cohen, D.E.; Fisher, E.A. Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Semin. Liver Dis. 2013, 33, 380–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corey, K.E.; Misdraji, J.; Gelrud, L.; Zheng, H.; Chung, R.T.; Krauss, R.M. Nonalcoholic steatohepatitis is associated with an atherogenic lipoprotein subfraction profile. Lipids Health Dis. 2014, 13, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH drug development hitches a lift on PPAR agonism. Cells 2020, 9, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslam, M.; Sanyal, A.J.; George, J.; Sanyal, A.; Neuschwander-Tetri, B.; Tiribelli, C.; Kleiner, D.E.; Brunt, E.; Bugianesi, E.; Yki-Järvinen, H.; et al. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020, 158, 1999–2014. [Google Scholar] [CrossRef] [PubMed]
- Han, M.A.T.; Altayar, O.; Hamdeh, S.; Takyar, V.; Rotman, Y.; Etzion, O.; Lefebvre, E.; Safadi, R.; Ratziu, V.; Prokop, L.J.; et al. Rates of and factors associated with placebo response in trials of pharmacotherapies for nonalcoholic steatohepatitis: Systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2019, 17, 616–629.e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015, 149, 367–378. [Google Scholar] [CrossRef]
- Glass, O.; Filozof, C.; Noureddin, M.; Berner-Hansen, M.; Schabel, E.; Omokaro, S.O.; Schattenberg, J.M.; Barradas, K.; Miller, V.; Francque, S.; et al. Standardisation of diet and exercise in clinical trials of NAFLD-NASH: Recommendations from the Liver Forum. J. Hepatol. 2020, 73, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F. Nonalcoholic fatty liver disease: Another leap forward. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Kumar, R.; Wang, M.; Zhu, Y.; Lin, S. MAFLD criteria overlooks a number of patients with severe steatosis: Is it clinically relevant? J. Hepatol. 2020, 73, 1265–1267. [Google Scholar] [CrossRef] [PubMed]
- Ciardullo, S.; Perseghin, G. Prevalence of NAFLD, MAFLD and associated advanced fibrosis in the contemporary United States population. Liver Int. 2021, 41, 1290–1293. [Google Scholar] [CrossRef]
- Kostrzewski, T.; Cornforth, T.; Snow, S.A.; Ouro-Gnao, L.; Rowe, C.; Large, E.M.; Hughes, D.J. Three-dimensional perfused human in vitro model of non-alcoholic fatty liver disease. World J. Gastroenterol. 2017, 23, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Cami, B.; De Boe, V.; Heymans, A.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; et al. Human hepatic in vitro models reveal distinct anti-NASH potencies of PPAR agonists. Cell Biol. Toxicol. 2021, 37, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Feaver, R.E.; Cole, B.K.; Lawson, M.J.; Hoang, S.A.; Marukian, S.; Blackman, B.R.; Figler, R.A.; Sanyal, A.J.; Wamhoff, B.R.; Dash, A. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. J. Clin. Invest. 2016, 1, e90954. [Google Scholar] [CrossRef] [PubMed]
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2020, 2, 1034–1045. [Google Scholar] [CrossRef]
- Boeckmans, J.; Buyl, K.; Natale, A.; Vandenbempt, V.; Branson, S.; De Boe, V.; Rogiers, V.; De Kock, J.; Rodrigues, R.M.; Vanhaecke, T. Elafibranor restricts lipogenic and inflammatory responses in a human skin stem cell-derived model of NASH. Pharmacol. Res. 2019, 144, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Sinton, M.C.; Meseguer-Ripolles, J.; Lucendo-Villarin, B.; Wernig-Zorc, S.; Thomson, J.P.; Carter, R.N.; Lyall, M.J.; Walker, P.D.; Thakker, A.; Meehan, R.R.; et al. A human pluripotent stem cell model for the analysis of metabolic dysfunction in hepatic steatosis. iScience 2021, 24, 101931. [Google Scholar] [CrossRef]
- Graffmann, N.; Ncube, A.; Martins, S.; Fiszl, A.R.; Reuther, P.; Bohndorf, M.; Wruck, W.; Beller, M.; Czekelius, C.; Adjaye, J. A stem cell based in vitro model of NAFLD enables the analysis of patient specific individual metabolic adaptations in response to a high fat diet and AdipoRon interference. Biol. Open 2021, 10, bio054189. [Google Scholar] [CrossRef]
- Gurevich, I.; Burton, S.A.; Munn, C.; Ohshima, M.; Goedland, M.E.; Czysz, K.; Rajesh, D. iPSC-derived hepatocytes generated from NASH donors provide a valuable platform for disease modeling and drug discovery. Biol. Open 2020, 9, 1–9. [Google Scholar] [CrossRef]
- Duwaerts, C.C.; Le Guillou, D.; Her, C.L.; Phillips, N.J.; Willenbring, H.; Mattis, A.N.; Maher, J.J. Induced pluripotent stem cell–derived hepatocytes from patients with nonalcoholic fatty liver disease display a disease-specific gene expression profile. Gastroenterology 2021, 160, 2591–2594. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling steatohepatitis in humans with pluripotent stem cell-derived organoids. Cell Metab. 2019, 30, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Rogue, A.; Anthérieu, S.; Vluggens, A.; Umbdenstock, T.; Claude, N.; De la Moureyre-Spire, C.; Weaver, R.J.; Guillouzo, A. PPAR agonists reduce steatosis in oleic acid-overloaded HepaRG cells. Toxicol. Appl. Pharmacol. 2014, 276, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbero-Becerra, V.J.; Giraudi, P.J.; Chávez-Tapia, N.C.; Uribe, M.; Tiribelli, C.; Rosso, N. The interplay between hepatic stellate cells and hepatocytes in an in vitro model of NASH. Toxicol. Vitr. 2015, 29, 1753–1758. [Google Scholar] [CrossRef] [PubMed]
- Lauschke, V.M.; Hendriks, D.F.G.; Bell, C.C.; Andersson, T.B.; Ingelman-Sundberg, M. Novel 3D culture systems for studies of human liver function and assessments of the hepatotoxicity of drugs and drug candidates. Chem. Res. Toxicol. 2016, 29, 1936–1955. [Google Scholar] [CrossRef] [PubMed]
- Polidoro, M.A.; Ferrari, E.; Marzorati, S.; Lleo, A.; Rasponi, M. Experimental liver models: From cell culture techniques to microfluidic organs-on-chip. Liver Int. 2021, 41, 1744–1761. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Dankers, A.C.A.; Lauschke, V.M.; Sison-Young, R.; Jenkins, R.; Rowe, C.; Goldring, C.E.; Park, K.; Regan, S.L.; Walker, T.; et al. Comparison of hepatic 2D sandwich cultures and 3D spheroids for long-term toxicity applications: A multicenter study. Toxicol. Sci. 2018, 162, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Kaji, H.; Camci-Unal, G.; Langer, R.; Khademhosseini, A. Engineering systems for the generation of patterned co-cultures for controlling cell–cell interactions. Biochim. Biophys. Acta Gen. Subj. 2011, 1810, 239–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, C.C.; Chouhan, B.; Andersson, L.C.; Andersson, H.; Dear, J.W.; Williams, D.P.; Söderberg, M. Functionality of primary hepatic non-parenchymal cells in a 3D spheroid model and contribution to acetaminophen hepatotoxicity. Arch. Toxicol. 2020, 94, 1251–1263. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Vyas, D.; Udyawar, D. A review on current state of art of bioprinting. In 3D Printing and Additive Manufacturing Technologies; Kumar, L., Pandey, P., Wimpenny, D., Eds.; Springer: Singapore, 2019; pp. 195–201. ISBN 9789811303050. [Google Scholar]
- Chella Krishnan, K.; Floyd, R.R.; Sabir, S.; Jayasekera, D.W.; Leon-Mimila, P.V.; Jones, A.E.; Cortez, A.A.; Shravah, V.; Péterfy, M.; Stiles, L.; et al. Liver pyruvate kinase promotes NAFLD/NASH in both mice and humans in a sex-specific manner. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Vandel, J.; Dubois-Chevalier, J.; Gheeraert, C.; Derudas, B.; Raverdy, V.; Thuillier, D.; Gaal, L.; Francque, S.; Pattou, F.; Staels, B.; et al. Hepatic molecular signatures highlight the sexual dimorphism of nonalcoholic steatohepatitis (NASH). Hepatology 2021, 73, 920–936. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shadid, S.; Abosi-Appeadu, K.; De Maertelaere, A.S.; Defreyne, J.; Veldeman, L.; Holst, J.J.; Lapauw, B.; Vilsbøll, T.; T’Sjoen, G. Effects of gender-affirming hormone therapy on insulin sensitivity and incretin responses in transgender people. Diabetes Care 2020, 43, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Shi, H. Sex hormones and their receptors regulate liver energy homeostasis. Int. J. Endocrinol. 2015, 2015, 294278. [Google Scholar] [CrossRef]
- Aden, D.P.; Fogel, A.; Plotkin, S.; Damjanov, I.; Knowles, B.B. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 1979, 282, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Aninat, C.; Piton, A.; Glaise, D.; Le Charpentier, T.; Langouët, S.; Morel, F.; Guguen-Guillouzo, C.; Guillouzo, A. Expression of cytochromes P450, conjugating enzymes and nuclear receptors in human hepatoma HepaRG cells. Drug Metab. Dispos. 2006, 34, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwimmer, J.B.; Behling, C.; Newbury, R.; Deutsch, R.; Nievergelt, C.; Schork, N.J.; Lavine, J.E. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005, 42, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.K.; Perito, E.R. Nonalcoholic liver disease in children and adolescents. Clin. Liver Dis. 2018, 22, 723–733. [Google Scholar] [CrossRef]
- Nobili, V.; Mosca, A.; De Vito, R.; Raponi, M.; Scorletti, E.; Byrne, C.D. Liver zonation in children with non-alcoholic fatty liver disease: Associations with dietary fructose and uric acid concentrations. Liver Int. 2018, 38, 1102–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Africa, J.A.; Behling, C.A.; Brunt, E.M.; Zhang, N.; Luo, Y.; Wells, A.; Hou, J.; Belt, P.H.; Kohil, R.; Lavine, J.E.; et al. In children with nonalcoholic fatty liver disease, zone 1 steatosis is associated with advanced fibrosis. Clin. Gastroenterol. Hepatol. 2018, 16, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.L.; Howe, L.D.; Jones, H.E.; Higgins, J.P.T.; Lawlor, D.A.; Fraser, A. The prevalence of non-alcoholic fatty liver disease in children and adolescents: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0140908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doycheva, I.; Watt, K.D.; Alkhouri, N. Nonalcoholic fatty liver disease in adolescents and young adults: The next frontier in the epidemic. Hepatology 2017, 65, 2100–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wu, M.; Liu, Z.; Yuan, H.; Wu, X.; Shi, T.; Chen, X.; Zhang, T. Increasing prevalence of NAFLD/NASH among children, adolescents and young adults from 1990 to 2017: A population-based observational study. BMJ Open 2021, 11, 42843. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Benda, C.; Duzinger, S.; Huang, Y.; Li, X.; Li, Y.; Guo, X.; Cao, G.; Chen, S.; Hao, L.; et al. Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 2011, 22, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Mulder, J.; Sharmin, S.; Chow, T.; Rodrigues, D.C.; Hildebrandt, M.R.; D’Cruz, R.; Rogers, I.; Ellis, J.; Rosenblum, N.D. Generation of infant- and pediatric-derived urinary induced pluripotent stem cells competent to form kidney organoids. Pediatr. Res. 2020, 87, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Assy, N.; Nasser, G.; Kamayse, I.; Nseir, W.; Beniashvili, Z.; Djibre, A.; Grosovski, M. Soft drink consumation linked with fatty liver in the absence of traditional risk factors. Can. J. Gastroenterol. 2008, 22, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Abid, A.; Taha, O.; Nseir, W.; Farah, R.; Grosovski, M.; Assy, N. Soft drink consumption is associated with fatty liver disease independent of metabolic syndrome. J. Hepatol. 2009, 51, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Ren. Physiol. 2006, 290, 625–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tappy, L.; Rosset, R. Health outcomes of a high fructose intake: The importance of physical activity. J. Physiol. 2019, 597, 3561–3571. [Google Scholar] [CrossRef] [Green Version]
- Steinmann, B.; Ranter, R. Disorders of fructose metabolism. In Inborn Metabolic Diseases; Saudubray, J.M., van den Berghe, G., Walter, J.H., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 158–165. [Google Scholar]
- Shepherd, E.L.; Saborano, R.; Northall, E.; Matsuda, K.; Ogino, H.; Yashiro, H.; Pickens, J.; Feaver, R.E.; Cole, B.K.; Hoang, S.A.; et al. Ketohexokinase inhibition improves NASH by reducing fructose-induced steatosis and fibrogenesis. JHEP Rep. 2021, 3, 100217. [Google Scholar] [CrossRef] [PubMed]
- Samji, N.S.; Snell, P.D.; Singal, A.K.; Satapathy, S.K. Racial disparities in diagnosis and prognosis of nonalcoholic fatty liver disease. Clin. Liver Dis. 2020, 16, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Rich, N.E.; Oji, S.; Mufti, A.R.; Browning, J.D.; Parikh, N.D.; Odewole, M.; Mayo, H.; Singal, A.G. Racial and ethnic disparities in non-alcoholic fatty liver disease prevalence, severity, and outcomes in the United States: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2018, 16, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 2016, 150, 1219–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Perry, R.C.; Ngai, C.; Ginsberg, H.N. Abstract 682: TM6SF2 is necessary for the late addition of lipid and secretion of fully-lipidated very low density lipoproteins from Hepg2 cell. Arterioscler. Thromb. Vasc. Biol. 2018, 36, A682. [Google Scholar]
- Borén, J.; Adiels, M.; Björnson, E.; Matikainen, N.; Söderlund, S.; Rämö, J.; Ståhlman, M.; Ripatti, P.; Ripatti, S.; Palotie, A.; et al. Effects of TM6SF2 E167K on hepatic lipid and very low-density lipoprotein metabolism in humans. JCI Insight 2020, 5, e144079. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Nick, A.; Hölttä-Vuori, M.; Thiele, C.; Isokuortti, E.; Lallukka-Brück, S.; Zhou, Y.; Hakkarainen, A.; Lundbom, N.; Peltonen, M.; et al. Human PNPLA3-I148M variant increases hepatic retention of polyunsaturated fatty acids. JCI Insight 2019, 4, e127902. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, F.V.; Claudel, T.; Caligiuri, A.; Marra, F.; Trauner, M.H. The I148M PNPLA3 variant is a novel key player modulating the pro-fibrogenic phenotype of human hepatic stellate cells. Hepatology 2017, 65, 1875–1890. [Google Scholar] [CrossRef] [Green Version]
- Tilson, S.G.; Morell, C.M.; Lenaerts, A.-S.; Park, S.B.; Hu, Z.; Jenkins, B.; Koulman, A.; Liang, T.J.; Vallier, L. Modeling PNPLA3-associated NAFLD using human-induced pluripotent stem cells. Hepatology 2021, 74, 2998–3017. [Google Scholar] [CrossRef]
- Sodum, N.; Kumar, G.; Bojja, S.L.; Kumar, N.; Rao, C.M. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol. Res. 2021, 167, 105484. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Gianotti, T.F.; Castaño, G.O.; Mallardi, P.; San Martino, J.; Ledesma, M.M.; Flichman, D.; Mirshahi, F.; Sanyal, A.J.; Sookoian, S. Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNAs to liver histology and disease pathogenesis. Gut 2015, 64, 800–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Hwang, S.; Cai, Y.; Kim, S.J.; Xu, M.; Yang, D.; Guillot, A.; Feng, D.; Seo, W.; Hou, X.; et al. MicroRNA-223 ameliorates nonalcoholic steatohepatitis and cancer by targeting multiple inflammatory and oncogenic genes in hepatocytes. Hepatology 2019, 70, 1150–1167. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Gianotti, T.F.; Burgueño, A.L.; Rey-Funes, M.; Loidl, C.F. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2012, 62, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Page, A.; Pauli, P.; Morán-Salvador, E.; White, S.; French, J.; Mann, J. Hepatic stellate cell transdifferentiation involves genome-wide remodeling of the DNA methylation landscape. Physiol. Behav. 2017, 64, 661–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Wong, V.W.-S.; Chan, H.L.-Y.; Cheng, A.S.-L. Epigenetic regulation of hepatocellular carcinoma in non-alcoholic fatty liver disease. Semin. Cancer Biol. 2013, 23, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Dreval, K.; Tryndyak, V.; de Conti, A.; Beland, F.A.; Pogribny, I.P. Gene expression and DNA methylation alterations during nonalcoholic steatohepatitis-associated liver carcinogenesis. Front. Genet. 2019, 10, 486. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, S.R.; Kasmi, K.C.E.; Jonscher, K.R.; Friedman, J.E. Developmental origins of NAFLD: A womb with a clue. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 81–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, K.R.; White, F.V.; Deutsch, G.H. Hepatic steatosis is prevalent in stillborns delivered to women with diabetes mellitus. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 152–158. [Google Scholar] [CrossRef] [PubMed]
- de Jesus, D.F.; Orime, K.; Kaminska, D.; Kimura, T.; Basile, G.; Wang, C.H.; Haertle, L.; Riemens, R.; Brown, N.K.; Hu, J.; et al. Parental metabolic syndrome epigenetically reprograms offspring hepatic lipid metabolism in mice. J. Clin. Invest. 2020, 130, 2391–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vliet-Ostaptchouk, J.V.; Nuotio, M.L.; Slagter, S.N.; Doiron, D.; Fischer, K.; Foco, L.; Gaye, A.; Gögele, M.; Heier, M.; Hiekkalinna, T.; et al. The prevalence of metabolic syndrome and metabolically healthy obesity in Europe: A collaborative analysis of ten large cohort studies. BMC Endocr. Disord. 2014, 14, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blüher, M. Metabolically healthy obesity. Endocr. Rev. 2020, 41, bnaa004. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zhuang, R.; Luo, X.; Yin, L.; Pang, C.; Feng, T.; You, H.; Zhai, Y.; Ren, Y.; Zhang, L.; et al. Prevalence of metabolically healthy obese and metabolically obese but normal weight in adults worldwide: A meta-analysis. Horm. Metab. Res. 2015, 47, 839–845. [Google Scholar] [CrossRef]
- Zheng, Q.; Lin, W.; Liu, C.; Zhou, Y.; Chen, T.; Zhang, L.; Zhang, X.; Yu, S.; Wu, Q.; Jin, Z.; et al. Prevalence and epidemiological determinants of metabolically obese but normal-weight in Chinese population. BMC Public Health 2020, 20, 487. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Chung, G.E.; Kwak, M.S.; Seo, H.B.; Kang, J.H.; Kim, W.; Kim, Y.J.; Yoon, J.H.; Lee, H.S.; Kim, C.Y. Body fat distribution and risk of incident and regressed nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2016, 14, 132–138. [Google Scholar] [CrossRef]
- van der Poorten, D.; Milner, K.-L.; Hui, J.; Hodge, A.; Trenell, M.I.; Kench, J.G.; London, R.; Peduto, T.; Chisholm, D.J.; George, J. Visceral fat: A key mediator of steatohepatitis in metabolic liver disease. Hepatology 2008, 48, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.J.; Kim, W.; Kim, D.; Yoon, J.H.; Lee, K.; Kim, J.H.; Cho, E.J.; Lee, J.H.; Kim, H.Y.; Kim, Y.J.; et al. Visceral obesity predicts significant fibrosis in patients with nonalcoholic fatty liver disease. Medicine 2015, 94, e2159. [Google Scholar] [CrossRef]
- Cho, S.A.; Joo, H.J.; Cho, J.Y.; Lee, S.H.; Park, J.H.; Hong, S.J.; Yu, C.W.; Lim, D.S. Visceral fat area and serum adiponectin level predict the development of metabolic syndrome in a community-based asymptomatic population. PLoS ONE 2017, 12, e0169289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, N.C.; Salles, G.F.; Cardoso, C.R.L.; Villela-Nogueira, C.A. Serum biomarkers in type 2 diabetic patients with non-alcoholic steatohepatitis and advanced fibrosis. Hepatol. Res. 2013, 43, 508–515. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Jonas, M.I.; Kurylowicz, A.; Bartoszewicz, Z.; Lisik, W.; Jonas, M.; Wierzbicki, Z.; Chmura, A.; Pruszczyk, P.; Puzianowska-Kuznicka, M. Interleukins 6 and 15 levels are higher in subcutaneous adipose tissue, but obesity is associated with their increased content in visceral fat depots. Int. J. Mol. Sci. 2015, 16, 25817–25830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.S.; Park, J.Y.; Yu, R. Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-alpha and IL-6. Diabetes Res. Clin. Pract. 2005, 69, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Wieckowska, A.; Papouchado, B.G.; Li, Z.Z.; Lopez, R.; Zein, N.N.; Feldstein, A.E. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2008, 103, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, V.L.; Rumsey, J.W.; Boone, R.; Malik, D.; Cai, Y.; Sriram, N.N.; Long, C.J.; McAleer, C.W.; Lambert, S.; Shuler, M.L.; et al. Validation of an adipose-liver human-on-a-chip model of NAFLD for preclinical therapeutic efficacy evaluation. Sci. Rep. 2021, 11, 13156. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.B.; Zheng, K.I.; Rios, R.S.; Targher, G.; Byrne, C.D.; Zheng, M.H. Global epidemiology of lean non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2020, 35, 2041–2050. [Google Scholar] [CrossRef] [PubMed]
- Ayonrinde, O.T. Historical narrative from fatty liver in the nineteenth century to contemporary NAFLD—Reconciling the present with the past. JHEP Rep. 2021, 3, 100261. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Esmaili, S.; Rogers, G.; Bugianesi, E.; Petta, S.; Marchesini, G.; Bayoumi, A.; Metwally, M.; Azardaryany, M.K.; Coulter, S.; et al. Lean NAFLD: A distinct entity shaped by differential metabolic adaptation. Hepatology 2020, 71, 1213–1227. [Google Scholar] [CrossRef] [PubMed]
- Fracanzani, A.L.; Petta, S.; Lombardi, R.; Pisano, G.; Russello, M.; Consonni, D.; Di Marco, V.; Cammà, C.; Mensi, L.; Dongiovanni, P.; et al. Liver and cardiovascular damage in patients with lean nonalcoholic fatty liver disease, and association with visceral obesity. Clin. Gastroenterol. Hepatol. 2017, 15, 1604–1611. [Google Scholar] [CrossRef]
- Younes, R.; Govaere, O.; Petta, S.; Miele, L.; Tiniakos, D.; Burt, A.; David, E.; Vecchio, F.M.; Maggioni, M.; Cabibi, D.; et al. Caucasian lean subjects with non-alcoholic fatty liver disease share long-term prognosis of non-lean: Time for reappraisal of BMI-driven approach? Gut 2022, 71, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michail, S.; Lin, M.; Frey, M.R.; Fanter, R.; Paliy, O.; Hilbush, B.; Reo, N.V. Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol. Ecol. 2015, 91, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.N.; Li, S.; Xue, G.; et al. Fatty liver disease caused by high-alcohol-producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Kawada, N. Role of the gut-liver axis in liver inflammation, fibrosis, and cancer: A special focus on the gut microbiota relationship. Hepatol. Commun. 2019, 3, 456–470. [Google Scholar] [CrossRef] [Green Version]
- Jeschke, M.G.; Klein, D.; Thasler, W.E.; Bolder, U.; Schlitt, H.J.; Jauch, K.W.; Weiss, T.S. Insulin decreases inflammatory signal transcription factor expression in primary human liver cells after LPS challenge. Mol. Med. 2008, 14, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, S.; Panera, N.; Mina, M.; Gnani, D.; Stefanis, C.D.; Crudele, A.; Rychlicki, C.; Petrini, S.; Bruscalupi, G.; Agostinelli, L.; et al. LPS-induced TNF-α factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget 2015, 6, 41434–41452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheder, R.K.; Hobkirk, J.; Stover, C.M. In vitro modulation of the LPS-induced proinflammatory profile of hepatocytes and macrophages- approaches for intervention in obesity? Front. Cell Dev. Biol. 2016, 4, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragonès, G.; Colom-Pellicer, M.; Aguilar, C.; Guiu-Jurado, E.; Martínez, S.; Sabench, F.; Antonio Porras, J.; Riesco, D.; Del Castillo, D.; Richart, C.; et al. Circulating microbiota-derived metabolites: A “liquid biopsy? Int. J. Obes. 2020, 44, 875–885. [Google Scholar] [CrossRef] [Green Version]
- Jungermann, K.; Kietzmann, T. Oxygen: Modulator of metabolic zonation and disease of the liver. Hepatology 2000, 31, 255–260. [Google Scholar] [CrossRef]
- Hijmans, B.S.; Grefhorst, A.; Oosterveer, M.H.; Groen, A.K. Zonation of glucose and fatty acid metabolism in the liver: Mechanism and metabolic consequences. Biochimie 2014, 96, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.M.; Brunt, E.M. Pathological features of fatty liver disease. Gastroenterology 2014, 147, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Hall, Z.; Bond, N.J.; Ashmore, T.; Sanders, F.; Ament, Z.; Wang, X.; Murray, A.J.; Bellafante, E.; Virtue, S.; Vidal-Puig, A.; et al. Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology 2017, 65, 1165–1180. [Google Scholar] [CrossRef] [PubMed]
- Lee-Montiel, F.T.; George, S.M.; Gough, A.H.; Sharma, A.D.; Wu, J.; DeBiasio, R.; Vernetti, L.A.; Taylor, D.L. Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems. Exp. Biol. Med. 2017, 242, 1617–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.; Galea, P.; Latif, S. What is the appropriate oxygen tension for in vitro culture? Mol. Hum. Reprod. 2006, 12, 653. [Google Scholar] [CrossRef]
- Parafati, M.; Kirby, R.J.; Khorasanizadeh, S.; Rastinejad, F.; Malany, S. A nonalcoholic fatty liver disease model in human induced pluripotent stem cell-derived hepatocytes, created by endoplasmic reticulum stress-induced steatosis. Dis. Model. Mech. 2018, 11, dmm033530. [Google Scholar] [CrossRef] [Green Version]
- Bertot, L.C.; Adams, L.A. The natural course of non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2016, 17, 774. [Google Scholar] [CrossRef] [Green Version]
- Saphner, T.; Triest-Robertson, S.; Li, H.; Holzman, P. The association of nonalcoholic steatohepatitis and tamoxifen in patients with breast cancer. Cancer 2009, 115, 3189–3195. [Google Scholar] [CrossRef]
- Adinolfi, L.E.; Rinaldi, L.; Guerrera, B.; Restivo, L.; Marrone, A.; Giordano, M.; Zampino, R. NAFLD and NASH in HCV infection: Prevalence and significance in hepatic and extrahepatic manifestations. Int. J. Mol. Sci. 2016, 17, 803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miele, L.; Liguori, A.; Marrone, G.; Biolato, M.; Araneo, C.; Vaccaro, F.G.; Gasbarrini, A.; Grieco, A. Fatty liver and drugs: The two sides of the same coin. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 86–94. [Google Scholar] [PubMed]
- Choi, H.S.J.; Brouwer, W.P.; Zanjir, W.M.R.; de Man, R.A.; Feld, J.J.; Hansen, B.E.; Janssen, H.L.A.; Patel, K. Nonalcoholic steatohepatitis is associated with liver-related outcomes and all-cause mortality in chronic hepatitis B. Hepatology 2020, 71, 539–548. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Dash, A.; Figler, R.A.; Sanyal, A.J.; Wamhoff, B.R. Drug-induced steatohepatitis. Expert Opin. Drug Metab. Toxicol. 2017, 13, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.M.; Branson, S.; De Boe, V.; Sachinidis, A.; Rogiers, V.; De Kock, J.; Vanhaecke, T. In vitro assessment of drug-induced liver steatosis based on human dermal stem cell-derived hepatic cells. Arch. Toxicol. 2016, 90, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Cuykx, M.; Claes, L.; Rodrigues, R.M.; Vanhaecke, T.; Covaci, A. Metabolomics profiling of steatosis progression in HepaRG cells using sodium valproate. Toxicol. Lett. 2018, 286, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Vorrink, S.U.; Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Prediction of drug-induced hepatotoxicity using long-term stable primary hepatic 3D spheroid cultures in chemically defined conditions. Toxicol. Sci. 2018, 163, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Leite, S.B.; Roosens, T.; El Taghdouini, A.; Mannaerts, I.; Smout, A.J.; Najimi, M.; Sokal, E.; Noor, F.; Chesne, C.; Van Grunsven, L.A. Novel human hepatic organoid model enables testing of drug-induced liver fibrosis in vitro. Biomaterials 2016, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hajifathalian, K.; Sagvand, B.T.; Mccullough, A.J. Effect of alcohol consumption on survival in non-alcoholic fatty liver disease: A national prospective cohort study. Hepatology 2019, 70, 511–521. [Google Scholar] [CrossRef]
- Ronksley, P.E.; Brien, S.E.; Turner, B.J.; Mukamal, K.J.; Ghali, W.A. Association of alcohol consumption with selected cardiovascular disease outcomes: A systematic review and meta-analysis. BMJ 2011, 342, d671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslam, M.; Sanyal, A.J.; George, J. Toward more accurate nomenclature for fatty liver diseases. Gastroenterology 2019, 157, 590–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, M.; Arteel, G.E. Effect of ethanol on lipid metabolism. J. Hepatol. 2019, 70, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaya, T.; Tanaka, N.; Suzuki, T.; Sano, K.; Horiuchi, A.; Komatsu, M.; Nakajima, T.; Nishizawa, T.; Joshita, S.; Umemura, T.; et al. Down-regulation of SREBP-1c is associated with the development of burned-out NASH. J. Hepatol. 2010, 53, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Vecchione, G.; Grasselli, E.; Compalati, A.D.; Ragazzoni, M.; Cortese, K.; Gallo, G.; Voci, A.; Vergani, L. Ethanol and fatty acids impair lipid homeostasis in an in vitro model of hepatic steatosis. Food Chem. Toxicol. 2016, 90, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Tandra, S.; Yeh, M.M.; Brunt, E.M.; Vuppalanchi, R.; Cummings, O.W.; Ünalp-Arida, A.; Wilson, L.A.; Chalasani, N. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J. Hepatol. 2011, 55, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.A.; Brunt, E.M.; Qazi, R.A.; Oliver, D.A.; Neuschwander-Tetri, B.A.; Di Bisceglie, A.M.; Bacon, B.R. Effect of significant histologic steatosis or steatohepatitis on response to antiviral therapy in patients with chronic hepatitis C. Clin. Gastroenterol. Hepatol. 2005, 3, 604–609. [Google Scholar] [CrossRef]
- Bondini, S.; Younossi, Z.M. Non-alcoholic fatty liver disease and hepatitis C infection. Minerva Gastroenterol. Dietol. 2006, 52, 135–143. [Google Scholar] [PubMed]
- Stevenson, H.L.; Utay, N.S. Hepatic steatosis in HCV-infected persons in the direct-acting antiviral era. Trop. Dis. Travel Med. Vaccines 2016, 2, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogasawara, N.; Kobayashi, M.; Akuta, N.; Kominami, Y.; Fujiyama, S.; Kawamura, Y.; Sezaki, H.; Hosaka, T.; Suzuki, F.; Saitoh, S.; et al. Serial changes in liver stiffness and controlled attenuation parameter following direct-acting antiviral therapy against hepatitis C virus genotype 1b. J. Med. Virol. 2018, 90, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Siphepho, P.Y.; Liu, Y.T.; Shabangu, C.S.; Huang, J.F.; Huang, C.F.; Yeh, M.L.; Yu, M.L.; Wang, S.C. The impact of steatosis on chronic hepatitis c progression and response to antiviral treatments. Biomedicines 2021, 9, 1491. [Google Scholar] [CrossRef]
- Boeckmans, J.; Rombaut, M.; Demuyser, T.; Declerck, B.; Piérard, D.; Rogiers, V.; De Kock, J.; Waumans, L.; Magerman, K.; Cartuyvels, R.; et al. Infections at the nexus of metabolic-associated fatty liver disease. Arch. Toxicol. 2021, 95, 2235–2253. [Google Scholar] [CrossRef] [PubMed]
- Ashfaq, U.A.; Khan, S.N.; Nawaz, Z.; Riazuddin, S. In-vitro model systems to study hepatitis C virus. Genet. Vaccines Ther. 2011, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Shlomai, A.; Schwartz, R.E.; Ramanan, V.; Bhatta, A.; de Jong, Y.P.; Bhatia, S.N.; Rice, C.M. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc. Natl. Acad. Sci. USA 2014, 111, 12193–12198. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Poniachik, J.; Roblero, J.P.; Urzúa, A.; Cattaneo, M. A new definition for non-alcoholic fatty liver disease. J. Hepatol. 2021, 74, 982–983. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; George, J. MAFLD: A holistic view to redefining fatty liver disease. J. Hepatol. 2021, 74, 983–985. [Google Scholar] [CrossRef]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.; Abdelmalek, M.F.; Diehl, A.M.; Caldwell, S.; Shiffman, M.L.; Ghalib, R.; Lawitz, E.; Rockey, D.C.; Schall, R.A.; Jia, C.; et al. Efficacy and safety of simtuzumab for the treatment of nonalcoholic steatohepatitis with bridging fibrosis or cirrhosis: Results of two phase 2b, dose-ranging, randomized, placebo-controlled trials. J. Hepatol. 2017, 66, S54. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamura, S.; Eslam, M.; Kawaguchi, T.; Tsutsumi, T.; Nakano, D.; Yoshinaga, S.; Takahashi, H.; Anzai, K.; George, J.; Torimura, T. MAFLD identifies patients with significant hepatic fibrosis better than NAFLD. Liver Int. 2020, 40, 3018–3030. [Google Scholar] [CrossRef] [PubMed]
- Natale, A.; Vanmol, K.; Arslan, A.; Van Vlierberghe, S.; Dubruel, P.; Van Erps, J.; Thienpont, H.; Buzgo, M.; Boeckmans, J.; De Kock, J.; et al. Technological advancements for the development of stem cell - based models for hepatotoxicity testing. Arch. Toxicol. 2019, 93, 1789–1805. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Lewis, J.; Douglas, D.; Kneteman, N.M.; Vance, D.E. Characterization of lipid and lipoprotein metabolism in primary human hepatocytes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Kiamehr, M.; Alexanova, A.; Viiri, L.E.; Heiskanen, L.; Vihervaara, T.; Kauhanen, D.; Ekroos, K.; Laaksonen, R.; Käkelä, R.; Aalto-Setälä, K. hiPSC-derived hepatocytes closely mimic the lipid profile of primary hepatocytes: A future personalised cell model for studying the lipid metabolism of the liver. J. Cell. Physiol. 2019, 234, 3744–3761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvist, A.J.; Kanebratt, K.P.; Walentinsson, A.; Palmgren, H.; O’Hara, M.; Björkbom, A.; Andersson, L.C.; Ahlqvist, M.; Andersson, T.B. Critical differences in drug metabolic properties of human hepatic cellular models, including primary human hepatocytes, stem cell derived hepatocytes, and hepatoma cell lines. Biochem. Pharmacol. 2018, 155, 124–140. [Google Scholar] [CrossRef] [PubMed]
- Kammerer, S.; Küpper, J.-H. Human hepatocyte systems for in vitro toxicology analysis. J. Cell. Biotechnol. 2018, 3, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Rose, S.; Ezan, F.; Cuvellier, M.; Bruyère, A.; Legagneux, V.; Langouët, S.; Baffet, G. Generation of proliferating human adult hepatocytes using optimized 3D culture conditions. Sci. Rep. 2021, 11, 515. [Google Scholar] [CrossRef] [PubMed]
- Rombaut, M.; Boeckmans, J.; Rodrigues, R.M.; van Grunsven, L.A.; Vanhaecke, T.; De Kock, J. Direct reprogramming of somatic cells into induced hepatocytes: Cracking the Enigma code. J. Hepatol. 2021, 75, 690–705. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Cell Type(s) | In Vitro Disease Trigger(s) | Etiology | Pharmacological Intervention | Ref. |
|---|---|---|---|---|
| In vitro models using primary cells | ||||
| Primary hepatocytes | Oleic acid and palmitic acid | Diet | Pioglitazone and metformin | [14] |
| Glucose, insulin, free fatty acids, TNF-α, IL-1β and TGF-β | Diet Inflammation | PPAR-agonists | [15] | |
| Primary hepatocytes, stellate cells and macrophages | Glucose, insulin, free fatty acids | Diet Insulin resistance | Obeticholic acid | [16] |
| Fructose | Diet | No pharmacological intervention | [17] | |
| Stem cell-based in vitro models | ||||
| hSKP-HPC | Glucose, insulin, free fatty acids, TNF-α, IL-1β and TGF-β | Diet Inflammation | Elafibranor | [18] |
| iPSC-derived hepatocyte-like cells | Lactate, pyruvate and octanoate | Mitochondrial dysfunction | No pharmacological intervention | [19] |
| Oleic acid | Diet Genetics | No pharmacological intervention | [20] | |
| Donor NAFLD background | Genetics | No pharmacological intervention | [21] | |
| Donor NAFLD background | Genetics | No pharmacological intervention | [22] | |
| iPSC-derived hepatocyte-like, stellate cell-like and Kupffer cell-like cells | Oleic acid and LPS | Gut dysbiosis | Obeticholic acid | [23] |
| In vitro models using cell lines | ||||
| HepaRG | Oleic acid | Diet | PPAR-agonists | [24] |
| HepaRG and HepG2 | Glucose, insulin, free fatty acids, TNF-α, IL-1β and TGF-β | Diet Inflammation | PPAR-agonists | [15] |
| Huh7 and LX-2 cells | Oleic acid and palmitic acid | Diet-induced fibrosis | No pharmacological intervention | [25] |
| System | Strengths | Limitations |
|---|---|---|
| Monolayer cultures [26,27] | - Low cost - Relatively simple handling - Convenient for analysis - Characterized and benchmarked - Suitable for high-throughput purposes | - Abnormal cell morphology - Loss of polarization - Rapid dedifferentiation, not suitable for long-term studies - Hampered cell–cell contacts - No representation of interorgan crosstalk |
| Sandwich cultures [26,27,28] | - Longer lifespan and preservation of metabolic activity - Decreased cellular flattening - Improved cell–cell contacts | - Renewal of overlay is required every couple of days to decelerate dedifferentiation - Vulnerable to batch-to-batch variation of extracellular matrix substrates - Limited exchange of nutrients and compounds due to extracellular matrix overlay - Less suitable for high-throughput purposes - No representation of interorgan crosstalk |
| Micropatterned co-cultures [26,29] | - Phenotypic stability over several weeks - Controlled degree of homo- and heterotypic cell–cell contacts | - Higher cost - Murine supporting cell line (3T3-J2) often used is a concern for physiological relevance - Interference of supporting cells with read-outs - No representation of interorgan crosstalk |
| Spheroids [28,30] | - Cell–cell interactions - Preservation of functional and metabolic activity over several weeks - Do not require scaffolds - Suitable for high-throughput purposes | - Size heterogeneity (depending on procedure) - No representation of interorgan crosstalk |
| Organoids [31] | - Cell–cell interactions - Preservation of functional and metabolic activity over several weeks - Recapitulate developmental phases of the liver | - Higher cost - Require scaffolds - High variability due to lack of standardized protocols - Limited options for genetic modification - Limited representation of interorgan crosstalk |
| Microfluidic devices [26,27] | - In vitro physiological liver environment - Cell–cell interactions - Perfusion, shear stress - Zonation possible by managing flow rate of medium - Inclusion of interorgan crosstalk | - High cost - High complexity - Requires specialized equipment - Less suitable for high-throughput purposes |
| Bioprinting [26,32] | - In vitro physiological liver environment - Cell–cell interactions - Vascularization | - High cost - Complex experimental setup - Limitations concerning cell viability and structural integrity depending on printing method, requires large amounts of cells - Less suitable for high-throughput purposes |
| Strengths for MAFLD Modeling | Limitations for MAFLD Modeling | |
|---|---|---|
| Primary human hepatocytes [148,149,150,151] | - excellent drug-metabolizing capacity - preserved lipid and lipoprotein metabolism | - scarcity of donor material - uncertain dietary history - uncertain alcohol intake history - uncertain drug intake history - limited lifespan - uncertain presence of pathogen- and damage-associated molecular patterns - often already unhealthy donors |
| Hepatoma-derived cell lines [149,150,151] | - easy genetic adaptations - long-term culture possible | - lack of population diversity - sex-specific - went through multiple passages - poor drug-metabolizing capacity - altered lipid and lipoprotein metabolism |
| Stem cell-derived models [149,150,151] | - unlimited source of cells - no ethical concerns - studying epigenetics - easy genetic adaptations - long-term culture possible - can obtain comparable lipid and lipoprotein metabolism to primary cells - population diversity—can be obtained from different donors | - limited drug-metabolizing capacity - long differentiation protocols - cryopreservation difficulties |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gatzios, A.; Rombaut, M.; Buyl, K.; De Kock, J.; Rodrigues, R.M.; Rogiers, V.; Vanhaecke, T.; Boeckmans, J. From NAFLD to MAFLD: Aligning Translational In Vitro Research to Clinical Insights. Biomedicines 2022, 10, 161. https://doi.org/10.3390/biomedicines10010161
Gatzios A, Rombaut M, Buyl K, De Kock J, Rodrigues RM, Rogiers V, Vanhaecke T, Boeckmans J. From NAFLD to MAFLD: Aligning Translational In Vitro Research to Clinical Insights. Biomedicines. 2022; 10(1):161. https://doi.org/10.3390/biomedicines10010161
Chicago/Turabian StyleGatzios, Alexandra, Matthias Rombaut, Karolien Buyl, Joery De Kock, Robim M. Rodrigues, Vera Rogiers, Tamara Vanhaecke, and Joost Boeckmans. 2022. "From NAFLD to MAFLD: Aligning Translational In Vitro Research to Clinical Insights" Biomedicines 10, no. 1: 161. https://doi.org/10.3390/biomedicines10010161