
Transcriptome Profiling of Eutopic and Ectopic Endometrial Stromal Cells in Women with Endometriosis Based on High-Throughput Sequencing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of Eutopic and Ectopic Endometrial Stromal Cells
2.2. RNA Isolation and Quality Assessment
2.3. RNA Sequencing
2.4. Functional Analysis Using Different Bioinformatics Tools
3. Results
3.1. mRNA Filtering and Mapping
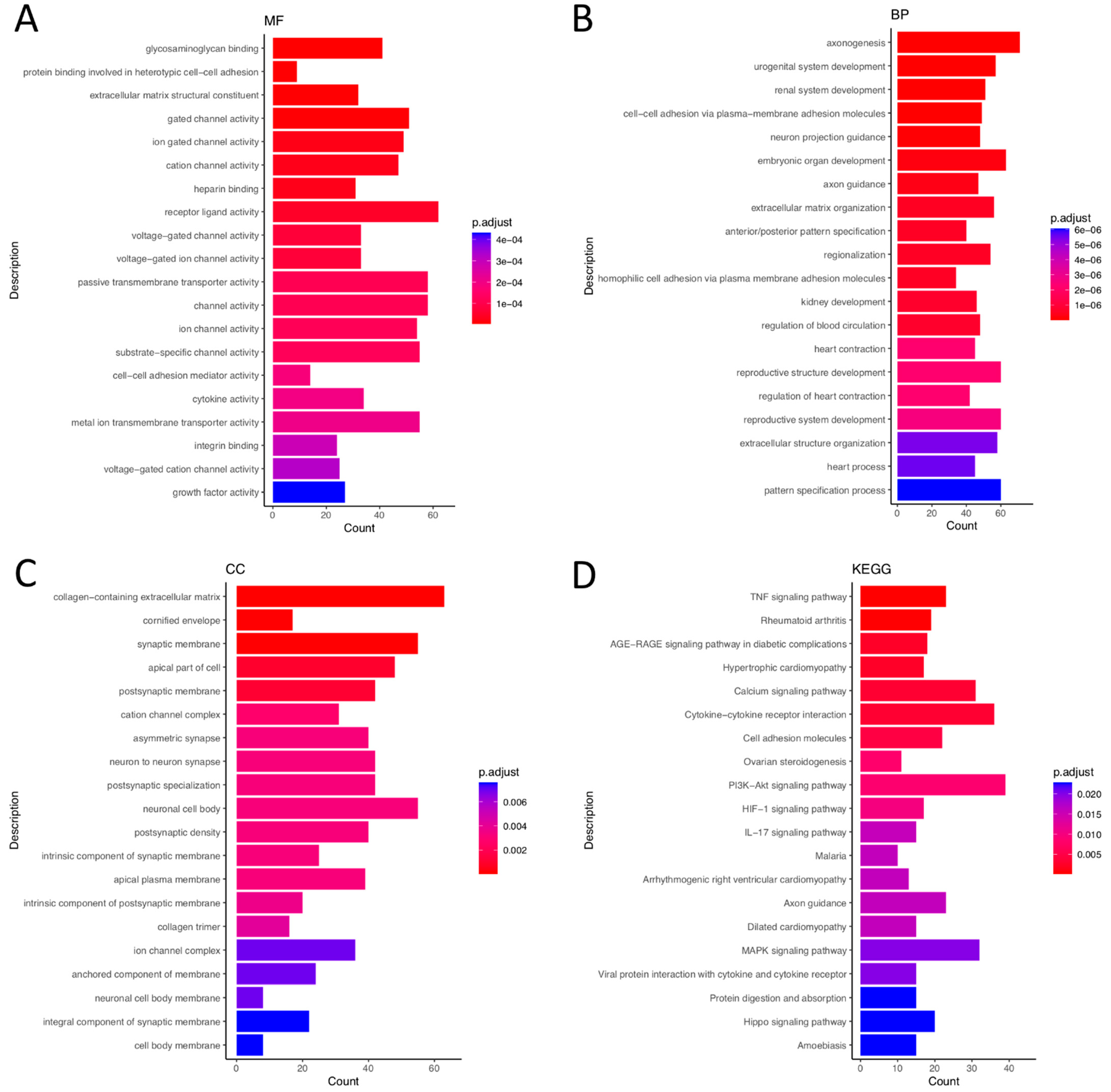
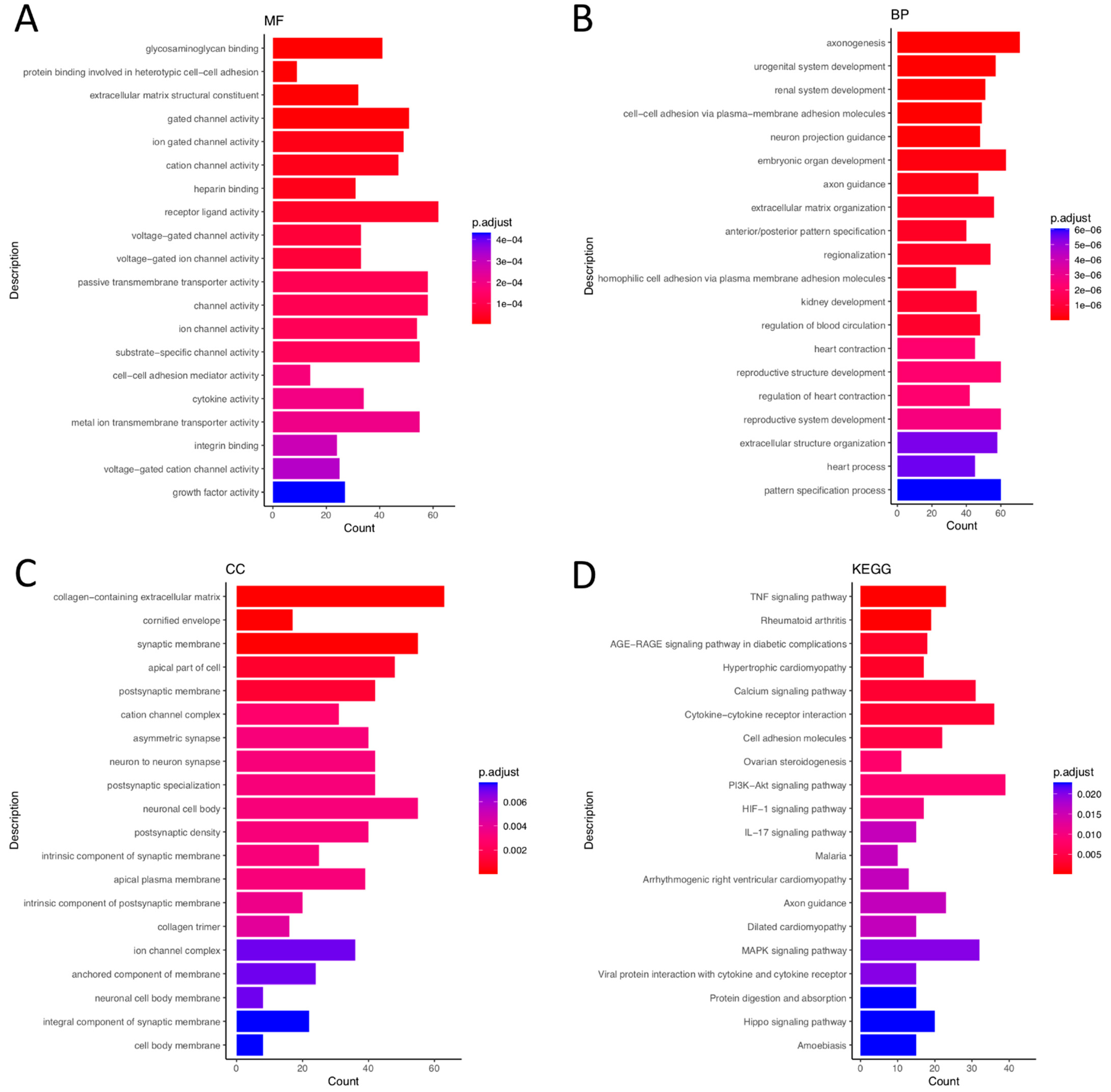
3.2. GO Analysis
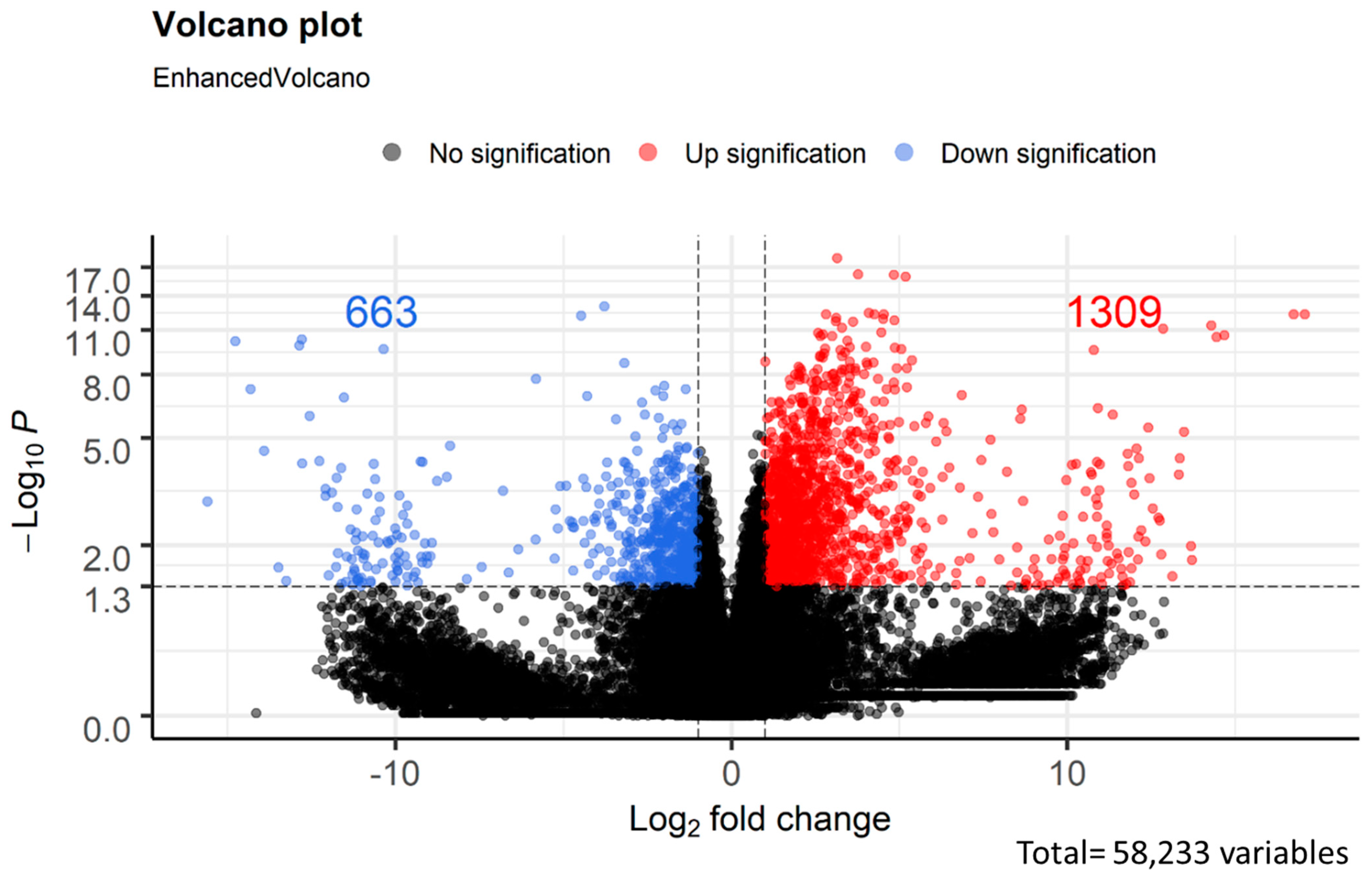
3.3. Analysis of Differentially Expressed Genes
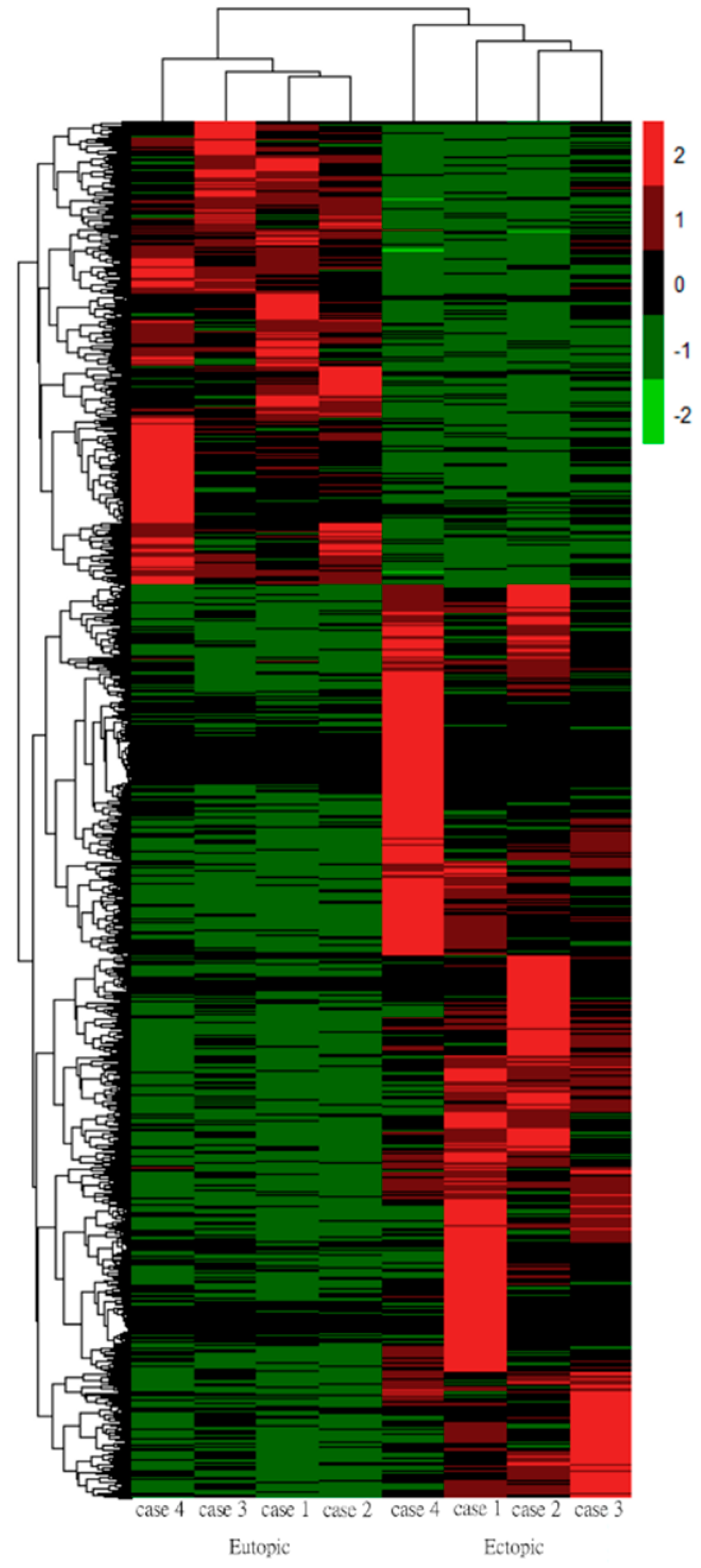
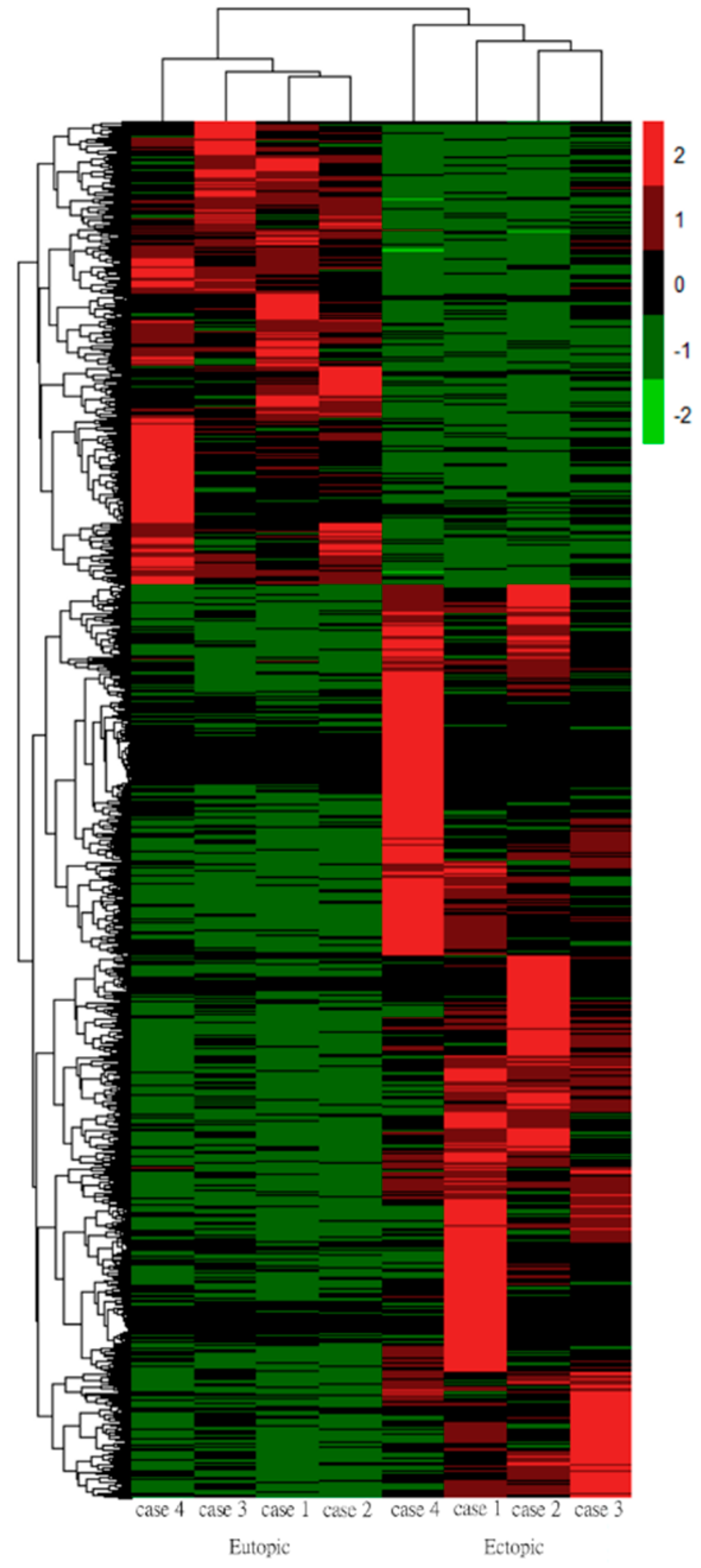
3.4. Hierarchical Clustering Analysis
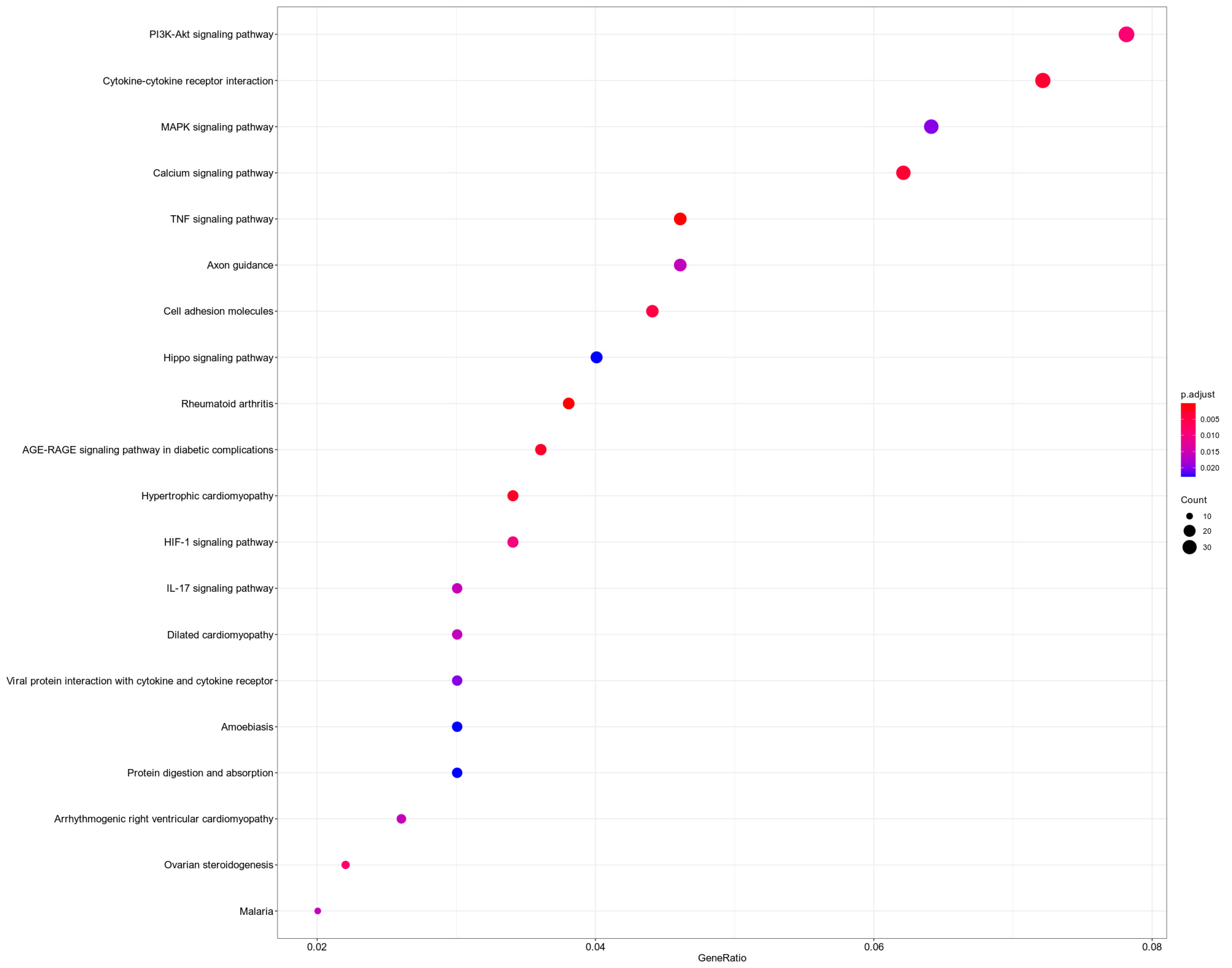
3.5. KEGG Analysis
3.6. PCA Plot of RNA-seq Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Somigliana, E.; Vigano, P.; Parazzini, F.; Stoppelli, S.; Giambattista, E.; Vercellini, P. Association between endometriosis and cancer: A comprehensive review and a critical analysis of clinical and epidemiological evidence. Gynecol. Oncol. 2006, 101, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Sourial, S.; Tempest, N.; Hapangama, D.K. Theories on the pathogenesis of endometriosis. Int. J. Reprod. Med. 2014, 2014, 179515. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.W.; Horng, H.C.; Ho, C.H.; Yen, M.S.; Chao, H.T.; Wang, P.H. Taiwan Association of Gynecology Systematic Review G: Women with endometriosis have higher comorbidities: Analysis of domestic data in Taiwan. J. Chin. Med. Assoc. 2016, 79, 577–582. [Google Scholar] [CrossRef]
- Gemmill, J.A.; Stratton, P.; Cleary, S.D.; Ballweg, M.L.; Sinaii, N. Cancers, infections, and endocrine diseases in women with endometriosis. Fertil. Steril. 2010, 94, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- Amayo, A.A.; Kirera, S. Comparison of calculated and direct low density lipoprotein cholesterol determinations in a routine laboratory. East Afr. Med. J. 2004, 81, 154–158. [Google Scholar] [CrossRef]
- Lattarulo, S.; Pezzolla, A.; Fabiano, G. Palasciano N: Intestinal endometriosis: Role of laparoscopy in diagnosis and treatment. Int. Surg. 2009, 94, 310–314. [Google Scholar]
- Romeo, A.; Fernandes, L.F.; Cervantes, G.V.; Botchorishvili, R.; Benedetto, C.; Adamyan, L.; Ussia, A.; Wattiez, A.; Kondo, W.; Koninckx, P.R. Which Knots Are Recommended in Laparoscopic Surgery and How to Avoid Insecure Knots. J. Minim. Invasive Gynecol. 2020, 27, 1395–1404. [Google Scholar] [CrossRef]
- Li, W.N.; Hsiao, K.Y.; Wang, C.A.; Chang, N.; Hsu, P.L.; Sun, C.H.; Wu, S.R.; Wu, M.H.; Tsai, S.J. Extracellular vesicle-associated VEGF-C promotes lymphangiogenesis and immune cells infiltration in endometriosis. Proc. Natl. Acad. Sci. USA 2020, 117, 25859–25868. [Google Scholar] [CrossRef]
- Meldrum, C.; Doyle, M.A.; Tothill, R.W. Next-generation sequencing for cancer diagnostics: A practical perspective. Clin. Biochem. Rev. 2011, 32, 177–195. [Google Scholar]
- Herreros-Villanueva, M.; Chen, C.C.; Tsai, E.M.; Er, T.K. Endometriosis-associated ovarian cancer: What have we learned so far? Clin. Chim. Acta 2019, 493, 63–72. [Google Scholar] [CrossRef]
- Yin, M.; Zhai, L.; Wang, J.; Yu, Q.; Li, T.; Xu, X.; Guo, X.; Mao, X.; Zhou, J.; Zhang, X. Comprehensive Analysis of RNA-Seq in Endometriosis Reveals Competing Endogenous RNA Network Composed of circRNA, lncRNA and mRNA. Front. Genet. 2022, 13, 828238. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, L.; Zhan, H.; Mo, Y.; Ren, Z.; Shao, A.; Lin, J. Single-cell transcriptomic analysis of endometriosis provides insights into fibroblast fates and immune cell heterogeneity. Cell Biosci. 2021, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Zou, G.; Wang, J.; Xu, X.; Xu, P.; Zhu, L.; Yu, Q.; Peng, Y.; Guo, X.; Li, T.; Zhang, X. Cell subtypes and immune dysfunction in peritoneal fluid of endometriosis revealed by single-cell RNA-sequencing. Cell Biosci. 2021, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Flynn, W.F.; Sivajothi, S.; Luo, D.; Bozal, S.B.; Dave, M.; Luciano, A.A.; Robson, P.; Luciano, D.E.; Courtois, E.T. Single-cell analysis of endometriosis reveals a coordinated transcriptional programme driving immunotolerance and angiogenesis across eutopic and ectopic tissues. Nat. Cell Biol. 2022, 24, 1306–1318. [Google Scholar] [CrossRef]
- Lin, S.C.; Lee, H.C.; Hsu, C.T.; Huang, Y.H.; Li, W.N.; Hsu, P.L.; Wu, M.H.; Tsai, S.J. Targeting Anthrax Toxin Receptor 2 Ameliorates Endometriosis Progression. Theranostics 2019, 9, 620–632. [Google Scholar] [CrossRef]
- Kao, A.P.; Wang, K.H.; Chang, C.C.; Lee, J.N.; Long, C.Y.; Chen, H.S.; Tsai, C.F.; Hsieh, T.H.; Tsai, E.M. Comparative study of human eutopic and ectopic endometrial mesenchymal stem cells and the development of an in vivo endometriotic invasion model. Fertil. Steril. 2011, 95, 1308–1315.e1301. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Morozova, O.; Hirst, M.; Marra, M.A. Applications of new sequencing technologies for transcriptome analysis. Annu. Rev. Genom. Hum. Genet. 2009, 10, 135–151. [Google Scholar] [CrossRef]
- Zubovic, L.; Piazza, S.; Tebaldi, T.; Cozzuto, L.; Palazzo, G.; Sidarovich, V.; De Sanctis, V.; Bertorelli, R.; Lammens, T.; Hofmans, M.; et al. The altered transcriptome of pediatric myelodysplastic syndrome revealed by RNA sequencing. J. Hematol. Oncol. 2020, 13, 135. [Google Scholar] [CrossRef]
- Hong, M.; Tao, S.; Zhang, L.; Diao, L.T.; Huang, X.; Huang, S.; Xie, S.J.; Xiao, Z.D.; Zhang, H. RNA sequencing: New technologies and applications in cancer research. J. Hematol. Oncol. 2020, 13, 166. [Google Scholar] [CrossRef] [PubMed]
- Oshlack, A.; Robinson, M.D.; Young, M.D. From RNA-seq reads to differential expression results. Genom. Biol. 2010, 11, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindarajan, M.; Wohlmuth, C.; Waas, M.; Bernardini, M.Q.; Kislinger, T. High-throughput approaches for precision medicine in high-grade serous ovarian cancer. J. Hematol. Oncol. 2020, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Adamyan, L.; Aznaurova, Y.; Stepanian, A.; Nikitin, D.; Garazha, A.; Suntsova, M.; Sorokin, M.; Buzdin, A. Gene Expression Signature of Endometrial Samples from Women with and without Endometriosis. J. Minim. Invasive Gynecol. 2021, 28, 1774–1785. [Google Scholar] [CrossRef]
- Zhao, L.; Gu, C.; Ye, M.; Zhang, Z.; Han, W.; Fan, W.; Meng, Y. Identification of global transcriptome abnormalities and potential biomarkers in eutopic endometria of women with endometriosis: A preliminary study. Biomed Rep. 2017, 6, 654–662. [Google Scholar] [CrossRef]
- Queckborner, S.; von Grothusen, C.; Boggavarapu, N.R.; Francis, R.M.; Davies, L.C.; Gemzell-Danielsson, K. Stromal Heterogeneity in the Human Proliferative Endometrium-A Single-Cell RNA Sequencing Study. J. Pers. Med. 2021, 11, 448. [Google Scholar] [CrossRef]
- Rekker, K.; Saare, M.; Eriste, E.; Tasa, T.; Kukuskina, V.; Roost, A.M.; Anderson, K.; Samuel, K.; Karro, H.; Salumets, A.; et al. High-throughput mRNA sequencing of stromal cells from endometriomas and endometrium. Reproduction 2017, 154, 93–100. [Google Scholar] [CrossRef]
- Che, X.H.; Chen, Y.C.; Chen, C.L.; Ye, X.L.; Zhu, H. Non-hormonal targets underlying endometriosis: A focus on molecular mechanisms. Mol. Reprod. Dev. 2015, 82, 410–431. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. The PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2015, 7, a011189. [Google Scholar] [CrossRef]
- McKinnon, B.D.; Kocbek, V.; Nirgianakis, K.; Bersinger, N.A.; Mueller, M.D. Kinase signalling pathways in endometriosis: Potential targets for non-hormonal therapeutics. Hum. Reprod. Update 2016, 22, 382–403. [Google Scholar] [CrossRef]
- Madanes, D.; Bilotas, M.A.; Baston, J.I.; Singla, J.J.; Meresman, G.F.; Baranao, R.I.; Ricci, A.G. PI3K/AKT pathway is altered in the endometriosis patient’s endometrium and presents differences according to severity stage. Gynecol. Endocrinol. 2020, 36, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Pavone, M.E.; Lu, Z.; Wei, J.; Kim, J.J. Increased activation of the PI3K/AKT pathway compromises decidualization of stromal cells from endometriosis. J. Clin. Endocrinol. Metab. 2012, 97, E35–E43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.H.; Khalaj, K.; Young, S.L.; Lessey, B.A.; Koti, M.; Tayade, C. Immune-inflammation gene signatures in endometriosis patients. Fertil. Steril. 2016, 106, 1420–1431.e1427. [Google Scholar] [CrossRef] [PubMed]
- Aksak, T.; Gumurdulu, D.; Cetin, M.T.; Polat, S. Expression of monocyte chemotactic protein 2 and tumor necrosis factor alpha in human normal endometrium and endometriotic tissues. J. Gynecol. Obstet. Hum. Reprod. 2021, 50, 101971. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.J.; Yang, H.L.; Shao, J.; Mei, J.; Chang, K.K.; Zhu, R.; Li, M.Q. Anti-inflammatory cytokines in endometriosis. Cell Mol. Life Sci. 2019, 76, 2111–2132. [Google Scholar] [CrossRef]
- Li, S.; Fu, X.; Wu, T.; Yang, L.; Hu, C.; Wu, R. Role of Interleukin-6 and Its Receptor in Endometriosis. Med. Sci. Monit. 2017, 23, 3801–3807. [Google Scholar] [CrossRef]
- Suen, J.L.; Chang, Y.; Chiu, P.R.; Hsieh, T.H.; His, E.; Chen, Y.C.; Chen, Y.F.; Tsai, E.M. Serum level of IL-10 is increased in patients with endometriosis, and IL-10 promotes the growth of lesions in a murine model. Am. J. Pathol. 2014, 184, 464–471. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Bora, G.; Yaba, A. The role of mitogen-activated protein kinase signaling pathway in endometriosis. J. Obstet. Gynaecol. Res. 2021, 47, 1610–1623. [Google Scholar] [CrossRef]
- Liu, Z.; Yi, L.; Du, M.; Gong, G.; Zhu, Y. Overexpression of TGF-beta enhances the migration and invasive ability of ectopic endometrial cells via ERK/MAPK signaling pathway. Exp. Ther. Med. 2019, 17, 4457–4464. [Google Scholar]
- Uimari, O.; Rahmioglu, N.; Nyholt, D.R.; Vincent, K.; Missmer, S.A.; Becker, C.; Morris, A.P.; Montgomery, G.W.; Zondervan, K.T. Genome-wide genetic analyses highlight mitogen-activated protein kinase (MAPK) signaling in the pathogenesis of endometriosis. Hum. Reprod. 2017, 32, 780–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-C.; Chou, Y.-C.; Hsu, C.-Y.; Tsai, E.-M.; Er, T.-K. Transcriptome Profiling of Eutopic and Ectopic Endometrial Stromal Cells in Women with Endometriosis Based on High-Throughput Sequencing. Biomedicines 2022, 10, 2432. https://doi.org/10.3390/biomedicines10102432
Chen C-C, Chou Y-C, Hsu C-Y, Tsai E-M, Er T-K. Transcriptome Profiling of Eutopic and Ectopic Endometrial Stromal Cells in Women with Endometriosis Based on High-Throughput Sequencing. Biomedicines. 2022; 10(10):2432. https://doi.org/10.3390/biomedicines10102432
Chicago/Turabian StyleChen, Chih-Chieh, Yung-Che Chou, Chia-Yi Hsu, Eing-Mei Tsai, and Tze-Kiong Er. 2022. "Transcriptome Profiling of Eutopic and Ectopic Endometrial Stromal Cells in Women with Endometriosis Based on High-Throughput Sequencing" Biomedicines 10, no. 10: 2432. https://doi.org/10.3390/biomedicines10102432
APA StyleChen, C.-C., Chou, Y.-C., Hsu, C.-Y., Tsai, E.-M., & Er, T.-K. (2022). Transcriptome Profiling of Eutopic and Ectopic Endometrial Stromal Cells in Women with Endometriosis Based on High-Throughput Sequencing. Biomedicines, 10(10), 2432. https://doi.org/10.3390/biomedicines10102432