
Self-Renewal of Macrophages: Tumor-Released Factors and Signaling Pathways
 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Macrophages Proliferation in Health and Disease
3. Tissue Microenvironment and Self-Renewal of Macrophages
Macrophages Proliferation in the Tumor Microenvironment (TME)
4. Self-Renewal of Macrophages: Molecular Mechanisms
5. Metabolism Signature Associated to Self-Renewal of Macrophages
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Franken, L.; Schiwon, M.; Kurts, C. Macrophages: Sentinels and regulators of the immune system. Cell. Microbiol. 2016, 18, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The metabolic signature of macrophage responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komohara, Y.; Jinushi, M.; Takeya, M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014, 105, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bugler-Lamb, A.; Guilliams, M. Myeloid Cells TREM Down Anti-tumor Responses. Cell 2020, 182, 796–798. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A. The mononuclear phagocyte system. Curr. Opin. Immunol. 2006, 18, 49–53. [Google Scholar] [CrossRef]
- Hume, D.A.; Irvine, K.M.; Pridans, C. The Mononuclear Phagocyte System: The Relationship between Monocytes and Macrophages. Trends Immunol. 2019, 40, 98–112. [Google Scholar] [CrossRef]
- van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, S.J.; Ruckerl, D.; Cook, P.C.; Jones, L.H.; Finkelman, F.D.; van Rooijen, N.; MacDonald, A.S.; Allen, J.E. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 2011, 332, 1284–1288. [Google Scholar] [CrossRef] [Green Version]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Yona, S.; Kim, K.-W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieweke, M.H.; Allen, J.E. Beyond stem cells: Self-renewal of differentiated macrophages. Science 2013, 342, 1242974. [Google Scholar] [CrossRef] [PubMed]
- Nasser, H.; Adhikary, P.; Abdel-Daim, A.; Noyori, O.; Panaampon, J.; Kariya, R.; Okada, S.; Ma, W.; Baba, M.; Takizawa, H.; et al. Establishment of bone marrow-derived M-CSF receptor-dependent self-renewing macrophages. Cell Death Discov. 2020, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Soucie, E.L.; Weng, Z.; Geirsdóttir, L.; Molawi, K.; Maurizio, J.; Fenouil, R.; Mossadegh-Keller, N.; Gimenez, G.; VanHille, L.; Beniazza, M.; et al. Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science 2016, 351, aad5510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, A.; Soucie, E.; Sarrazin, S.; Sieweke, M.H. MafB/c-Maf deficiency enables self-renewal of differentiated functional macrophages. Science 2009, 326, 867–871. [Google Scholar] [CrossRef]
- Davies, L.C.; Rosas, M.; Jenkins, S.J.; Liao, C.-T.; Scurr, M.J.; Brombacher, F.; Fraser, D.J.; Allen, J.E.; Jones, S.A.; Taylor, P.R. Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat. Commun. 2013, 4, 1886. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef] [Green Version]
- Bain, C.C.; Hawley, C.A.; Garner, H.; Scott, C.L.; Schridde, A.; Steers, N.J.; Mack, M.; Joshi, A.; Guilliams, M.; Mowat, A.M.I.; et al. Long-lived self-renewing bone marrow-derived macrophages displace embryo-derived cells to inhabit adult serous cavities. Nat. Commun. 2016, 7, ncomms11852. [Google Scholar] [CrossRef] [Green Version]
- Ensan, S.; Li, A.; Besla, R.; Degousee, N.; Cosme, J.; Roufaiel, M.; Shikatani, E.A.; El-Maklizi, M.; Williams, J.W.; Robins, L.; et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat. Immunol. 2016, 17, 159–168. [Google Scholar] [CrossRef]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laviron, M.; Boissonnas, A. Ontogeny of Tumor-Associated Macrophages. Front. Immunol. 2019, 10, 1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loyher, P.-L.; Hamon, P.; Laviron, M.; Meghraoui-Kheddar, A.; Goncalves, E.; Deng, Z.; Torstensson, S.; Bercovici, N.; Baudesson de Chanville, C.; Combadière, B.; et al. Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med. 2018, 215, 2536–2553. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.-W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef]
- Müller, A.; Brandenburg, S.; Turkowski, K.; Müller, S.; Vajkoczy, P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 2015, 137, 278–288. [Google Scholar] [CrossRef]
- Evans, R.; Cullen, R.T. In situ proliferation of intratumor macrophages. J. Leukoc. Biol. 1984, 35, 561–572. [Google Scholar] [CrossRef]
- Bottazzi, B.; Erba, E.; Nobili, N.; Fazioli, F.; Rambaldi, A.; Mantovani, A. A paracrine circuit in the regulation of the proliferation of macrophages infiltrating murine sarcomas. J. Immunol. 1990, 144, 2409–2412. [Google Scholar]
- Campbell, M.J.; Tonlaar, N.Y.; Garwood, E.R.; Huo, D.; Moore, D.H.; Khramtsov, A.I.; Au, A.; Baehner, F.; Chen, Y.; Malaka, D.O.; et al. Proliferating macrophages associated with high grade, hormone receptor negative breast cancer and poor clinical outcome. Breast Cancer Res. Treat. 2011, 128, 703–711. [Google Scholar] [CrossRef] [Green Version]
- Tymoszuk, P.; Evens, H.; Marzola, V.; Wachowicz, K.; Wasmer, M.-H.; Datta, S.; Müller-Holzner, E.; Fiegl, H.; Böck, G.; van Rooijen, N.; et al. In situ proliferation contributes to accumulation of tumor-associated macrophages in spontaneous mammary tumors. Eur. J. Immunol. 2014, 44, 2247–2262. [Google Scholar] [CrossRef]
- de Sousa, J.R.; Da Costa Vasconcelos, P.F.; Quaresma, J.A.S. Functional aspects, phenotypic heterogeneity, and tissue immune response of macrophages in infectious diseases. Infect. Drug Resist. 2019, 12, 2589–2611. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covarrubias, A.J.; Aksoylar, H.I.; Horng, T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin. Immunol. 2015, 27, 286–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.C.-C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Schilling, J.D.; Pearce, E.J. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 2016, 45, 817–830. [Google Scholar] [CrossRef] [Green Version]
- Byles, V.; Covarrubias, A.J.; Ben-Sahra, I.; Lamming, D.W.; Sabatini, D.M.; Manning, B.D.; Horng, T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013, 4, 2834. [Google Scholar] [CrossRef] [Green Version]
- Blériot, C.; Chakarov, S.; Ginhoux, F. Determinants of resident tissue macrophage identity and function. Immunity 2020, 52, 957–970. [Google Scholar] [CrossRef]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef]
- Belhareth, R. Macrophage populations and self-renewal: Changing the paradigm. World J. Immunol. 2015, 5, 131. [Google Scholar] [CrossRef]
- Buchrieser, J.; James, W.; Moore, M.D. Human Induced Pluripotent Stem Cell-Derived Macrophages Share Ontogeny with MYB-Independent Tissue-Resident Macrophages. Stem Cell Rep. 2017, 8, 334–345. [Google Scholar] [CrossRef]
- Ying, W.; Lee, Y.S.; Dong, Y.; Seidman, J.S.; Yang, M.; Isaac, R.; Seo, J.B.; Yang, B.-H.; Wollam, J.; Riopel, M.; et al. Expansion of Islet-Resident Macrophages Leads to Inflammation Affecting β Cell Proliferation and Function in Obesity. Cell Metab. 2019, 29, 457–474.e5. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Yang, Q.; Cao, J.; Xie, N.; Liu, K.; Shou, P.; Qian, F.; Wang, Y.; Shi, Y. Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death Dis. 2016, 7, e2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butcher, M.J.; Hallinger, D.; Garcia, E.; Machida, Y.; Chakrabarti, S.; Nadler, J.; Galkina, E.V.; Imai, Y. Association of proinflammatory cytokines and islet resident leucocytes with islet dysfunction in type 2 diabetes. Diabetologia 2014, 57, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donath, M.Y.; Schumann, D.M.; Faulenbach, M.; Ellingsgaard, H.; Perren, A.; Ehses, J.A. Islet inflammation in type 2 diabetes: From metabolic stress to therapy. Diabetes Care 2008, 31 (Suppl. 2), S161–S164. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, W.; Fu, W.; Lee, Y.S.; Olefsky, J.M. The role of macrophages in obesity-associated islet inflammation and β-cell abnormalities. Nat. Rev. Endocrinol. 2020, 16, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K.; Cheng, T.; Zhan, J.; Peng, X.; Zhang, Y.; Wen, J.; Chen, X.; Ying, M. Targeting tumor-associated macrophages in the tumor microenvironment. Oncol. Lett. 2020, 20, 234. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Yrigoyen, M.; Cassetta, L.; Pollard, J.W. Macrophage targeting in cancer. Ann. N. Y. Acad. Sci. 2020, 1499, 18–41. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, J.; Huang, L. Tackling tams for cancer immunotherapy: It’s nano time. Trends Pharmacol. Sci. 2020, 41, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Mandal, G.; Roy Chowdhury, S.; Purohit, S.; Payne, K.K.; Anadon, C.; Gupta, A.; Swanson, P.; Yu, X.; Conejo-Garcia, J.R.; et al. Exosomes Produced by Mesenchymal Stem Cells Drive Differentiation of Myeloid Cells into Immunosuppressive M2-Polarized Macrophages in Breast Cancer. J. Immunol. 2019, 203, 3447–3460. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stangé, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [Green Version]
- Katzenelenbogen, Y.; Sheban, F.; Yalin, A.; Yofe, I.; Svetlichnyy, D.; Jaitin, D.A.; Bornstein, C.; Moshe, A.; Keren-Shaul, H.; Cohen, M.; et al. Coupled scRNA-Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. Cell 2020, 182, 872–885.e19. [Google Scholar] [CrossRef]
- Sathe, A.; Grimes, S.M.; Lau, B.T.; Chen, J.; Suarez, C.; Huang, R.J.; Poultsides, G.; Ji, H.P. Single-Cell Genomic Characterization Reveals the Cellular Reprogramming of the Gastric Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 2640–2653. [Google Scholar] [CrossRef] [Green Version]
- Mulder, K.; Patel, A.A.; Kong, W.T.; Piot, C.; Halitzki, E.; Dunsmore, G.; Khalilnezhad, S.; Irac, S.E.; Dubuisson, A.; Chevrier, M.; et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity 2021, 54, 1883–1900.e5. [Google Scholar] [CrossRef]
- Tuong, Z.K.; Loudon, K.W.; Berry, B.; Richoz, N.; Jones, J.; Tan, X.; Nguyen, Q.; George, A.; Hori, S.; Field, S.; et al. Resolving the immune landscape of human prostate at a single-cell level in health and cancer. Cell Rep. 2021, 37, 110132. [Google Scholar] [CrossRef]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e10. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.K.; Lee, K.; Hong, Y.; Cho, J.H.; Choi, J.W.; Lee, J.-I.; Suh, Y.-L.; Ku, B.M.; Eum, H.H.; et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef]
- Zhao, H.; Teng, Y.; Hao, W.; Li, J.; Li, Z.; Chen, Q.; Yin, C.; Yue, W. Single-cell analysis revealed that IL4I1 promoted ovarian cancer progression. J. Transl. Med. 2021, 19, 454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Cheng, S.; Wang, Y.; Wang, M.; Lu, Y.; Wen, Z.; Ge, Y.; Ma, Q.; Chen, Y.; Zhang, Y.; et al. Interrogation of the microenvironmental landscape in spinal ependymomas reveals dual functions of tumor-associated macrophages. Nat. Commun. 2021, 12, 6867. [Google Scholar] [CrossRef] [PubMed]
- Giurisato, E.; Lonardi, S.; Telfer, B.; Lussoso, S.; Risa-Ebrí, B.; Zhang, J.; Russo, I.; Wang, J.; Santucci, A.; Finegan, K.G.; et al. Extracellular-Regulated Protein Kinase 5-Mediated Control of p21 Expression Promotes Macrophage Proliferation Associated with Tumor Growth and Metastasis. Cancer Res. 2020, 80, 3319–3330. [Google Scholar] [CrossRef] [PubMed]
- Motta, J.M.; Rumjanek, V.M.; Mantovani, A.; Locati, M. Tumor-Released Products Promote Bone Marrow-Derived Macrophage Survival and Proliferation. Biomedicines 2021, 9, 1387. [Google Scholar] [CrossRef]
- Ginhoux, F.; Tacke, F.; Angeli, V.; Bogunovic, M.; Loubeau, M.; Dai, X.-M.; Stanley, E.R.; Randolph, G.J.; Merad, M. Langerhans cells arise from monocytes in vivo. Nat. Immunol. 2006, 7, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Röszer, T. Understanding the Biology of Self-Renewing Macrophages. Cells 2018, 7, 103. [Google Scholar] [CrossRef] [Green Version]
- Fejer, G.; Wegner, M.D.; Györy, I.; Cohen, I.; Engelhard, P.; Voronov, E.; Manke, T.; Ruzsics, Z.; Dölken, L.; Prazeres da Costa, O.; et al. Nontransformed, GM-CSF-dependent macrophage lines are a unique model to study tissue macrophage functions. Proc. Natl. Acad. Sci. USA 2013, 110, E2191–E2198. [Google Scholar] [CrossRef] [Green Version]
- Draijer, C.; Penke, L.R.K.; Peters-Golden, M. Distinctive Effects of GM-CSF and M-CSF on Proliferation and Polarization of Two Major Pulmonary Macrophage Populations. J. Immunol. 2019, 202, 2700–2709. [Google Scholar] [CrossRef]
- Braune, J.; Weyer, U.; Hobusch, C.; Mauer, J.; Brüning, J.C.; Bechmann, I.; Gericke, M. IL-6 Regulates M2 Polarization and Local Proliferation of Adipose Tissue Macrophages in Obesity. J. Immunol. 2017, 198, 2927–2934. [Google Scholar] [CrossRef] [Green Version]
- Amano, S.U.; Cohen, J.L.; Vangala, P.; Tencerova, M.; Nicoloro, S.M.; Yawe, J.C.; Shen, Y.; Czech, M.P.; Aouadi, M. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 2014, 19, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Tardelli, M.; Zeyda, K.; Moreno-Viedma, V.; Wanko, B.; Grün, N.G.; Staffler, G.; Zeyda, M.; Stulnig, T.M. Osteopontin is a key player for local adipose tissue macrophage proliferation in obesity. Mol. Metab. 2016, 5, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Maienschein-Cline, M.; Koh, T.J. Enhanced proliferation of ly6c+ monocytes/macrophages contributes to chronic inflammation in skin wounds of diabetic mice. J. Immunol. 2021, 206, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-L.; Wang, C.-J.; Lai, Z.-Z.; Yang, S.-L.; Zheng, Z.-M.; Shi, J.-W.; Li, M.-Q.; Shao, J. Decidual stromal cells maintain decidual macrophage homeostasis by secreting IL-24 in early pregnancy. Am. J. Reprod. Immunol. 2020, 84, e13261. [Google Scholar] [CrossRef] [PubMed]
- Linde, N.; Casanova-Acebes, M.; Sosa, M.S.; Mortha, A.; Rahman, A.; Farias, E.; Harper, K.; Tardio, E.; Reyes Torres, I.; Jones, J.; et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepe, G.; Braga, D.; Renzi, T.A.; Villa, A.; Bolego, C.; D’Avila, F.; Barlassina, C.; Maggi, A.; Locati, M.; Vegeto, E. Self-renewal and phenotypic conversion are the main physiological responses of macrophages to the endogenous estrogen surge. Sci. Rep. 2017, 7, 44270. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Guo, C.; Hu, P.; Chen, J.; Liu, Q.; Wu, X.; Cao, Y.; Wu, J. CSF1 is involved in breast cancer progression through inducing monocyte differentiation and homing. Int. J. Oncol. 2016, 49, 2064–2074. [Google Scholar] [CrossRef] [Green Version]
- Pixley, F.J. Macrophage Migration and Its Regulation by CSF-1. Int. J. Cell Biol. 2012, 2012, 501962. [Google Scholar] [CrossRef] [Green Version]
- Richardsen, E.; Uglehus, R.D.; Johnsen, S.H.; Busund, L.-T. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. Anticancer Res. 2015, 35, 865–874. [Google Scholar]
- Richardsen, E.; Uglehus, R.D.; Due, J.; Busch, C.; Busund, L.T.R. The prognostic impact of M-CSF, CSF-1 receptor, CD68 and CD3 in prostatic carcinoma. Histopathology 2008, 53, 30–38. [Google Scholar] [CrossRef]
- Kuropkat, C.; Dünne, A.A.; Plehn, S.; Ossendorf, M.; Herz, U. Macrophage colony-stimulating factor as a tumor marker for squamous cell carcinoma of the head and neck. Tumor Biol. 2003, 24, 236–240. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruessner, C.; Gruessner, A.; Glaser, K.; Abushahin, N.; Laughren, C.; Zheng, W.; Chambers, S.K. Biomarkers and endosalpingiosis in the ovarian and tubal microenvironment of women at high-risk for pelvic serous carcinoma. Am. J. Cancer Res. 2014, 4, 61–72. [Google Scholar] [PubMed]
- Li, H.-W.; Tang, S.-L. Colony Stimulating Factor-1 and its Receptor in Gastrointestinal Malignant Tumors. J. Cancer 2021, 12, 7111–7119. [Google Scholar] [CrossRef] [PubMed]
- Mroczko, B.; Szmitkowski, M.; Okulczyk, B. Hematopoietic growth factors in colorectal cancer patients. Clin. Chem. Lab. Med. 2003, 41, 646–651. [Google Scholar] [CrossRef]
- Wang, H.; Shao, Q.; Sun, J.; Ma, C.; Gao, W.; Wang, Q.; Zhao, L.; Qu, X. Interactions between colon cancer cells and tumor-infiltrated macrophages depending on cancer cell-derived colony stimulating factor 1. Oncoimmunology 2016, 5, e1122157. [Google Scholar] [CrossRef] [Green Version]
- Mroczko, B.; Szmitkowski, M.; Wereszczynska-Siemiatkowska, U.; Jurkowska, G. Stem cell factor and macrophage-colony stimulating factor in patients with pancreatic cancer. Clin. Chem. Lab. Med. 2004, 42, 256–260. [Google Scholar] [CrossRef]
- Blondy, T.; d Almeida, S.M.; Briolay, T.; Tabiasco, J.; Meiller, C.; Chéné, A.-L.; Cellerin, L.; Deshayes, S.; Delneste, Y.; Fonteneau, J.-F.; et al. Involvement of the M-CSF/IL-34/CSF-1R pathway in malignant pleural mesothelioma. J. Immunother. Cancer 2020, 8, e000182. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Xu, D.; Austin, C.D.; Caplazi, P.; Senger, K.; Sun, Y.; Jeet, S.; Young, J.; Delarosa, D.; Suto, E.; et al. Function of CSF1 and IL34 in macrophage homeostasis, inflammation, and cancer. Front. Immunol. 2019, 10, 2019. [Google Scholar] [CrossRef]
- Anfray, C.; Ummarino, A.; Andón, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Baud’huin, M.; Renault, R.; Charrier, C.; Riet, A.; Moreau, A.; Brion, R.; Gouin, F.; Duplomb, L.; Heymann, D. Interleukin-34 is expressed by giant cell tumours of bone and plays a key role in RANKL-induced osteoclastogenesis. J. Pathol. 2010, 221, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Baghdadi, M.; Endo, H.; Takano, A.; Ishikawa, K.; Kameda, Y.; Wada, H.; Miyagi, Y.; Yokose, T.; Ito, H.; Nakayama, H.; et al. High co-expression of IL-34 and M-CSF correlates with tumor progression and poor survival in lung cancers. Sci. Rep. 2018, 8, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillonneau, C.; Bézie, S.; Anegon, I. Immunoregulatory properties of the cytokine IL-34. Cell Mol. Life Sci. 2017, 74, 2569–2586. [Google Scholar] [CrossRef]
- Wang, B.; Xu, W.; Tan, M.; Xiao, Y.; Yang, H.; Xia, T.-S. Integrative genomic analyses of a novel cytokine, interleukin-34 and its potential role in cancer prediction. Int. J. Mol. Med. 2015, 35, 92–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greter, M.; Lelios, I.; Pelczar, P.; Hoeffel, G.; Price, J.; Leboeuf, M.; Kündig, T.M.; Frei, K.; Ginhoux, F.; Merad, M.; et al. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 2012, 37, 1050–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preisser, L.; Miot, C.; Le Guillou-Guillemette, H.; Beaumont, E.; Foucher, E.D.; Garo, E.; Blanchard, S.; Frémaux, I.; Croué, A.; Fouchard, I.; et al. IL-34 and macrophage colony-stimulating factor are overexpressed in hepatitis C virus fibrosis and induce profibrotic macrophages that promote collagen synthesis by hepatic stellate cells. Hepatology 2014, 60, 1879–1890. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Szretter, K.J.; Vermi, W.; Gilfillan, S.; Rossini, C.; Cella, M.; Barrow, A.D.; Diamond, M.S.; Colonna, M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 2012, 13, 753–760. [Google Scholar] [CrossRef] [Green Version]
- Baghdadi, M.; Wada, H.; Nakanishi, S.; Abe, H.; Han, N.; Putra, W.E.; Endo, D.; Watari, H.; Sakuragi, N.; Hida, Y.; et al. Chemotherapy-Induced IL34 Enhances Immunosuppression by Tumor-Associated Macrophages and Mediates Survival of Chemoresistant Lung Cancer Cells. Cancer Res. 2016, 76, 6030–6042. [Google Scholar] [CrossRef] [Green Version]
- Montalbán Del Barrio, I.; Penski, C.; Schlahsa, L.; Stein, R.G.; Diessner, J.; Wöckel, A.; Dietl, J.; Lutz, M.B.; Mittelbronn, M.; Wischhusen, J.; et al. Adenosine-generating ovarian cancer cells attract myeloid cells which differentiate into adenosine-generating tumor associated macrophages-a self-amplifying, CD39- and CD73-dependent mechanism for tumor immune escape. J. Immunother. Cancer 2016, 4, 49. [Google Scholar] [CrossRef] [Green Version]
- Strakhova, R.; Cadassou, O.; Cros-Perrial, E.; Jordheim, L.P. Regulation of tumor infiltrated innate immune cells by adenosine. Purinergic Signal. 2020, 16, 289–295. [Google Scholar] [CrossRef]
- Cekic, C. Modulation of myeloid cells by adenosine signaling. Curr. Opin. Pharmacol. 2020, 53, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Caorsi, R.; Penco, F.; Schena, F.; Gattorno, M. Monogenic polyarteritis: The lesson of ADA2 deficiency. Pediatr. Rheumatol. 2016, 14, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zavialov, A.V.; Gracia, E.; Glaichenhaus, N.; Franco, R.; Zavialov, A.V.; Lauvau, G. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J. Leukoc. Biol. 2010, 88, 279–290. [Google Scholar] [CrossRef]
- Kutryb-Zajac, B.; Harasim, G.; Jedrzejewska, A.; Krol, O.; Braczko, A.; Jablonska, P.; Mierzejewska, P.; Zielinski, J.; Slominska, E.M.; Smolenski, R.T. Macrophage-Derived Adenosine Deaminase 2 Correlates with M2 Macrophage Phenotype in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 3764. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Chu, Y.; Li, Z.; Yu, X.; Huang, Z.; Xu, J.; Zheng, L. Tumor-derived adenosine promotes macrophage proliferation in human hepatocellular carcinoma. J. Hepatol. 2021, 74, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Rauschmeier, R.; Gustafsson, C.; Reinhardt, A.; A-Gonzalez, N.; Tortola, L.; Cansever, D.; Subramanian, S.; Taneja, R.; Rossner, M.J.; Sieweke, M.H.; et al. Bhlhe40 and Bhlhe41 transcription factors regulate alveolar macrophage self-renewal and identity. EMBO J. 2019, 38, e101233. [Google Scholar] [CrossRef] [PubMed]
- Jarjour, N.N.; Schwarzkopf, E.A.; Bradstreet, T.R.; Shchukina, I.; Lin, C.-C.; Huang, S.C.-C.; Lai, C.-W.; Cook, M.E.; Taneja, R.; Stappenbeck, T.S.; et al. Bhlhe40 mediates tissue-specific control of macrophage proliferation in homeostasis and type 2 immunity. Nat. Immunol. 2019, 20, 687–700. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Aksoylar, H.I.; Yu, J.; Snyder, N.W.; Worth, A.J.; Iyer, S.S.; Wang, J.; Ben-Sahra, I.; Byles, V.; Polynne-Stapornkul, T.; et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife 2016, 5, e11612. [Google Scholar] [CrossRef]
- Molgora, M.; Esaulova, E.; Vermi, W.; Hou, J.; Chen, Y.; Luo, J.; Brioschi, S.; Bugatti, M.; Omodei, A.S.; Ricci, B.; et al. TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 2020, 182, 886–900.e17. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Z.; Wen, H.; Guo, Y.; Xu, F.; Zhu, Q.; Yuan, W.; Luo, R.; Lu, C.; Liu, R.; et al. Immunosuppressive TREM2(+) macrophages are associated with undesirable prognosis and responses to anti-PD-1 immunotherapy in non-small cell lung cancer. Cancer Immunol. Immunother. 2022, 71, 2511–2522. [Google Scholar] [CrossRef]
- Ulland, T.K.; Colonna, M. TREM2-a key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef]
- Coelho, I.; Duarte, N.; Macedo, M.P.; Penha-Gonçalves, C. Insights into Macrophage/Monocyte-Endothelial Cell Crosstalk in the Liver: A Role for Trem-2. J. Clin. Med. 2021, 10, 1248. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Chen, X.-F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Painter, M.M.; Atagi, Y.; Liu, C.-C.; Rademakers, R.; Xu, H.; Fryer, J.D.; Bu, G. TREM2 in CNS homeostasis and neurodegenerative disease. Mol. Neurodegener. 2015, 10, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, S.B.; Steele, N.G.; Carpenter, E.S.; Donahue, K.L.; Bushnell, G.G.; Morris, A.H.; The, S.; Orbach, S.M.; Sirihorachai, V.R.; Nwosu, Z.C.; et al. Pancreatic cancer is marked by complement-high blood monocytes and tumor-associated macrophages. Life Sci. Alliance 2021, 4, e202000935. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Reeve, J.L.; Liu, Y.; Teitelbaum, S.L.; Ross, F.P. DAP12 couples c-Fms activation to the osteoclast cytoskeleton by recruitment of Syk. Mol. Cell 2008, 31, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Otero, K.; Turnbull, I.R.; Poliani, P.L.; Vermi, W.; Cerutti, E.; Aoshi, T.; Tassi, I.; Takai, T.; Stanley, S.L.; Miller, M.; et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat. Immunol. 2009, 10, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Cella, M.; Buonsanti, C.; Strader, C.; Kondo, T.; Salmaggi, A.; Colonna, M. Impaired differentiation of osteoclasts in TREM-2-deficient individuals. J. Exp. Med. 2003, 198, 645–651. [Google Scholar] [CrossRef]
- Wu, K.; Byers, D.E.; Jin, X.; Agapov, E.; Alexander-Brett, J.; Patel, A.C.; Cella, M.; Gilfilan, S.; Colonna, M.; Kober, D.L.; et al. TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J. Exp. Med. 2015, 212, 681–697. [Google Scholar] [CrossRef] [Green Version]
- Valledor, A.F.; Comalada, M.; Xaus, J.; Celada, A. The differential time-course of extracellular-regulated kinase activity correlates with the macrophage response toward proliferation or activation. J. Biol. Chem. 2000, 275, 7403–7409. [Google Scholar] [CrossRef] [Green Version]
- Richardson, E.T.; Shukla, S.; Nagy, N.; Boom, W.H.; Beck, R.C.; Zhou, L.; Landreth, G.E.; Harding, C.V. ERK signaling is essential for macrophage development. PLoS ONE 2015, 10, e0140064. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, S.; Faccio, R.; Chappel, J.; Zheng, L.; Feng, X.; Weber, J.D.; Teitelbaum, S.L.; Ross, F.P. c-Fms tyrosine 559 is a major mediator of M-CSF-induced proliferation of primary macrophages. J. Biol. Chem. 2007, 282, 18980–18990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosas, M.; Davies, L.C.; Giles, P.J.; Liao, C.-T.; Kharfan, B.; Stone, T.C.; O’Donnell, V.B.; Fraser, D.J.; Jones, S.A.; Taylor, P.R. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science 2014, 344, 645–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imperatore, F.; Maurizio, J.; Vargas Aguilar, S.; Busch, C.J.; Favret, J.; Kowenz-Leutz, E.; Cathou, W.; Gentek, R.; Perrin, P.; Leutz, A.; et al. SIRT1 regulates macrophage self-renewal. EMBO J. 2017, 36, 2353–2372. [Google Scholar] [CrossRef] [PubMed]
- Rovida, E.; Spinelli, E.; Sdelci, S.; Barbetti, V.; Morandi, A.; Giuntoli, S.; Dello Sbarba, P. ERK5/BMK1 is indispensable for optimal colony-stimulating factor 1 (CSF-1)-induced proliferation in macrophages in a Src-dependent fashion. J. Immunol. 2008, 180, 4166–4172. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Chen, S.; Li, T.; Yang, Q.; Liu, J.; Tao, Y.; Meng, Y.; Chen, J.; Feng, X.; Han, Z.; et al. Critical Role of Lkb1 in the Maintenance of Alveolar Macrophage Self-Renewal and Immune Homeostasis. Front. Immunol. 2021, 12, 629281. [Google Scholar] [CrossRef]
- Deng, W.; Yang, J.; Lin, X.; Shin, J.; Gao, J.; Zhong, X.-P. Essential Role of mTORC1 in Self-Renewal of Murine Alveolar Macrophages. J. Immunol. 2017, 198, 492–504. [Google Scholar] [CrossRef] [Green Version]
- Bakopoulos, D.; Whisstock, J.C.; Warr, C.G.; Johnson, T.K. Macrophage self-renewal is regulated by transient expression of PDGF- and VEGF-related factor 2. FEBS J. 2022, 289, 3735–3751. [Google Scholar] [CrossRef]
- Ampem, G.; Junginger, A.; Yu, H.; Balogh, L.; Thuróczy, J.; Schneider, M.E.; Röszer, T. The environmental obesogen bisphenol A increases macrophage self-renewal. Cell Tissue Res. 2019, 378, 1–16. [Google Scholar] [CrossRef]
- Wang, T.; Liu, H.; Lian, G.; Zhang, S.-Y.; Wang, X.; Jiang, C. HIF1α-Induced Glycolysis Metabolism Is Essential to the Activation of Inflammatory Macrophages. Mediat. Inflamm. 2017, 2017, 9029327. [Google Scholar] [CrossRef] [Green Version]
- Meiser, J.; Krämer, L.; Sapcariu, S.C.; Battello, N.; Ghelfi, J.; D’Herouel, A.F.; Skupin, A.; Hiller, K. Pro-inflammatory Macrophages Sustain Pyruvate Oxidation through Pyruvate Dehydrogenase for the Synthesis of Itaconate and to Enable Cytokine Expression. J. Biol. Chem. 2016, 291, 3932–3946. [Google Scholar] [CrossRef] [PubMed]
- MacMicking, J.; Xie, Q.W.; Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.D.; Diotallevi, M.; Nicol, T.; McNeill, E.; Shaw, A.; Chuaiphichai, S.; Hale, A.; Starr, A.; Nandi, M.; Stylianou, E.; et al. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 2019, 28, 218–230.e7. [Google Scholar] [CrossRef] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, B.; O’Neill, L.A.J. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huangfu, N.; Zheng, W.; Xu, Z.; Wang, S.; Wang, Y.; Cheng, J.; Li, Z.; Cheng, K.; Zhang, S.; Chen, X.; et al. RBM4 regulates M1 macrophages polarization through targeting STAT1-mediated glycolysis. Int. Immunopharmacol. 2020, 83, 106432. [Google Scholar] [CrossRef]
- Roberts, J.; Fallon, P.G.; Hams, E. The pivotal role of macrophages in metabolic distress. In Macrophage Activation-Biology and Disease; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Liu, R.; Yu, Q.; Dong, L.; Bi, Y.; Liu, G. Metabolic reprogramming of macrophages during infections and cancer. Cancer Lett. 2019, 452, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Modolell, M.; Corraliza, I.M.; Link, F.; Soler, G.; Eichmann, K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur. J. Immunol. 1995, 25, 1101–1104. [Google Scholar] [CrossRef]
- Vadiveloo, P.K.; Keramidaris, E.; Morrison, W.A.; Stewart, A.G. Lipopolysaccharide-induced cell cycle arrest in macrophages occurs independently of nitric oxide synthase II induction. Biochim. Biophys. Acta 2001, 1539, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.A.; Vairo, G.; Knight, K.R.; Cocks, B.G. Activation and proliferation signals in murine macrophages. Biochemical signals controlling the regulation of macrophage urokinase-type plasminogen activator activity by colony-stimulating factors and other agents. Blood 1991, 77, 616–627. [Google Scholar] [CrossRef]
- Ayroldi, E.; Blasi, E.; Varesio, L.; Wiltrout, R.H. Inhibition of proliferation of retrovirus-immortalized macrophages by LPS and IFN-gamma: Possible autocrine down-regulation of cell growth by induction of IL1 and TNF. Biotherapy 1992, 4, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D. M1 and M2 Macrophages: Oracles of Health and Disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Lu, Y.; Martinez, J.; Bi, Y.; Lian, G.; Wang, T.; Milasta, S.; Wang, J.; Yang, M.; Liu, G.; et al. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1α-dependent. Proc. Natl. Acad. Sci. USA 2016, 113, 1564–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.C.-C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odegaard, J.I.; Vats, D.; Zhang, L.; Ricardo-Gonzalez, R.; Smith, K.L.; Sykes, D.B.; Kamps, M.P.; Chawla, A. Quantitative expansion of ES cell-derived myeloid progenitors capable of differentiating into macrophages. J. Leukoc. Biol. 2007, 81, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieler, M.; Hofmann, M.; Schabbauer, G. More than just protein building blocks: How amino acids and related metabolic pathways fuel macrophage polarization. FEBS J. 2021, 288, 3694–3714. [Google Scholar] [CrossRef]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odegaard, J.I.; Chawla, A. Alternative macrophage activation and metabolism. Annu. Rev. Pathol. 2011, 6, 275–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, S.M. Regulation of enzymes of the urea cycle and arginine metabolism. Annu. Rev. Nutr. 2002, 22, 87–105. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, S.; Vuckovic, I.; Jeon, R.; Lerman, A.; Folmes, C.D.; Dzeja, P.P.; Herrmann, J. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab. 2018, 28, 463–475.e4. [Google Scholar] [CrossRef]
- Hu, Q.; Shi, H.; Zeng, T.; Liu, H.; Su, Y.; Cheng, X.; Ye, J.; Yin, Y.; Liu, M.; Zheng, H.; et al. Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still’s disease. Arthritis Res. Ther. 2019, 21, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.; Park, P.S.U.; Lee, Y.; Mun, S.H.; Giannopoulou, E.; Fujii, T.; Lee, K.P.; Violante, S.N.; Cross, J.R.; Park-Min, K.-H. MYC-mediated early glycolysis negatively regulates proinflammatory responses by controlling IRF4 in inflammatory macrophages. Cell Rep. 2021, 35, 109264. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Wu, Y.; Huang, S.; Zhang, R.; Son, Y.M.; Li, C.; Cheon, I.S.; Gao, X.; Wang, M.; Chen, Y.; et al. Uncoupling of macrophage inflammation from self-renewal modulates host recovery from respiratory viral infection. Immunity 2021, 54, 1200–1218.e9. [Google Scholar] [CrossRef]
- Pereira, M.; Chen, T.-D.; Buang, N.; Olona, A.; Ko, J.-H.; Prendecki, M.; Costa, A.S.H.; Nikitopoulou, E.; Tronci, L.; Pusey, C.D.; et al. Acute iron deprivation reprograms human macrophage metabolism and reduces inflammation in vivo. Cell Rep. 2019, 28, 498–511.e5. [Google Scholar] [CrossRef] [Green Version]
- Wessendarp, M.; Watanabe-Chailland, M.; Liu, S.; Stankiewicz, T.; Ma, Y.; Kasam, R.K.; Shima, K.; Chalk, C.; Carey, B.; Rosendale, L.-R.; et al. Role of GM-CSF in regulating metabolism and mitochondrial functions critical to macrophage proliferation. Mitochondrion 2022, 62, 85–101. [Google Scholar] [CrossRef]
- Heaster, T.M.; Heaton, A.R.; Sondel, P.M.; Skala, M.C. Intravital metabolic autofluorescence imaging captures macrophage heterogeneity across normal and cancerous tissue. Front. Bioeng. Biotechnol. 2021, 9, 644648. [Google Scholar] [CrossRef]
- Boyer, S.; Lee, H.-J.; Steele, N.; Zhang, L.; Sajjakulnukit, P.; Andren, A.; Ward, M.H.; Singh, R.; Basrur, V.; Zhang, Y.; et al. Multiomic characterization of pancreatic cancer-associated macrophage polarization reveals deregulated metabolic programs driven by the GM-CSF-PI3K pathway. eLife 2022, 11, e73796. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Andrejeva, G.; Rathmell, J.C. Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef]
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; Van Eck, M.; Auphan-Anezin, N.; et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab. 2019, 29, 1376–1389.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puthenveetil, A.; Dubey, S. Metabolic reprograming of tumor-associated macrophages. Ann. Transl. Med. 2020, 8, 1030. [Google Scholar] [CrossRef] [PubMed]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark Res. 2021, 9, 1–17. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Soncin, I.; Sheng, J.; Chen, Q.; Foo, S.; Duan, K.; Lum, J.; Poidinger, M.; Zolezzi, F.; Karjalainen, K.; Ruedl, C. The tumour microenvironment creates a niche for the self-renewal of tumour-promoting macrophages in colon adenoma. Nat. Commun. 2018, 9, 582. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Allavena, P.; Mantovani, A. Cancer related inflammation: The macrophage connection. Cancer Lett. 2008, 267, 204–215. [Google Scholar] [CrossRef]
- Motta, J.M.; Rumjanek, V.M. Modulation of cytokine production by monocytes and developing-dendritic cells under the influence of leukemia and lymphoma cell products. Cell Biol. Int. 2021, 45, 890–897. [Google Scholar] [CrossRef]
- He, D.; Wang, D.; Lu, P.; Yang, N.; Xue, Z.; Zhu, X.; Zhang, P.; Fan, G. Single-cell RNA sequencing reveals heterogeneous tumor and immune cell populations in early-stage lung adenocarcinomas harboring EGFR mutations. Oncogene 2020, 40, 355–368. [Google Scholar] [CrossRef]
- Ma, R.-Y.; Black, A.; Qian, B.-Z. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 2022, 43, 546–563. [Google Scholar] [CrossRef]
- Thompson, T.W.; Jackson, B.T.; Li, P.J.; Wang, J.; Kim, A.B.; Huang, K.T.H.; Zhang, L.; Raulet, D.H. Tumor-derived CSF-1 induces the NKG2D ligand RAE-1δ on tumor-infiltrating macrophages. eLife 2018, 7, e32919. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeannin, P.; Paolini, L.; Adam, C.; Delneste, Y. The roles of CSFs on the functional polarization of tumor-associated macrophages. FEBS J. 2018, 285, 680–699. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Yang, Y.; Ma, P.; Huang, H.; Tang, Q.; Miao, H.; Fang, Y.; Jiang, N.; Li, Y.; Zhu, Q.; et al. Landscape and perspectives of macrophage -targeted cancer therapy in clinical trials. Mol. Ther. Oncolytics 2022, 24, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Johnson, K.C.C.; Gatti-Mays, M.E.; Li, Z. Emerging strategies in targeting tumor-resident myeloid cells for cancer immunotherapy. J. Hematol. Oncol. 2022, 15, 118. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
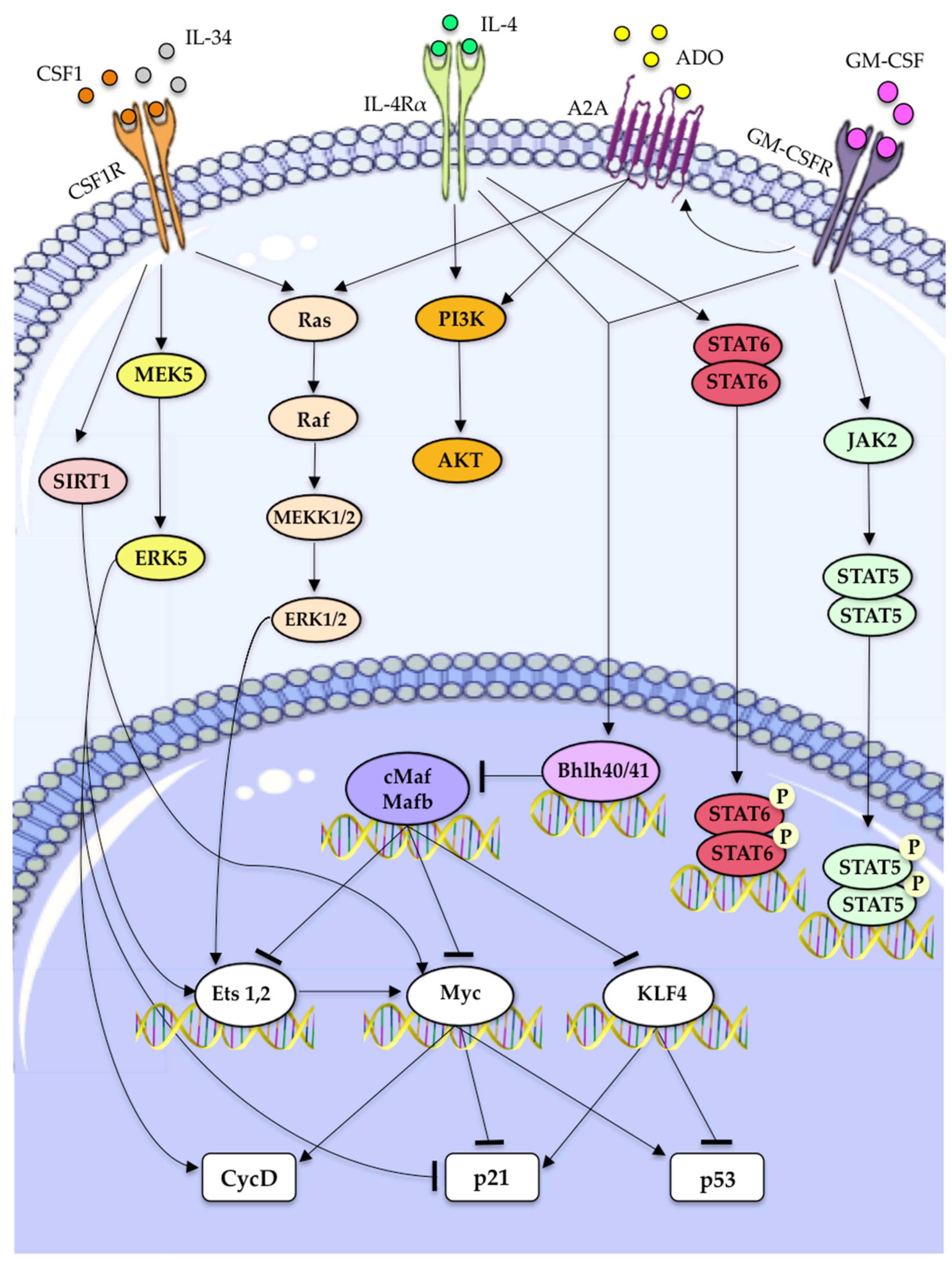
| Molecular Factor | Mechanism to Drive Macrophages Self-Renewal | References |
|---|---|---|
| CSF-1 | • ERK1/2 activation, inducing the activation of Ets transcription factors, thus increasing of cyclin-D expression. | [12,78]. |
| • ERK5 activation, downregulation of p21 expression and increased expression of Ets2 and Cyclin-D | [63] | |
| • KLF2 and KLF4 upregulation. | [13] | |
| • c-myc upregulation. | [13] | |
| IL-34 | • Partially affects TAMs proliferation through its ability to interact with CSF1R. | [99] |
| GM-CSF | • Activation of JAK2/STAT5 pathway. | [12,13,14] |
| • Activation of Bhlhe40/Bhlhe41 which inhibits c-Maf and Mafb transcription factors, thereby directly promoting expression of transcripts encoding cell cycle-related proteins. | [107,108] | |
| ADO-1 | • Working synergically with GM-CSF causes the upregulaion of PI3K/Akt and MEK/ERK signaling downstream of A2A activation. | [106] |
| • Promotes macrophage proliferation, in part, through the transcriptional regulation of A2A. | [106] | |
| IL-4 | • Induce the transcription of STAT6 and activates PI3K/AKT signaling. | [12] |
| • May stimulate macrophage proliferation in an Akt- and ACLY-dependent manner. | [109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filiberti, S.; Russo, M.; Lonardi, S.; Bugatti, M.; Vermi, W.; Tournier, C.; Giurisato, E. Self-Renewal of Macrophages: Tumor-Released Factors and Signaling Pathways. Biomedicines 2022, 10, 2709. https://doi.org/10.3390/biomedicines10112709
Filiberti S, Russo M, Lonardi S, Bugatti M, Vermi W, Tournier C, Giurisato E. Self-Renewal of Macrophages: Tumor-Released Factors and Signaling Pathways. Biomedicines. 2022; 10(11):2709. https://doi.org/10.3390/biomedicines10112709
Chicago/Turabian StyleFiliberti, Serena, Mariapia Russo, Silvia Lonardi, Mattia Bugatti, William Vermi, Cathy Tournier, and Emanuele Giurisato. 2022. "Self-Renewal of Macrophages: Tumor-Released Factors and Signaling Pathways" Biomedicines 10, no. 11: 2709. https://doi.org/10.3390/biomedicines10112709
APA StyleFiliberti, S., Russo, M., Lonardi, S., Bugatti, M., Vermi, W., Tournier, C., & Giurisato, E. (2022). Self-Renewal of Macrophages: Tumor-Released Factors and Signaling Pathways. Biomedicines, 10(11), 2709. https://doi.org/10.3390/biomedicines10112709