
Immune Checkpoint Molecules and Glucose Metabolism in HIV-Induced T Cell Exhaustion

 , ,
, ,  and
and 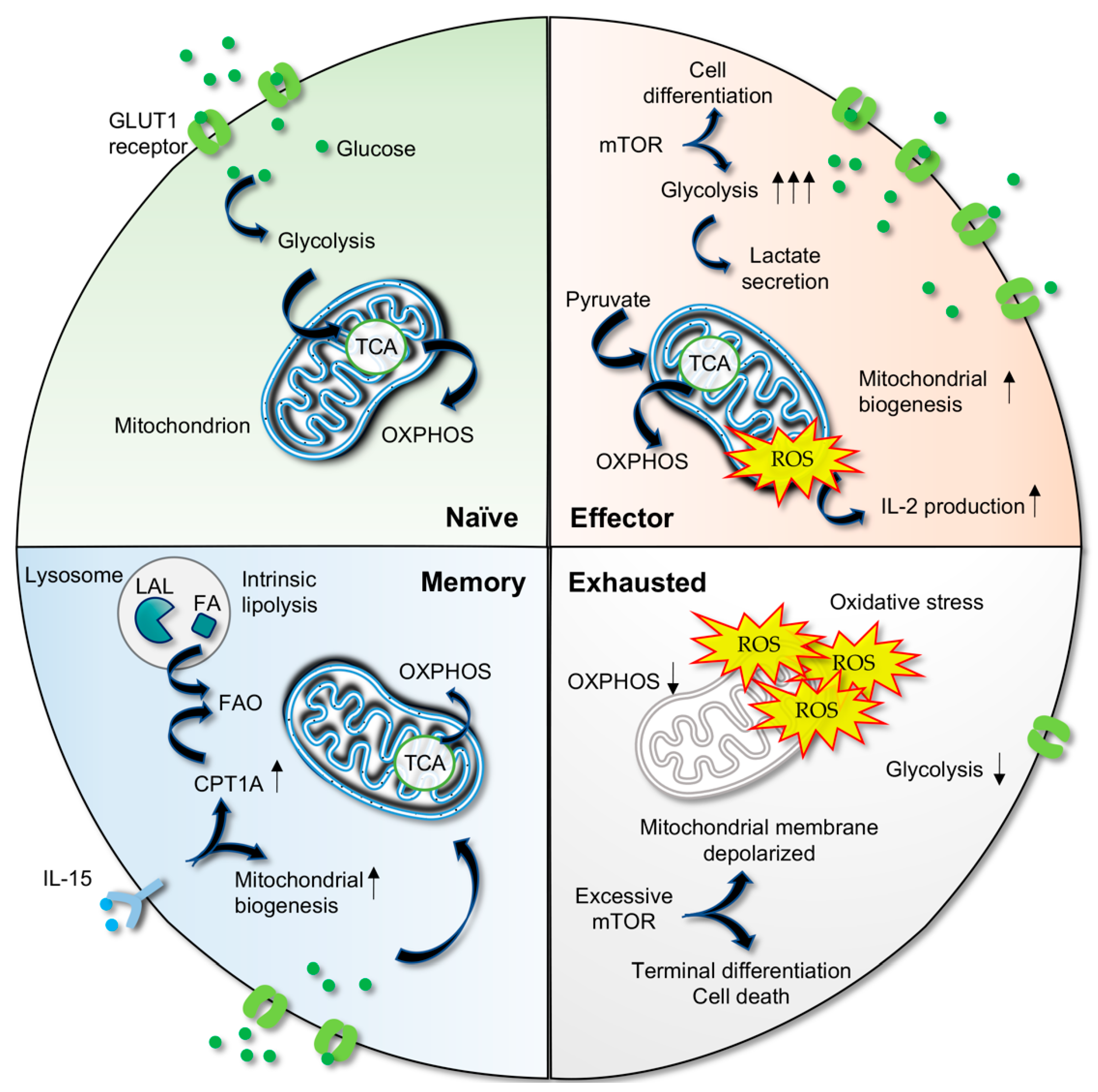{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Elevated Expression of T Cell Immune Checkpoint Molecules in HIV
2.1. PD-1
2.2. CTLA-4
2.3. TIM-3
2.4. LAG-3
2.5. TIGIT
3. Metabolic Plasticity in T Cells
3.1. Activated T Cells Utilize Aerobic Glycolysis
3.2. Metabolic Shift in Memory T Cells
3.3. Metabolic Deregulation Results in T Cell Exhaustion
4. Altered Glucose Metabolism in HIV Infection
4.1. Glucose Metabolism Enhances Cell Permissibility to HIV Entry
4.2. HIV Interferes T Cell Metabolism
4.3. Long-Term HAART Positively Influences T Cell Metabolism Fitness
4.4. T Cell Metabolism in Elite Controllers
5. Achieving HIV Remission through Immune Checkpoint Inhibitors and Glucose Metabolic Enhancement
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; Van Der Most, R.; Ahmed, R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.N.; Ahmed, R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2009, 106, 8623–8628. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.; Gordon, S.; Chan, C.N.; Wang, H.; Lindemuth, E.; Galardi, C.; Falcinelli, S.D.; Raines, S.L.; Read, J.L.; Nguyen, K. CTLA-4 and PD-1 dual blockade induces SIV reactivation without control of rebound after antiretroviral therapy interruption. Nat. Med. 2020, 26, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Le Garff, G.; Samri, A.; Lambert-Niclot, S.; Even, S.; Lavolé, A.; Cadranel, J.; Spano, J.-P.; Autran, B.; Marcelin, A.-G.; Guihot, A. Transient HIV-specific T cells increase and inflammation in an HIV-infected patient treated with nivolumab. Aids 2017, 31, 1048–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, J.K.; Cyktor, J.C.; Fyne, E.; Campellone, S.; Mason, S.W.; Mellors, J.W. Blockade of the PD-1 axis alone is not sufficient to activate HIV-1 virion production from CD4+ T cells of individuals on suppressive ART. PLoS ONE 2019, 14, e0211112. [Google Scholar] [CrossRef] [PubMed]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef] [Green Version]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [Green Version]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.-W.; Godec, J.; LaFleur, M.W.; Brown, F.D. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553. [Google Scholar] [CrossRef] [Green Version]
- Moskophidis, D.; Lechner, F.; Pircher, H.; Zinkernagel, R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993, 362, 758. [Google Scholar] [CrossRef]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.-R.; Delwart, E.; Sepulveda, H.; Balderas, R.S. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Migueles, S.A.; Laborico, A.C.; Shupert, W.L.; Sabbaghian, M.S.; Rabin, R.; Hallahan, C.W.; Van Baarle, D.; Kostense, S.; Miedema, F.; McLaughlin, M. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat. Immunol. 2002, 3, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhang, A.; Qiu, C.; Wang, W.; Yang, Y.; Qiu, C.; Liu, A.; Zhu, L.; Yuan, S.; Hu, H. The upregulation of LAG-3 on T cells defines a subpopulation with functional exhaustion and correlates with disease progression in HIV-infected subjects. J. Immunol. 2015, 194, 3873–3882. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, D.E.; Kavanagh, D.G.; Pereyra, F.; Zaunders, J.J.; Mackey, E.W.; Miura, T.; Palmer, S.; Brockman, M.; Rathod, A.; Piechocka-Trocha, A. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat. Immunol. 2007, 8, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; Ndhlovu, L.C.; Barbour, J.D.; Sheth, P.M.; Jha, A.R.; Long, B.R.; Wong, J.C.; Satkunarajah, M.; Schweneker, M.; Chapman, J.M. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008, 205, 2763–2779. [Google Scholar] [CrossRef] [PubMed]
- Chew, G.M.; Fujita, T.; Webb, G.M.; Burwitz, B.J.; Wu, H.L.; Reed, J.S.; Hammond, K.B.; Clayton, K.L.; Ishii, N.; Abdel-Mohsen, M. TIGIT marks exhausted T cells, correlates with disease progression, and serves as a target for immune restoration in HIV and SIV infection. PLoS Pathog. 2016, 12, e1005349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozkazanc, D.; Yoyen-Ermis, D.; Tavukcuoglu, E.; Buyukasik, Y.; Esendagli, G. Functional exhaustion of CD4+ T cells induced by co-stimulatory signals from myeloid leukaemia cells. Immunology 2016, 149, 460–471. [Google Scholar] [CrossRef] [Green Version]
- Fromentin, R.; Bakeman, W.; Lawani, M.B.; Khoury, G.; Hartogensis, W.; DaFonseca, S.; Killian, M.; Epling, L.; Hoh, R.; Sinclair, E. CD4+ T cells expressing PD-1, TIGIT and LAG-3 contribute to HIV persistence during ART. PLoS Pathog. 2016, 12, e1005761. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef]
- Sheppard, K.-A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3ζ signalosome and downstream signaling to PKCθ. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Planès, R.; BenMohamed, L.; Leghmari, K.; Delobel, P.; Izopet, J.; Bahraoui, E. HIV-1 Tat protein induces PD-L1 (B7-H1) expression on dendritic cells through tumor necrosis factor alpha-and toll-like receptor 4-mediated mechanisms. J. Virol. 2014, 88, 6672–6689. [Google Scholar] [CrossRef] [Green Version]
- Muthumani, K.; Choo, A.Y.; Shedlock, D.J.; Laddy, D.J.; Sundaram, S.G.; Hirao, L.; Wu, L.; Thieu, K.P.; Chung, C.W.; Lankaraman, K.M. Human immunodeficiency virus type 1 Nef induces programmed death 1 expression through a p38 mitogen-activated protein kinase-dependent mechanism. J. Virol. 2008, 82, 11536–11544. [Google Scholar] [CrossRef] [Green Version]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Zhang, Z.; Wang, X.; Fu, J.-L.; Yao, J.; Jiao, Y.; Chen, L.; Zhang, H.; Wei, J.; Jin, L. PD-1 up-regulation is correlated with HIV-specific memory CD8+ T-cell exhaustion in typical progressors but not in long-term nonprogressors. Blood 2007, 109, 4671–4678. [Google Scholar] [CrossRef]
- Petrovas, C.; Casazza, J.P.; Brenchley, J.M.; Price, D.A.; Gostick, E.; Adams, W.C.; Precopio, M.L.; Schacker, T.; Roederer, M.; Douek, D.C. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 2006, 203, 2281–2292. [Google Scholar] [CrossRef]
- Evans, V.A.; Van Der Sluis, R.M.; Solomon, A.; Dantanarayana, A.; McNeil, C.; Garsia, R.; Palmer, S.; Fromentin, R.; Chomont, N.; Sékaly, R.-P. PD-1 contributes to the establishment and maintenance of HIV-1 latency. AIDS 2018, 32, 1491. [Google Scholar] [CrossRef]
- Dafonseca, S.; Chomont, N.; El Far, M.; Boulassel, R.; Routy, J.; Sékaly, R. Purging the HIV-1 reservoir through the disruption of the PD-1 pathway. J. Int. AIDS Soc. 2010, 13, O15. [Google Scholar] [CrossRef]
- Guihot, A.; Marcelin, A.-G.; Massiani, M.-A.; Samri, A.; Soulié, C.; Autran, B.; Spano, J.-P. Drastic decrease of the HIV reservoir in a patient treated with nivolumab for lung cancer. Ann. Oncol. 2018, 29, 517–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [Green Version]
- Gay, C.L.; Bosch, R.J.; Ritz, J.; Hataye, J.M.; Aga, E.; Tressler, R.L.; Mason, S.W.; Hwang, C.K.; Grasela, D.M.; Ray, N. Clinical trial of the anti-PD-L1 antibody BMS-936559 in HIV-1 infected participants on suppressive antiretroviral therapy. J. Infect. Dis. 2017, 215, 1725–1733. [Google Scholar] [CrossRef] [Green Version]
- Spano, J.-P.; Veyri, M.; Gobert, A.; Guihot, A.; Perré, P.; Kerjouan, M.; Brosseau, S.; Cloarec, N.; Montaudié, H.; Helissey, C. Immunotherapy for cancer in people living with HIV: Safety with an efficacy signal from the series in real life experience. Aids 2019, 33, F13–F19. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Rajdev, L.; Rhodes, A.; Dantanarayana, A.; Tennakoon, S.; Chea, S.; Spelman, T.; Lensing, S.; Rutishauser, R.; Bakkour, S. Impact of Anti–PD-1 and Anti–CTLA-4 on the Human Immunodeficiency Virus (HIV) Reservoir in People Living With HIV With Cancer on Antiretroviral Therapy: The AIDS Malignancy Consortium 095 Study. Clin. Infect. Dis. 2021, 73, e1973–e1981. [Google Scholar] [CrossRef]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Leng, Q.; Bentwich, Z.; Magen, E.; Kalinkovich, A.; Borkow, G. CTLA-4 upregulation during HIV infection: Association with anergy and possible target for therapeutic intervention. Aids 2002, 16, 519–529. [Google Scholar] [CrossRef]
- El-Far, M.; Ancuta, P.; Routy, J.-P.; Zhang, Y.; Bakeman, W.; Bordi, R.; DaFonseca, S.; Said, E.A.; Gosselin, A.; Tep, T.-S. Nef promotes evasion of human immunodeficiency virus type 1-infected cells from the CTLA-4-mediated inhibition of T-cell activation. J. Gen. Virol. 2015, 96, 1463. [Google Scholar] [CrossRef] [Green Version]
- McGary, C.S.; Deleage, C.; Harper, J.; Micci, L.; Ribeiro, S.P.; Paganini, S.; Kuri-Cervantes, L.; Benne, C.; Ryan, E.S.; Balderas, R. CTLA-4+ PD-1− memory CD4+ T cells critically contribute to viral persistence in antiretroviral therapy-suppressed, SIV-infected rhesus macaques. Immunity 2017, 47, 776–788. e775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigam, P.; Velu, V.; Kannanganat, S.; Chennareddi, L.; Kwa, S.; Siddiqui, M.; Amara, R.R. Expansion of FOXP3+ CD8 T cells with suppressive potential in colorectal mucosa following a pathogenic simian immunodeficiency virus infection correlates with diminished antiviral T cell response and viral control. J. Immunol. 2010, 184, 1690–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yero, A.; Shi, T.; Routy, J.-P.; Tremblay, C.; Durand, M.; Costiniuk, C.T.; Jenabian, M.-A. FoxP3+ CD8 T-cells in acute HIV infection and following early antiretroviral therapy initiation. Front. Immunol. 2022, 13, 962912. [Google Scholar] [CrossRef] [PubMed]
- Cecchinato, V.; Tryniszewska, E.; Ma, Z.M.; Vaccari, M.; Boasso, A.; Tsai, W.-P.; Petrovas, C.; Fuchs, D.; Heraud, J.-M.; Venzon, D. Immune activation driven by CTLA-4 blockade augments viral replication at mucosal sites in simian immunodeficiency virus infection. J. Immunol. 2008, 180, 5439–5447. [Google Scholar] [CrossRef] [Green Version]
- Wightman, F.; Solomon, A.; Kumar, S.S.; Urriola, N.; Gallagher, K.; Hiener, B.; Palmer, S.; Mcneil, C.; Garsia, R.; Lewin, S.R. Effect of ipilimumab on the HIV reservoir in an HIV-infected individual with metastatic melanoma. AIDS 2015, 29, 504. [Google Scholar] [CrossRef] [Green Version]
- Elahi, S.; Shahbaz, S.; Houston, S. Selective upregulation of CTLA-4 on CD8+ T cells restricted by HLA-B* 35Px renders them to an exhausted phenotype in HIV-1 infection. PLoS Pathog. 2020, 16, e1008696. [Google Scholar] [CrossRef]
- Van der Sluis, R.M.; Kumar, N.A.; Pascoe, R.D.; Zerbato, J.M.; Evans, V.A.; Dantanarayana, A.I.; Anderson, J.L.; Sékaly, R.P.; Fromentin, R.; Chomont, N. Combination immune checkpoint blockade to reverse HIV latency. J. Immunol. 2020, 204, 1242–1254. [Google Scholar] [CrossRef]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef]
- Sabatos, C.A.; Chakravarti, S.; Cha, E.; Schubart, A.; Sánchez-Fueyo, A.; Zheng, X.X.; Coyle, A.J.; Strom, T.B.; Freeman, G.J.; Kuchroo, V.K. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol. 2003, 4, 1102–1110. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.A.; Edgar, C.R.; Prévost, J.; Trothen, S.M.; Lurie, A.; Mumby, M.J.; Galbraith, A.; Kirchhoff, F.; Haeryfar, S.M.; Finzi, A. The HIV-1 accessory protein Nef increases surface expression of the checkpoint receptor Tim-3 in infected CD4+ T cells. J. Biol. Chem. 2021, 297, 101042. [Google Scholar] [CrossRef] [PubMed]
- Prévost, J.; Edgar, C.R.; Richard, J.; Trothen, S.M.; Jacob, R.A.; Mumby, M.J.; Pickering, S.; Dubé, M.; Kaufmann, D.E.; Kirchhoff, F. HIV-1 Vpu downregulates Tim-3 from the surface of infected CD4+ T cells. J. Virol. 2020, 94, e01999-19. [Google Scholar] [CrossRef] [PubMed]
- Elahi, S.; Dinges, W.L.; Lejarcegui, N.; Laing, K.J.; Collier, A.C.; Koelle, D.M.; McElrath, M.J.; Horton, H. Protective HIV-specific CD8+ T cells evade Treg cell suppression. Nat. Med. 2011, 17, 989–995. [Google Scholar] [CrossRef]
- Sakhdari, A.; Mujib, S.; Vali, B.; Yue, F.Y.; MacParland, S.; Clayton, K.; Jones, R.B.; Liu, J.; Lee, E.Y.; Benko, E. Tim-3 negatively regulates cytotoxicity in exhausted CD8+ T cells in HIV infection. PLoS ONE 2012, 7, e40146. [Google Scholar] [CrossRef]
- Hannier, S.; Tournier, M.; Bismuth, G.; Triebel, F. CD3/TCR complex-associated lymphocyte activation gene-3 molecules inhibit CD3/TCR signaling. J. Immunol. 1998, 161, 4058–4065. [Google Scholar]
- Hoffmann, M.; Pantazis, N.; Martin, G.E.; Hickling, S.; Hurst, J.; Meyerowitz, J.; Willberg, C.B.; Robinson, N.; Brown, H.; Fisher, M.; et al. Exhaustion of Activated CD8 T Cells Predicts Disease Progression in Primary HIV-1 Infection. PLoS Pathog. 2016, 12, e1005661. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Grosso, J.F.; Goldberg, M.V.; Getnet, D.; Bruno, T.C.; Yen, H.-R.; Pyle, K.J.; Hipkiss, E.; Vignali, D.A.; Pardoll, D.M.; Drake, C.G. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J. Immunol. 2009, 182, 6659–6669. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Li, X.; Zhang, L.; Zhu, Q.; Chen, C.; Bao, J.; Chen, Y. CD4+ T cell exhaustion revealed by high PD-1 and LAG-3 expression and the loss of helper T cell function in chronic hepatitis B. BMC Immunol. 2019, 20, 27. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Blackburn, S.D.; Intlekofer, A.M.; Kao, C.; Angelosanto, J.M.; Reiner, S.L.; Wherry, E.J. A role for the transcriptional repressor Blimp-1 in CD8+ T cell exhaustion during chronic viral infection. Immunity 2009, 31, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Levin, S.D.; Taft, D.W.; Brandt, C.S.; Bucher, C.; Howard, E.D.; Chadwick, E.M.; Johnston, J.; Hammond, A.; Bontadelli, K.; Ardourel, D. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur. J. Immunol. 2011, 41, 902–915. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Liu, T.; Wang, Z.; Ma, M.; Lei, J.; Zhang, Z.; Fu, S.; Fu, Y.; Hu, Q.; Ding, H. Expression of the inhibitory receptor TIGIT is up-regulated specifically on NK cells with CD226 activating receptor from HIV-infected individuals. Front. Immunol. 2018, 9, 2341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48. [Google Scholar] [CrossRef]
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 2011, 186, 1338–1342. [Google Scholar] [CrossRef] [Green Version]
- Tauriainen, J.; Scharf, L.; Frederiksen, J.; Naji, A.; Ljunggren, H.-G.; Sönnerborg, A.; Lund, O.; Reyes-Terán, G.; Hecht, F.M.; Deeks, S.G. Perturbed CD8+ T cell TIGIT/CD226/PVR axis despite early initiation of antiretroviral treatment in HIV infected individuals. Sci. Rep. 2017, 7, 40354. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lu, X.; Cheung, A.K.L.; Zhang, Q.; Liu, Z.; Li, Z.; Yuan, L.; Wang, R.; Liu, Y.; Tang, B. Analysis of the Characteristics of TIGIT-Expressing CD3− CD56+ NK Cells in Controlling Different Stages of HIV-1 Infection. Front. Immunol. 2021, 12, 602492. [Google Scholar] [CrossRef]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 2018, 7, e1466769. [Google Scholar] [CrossRef]
- Ostroumov, D.; Duong, S.; Wingerath, J.; Woller, N.; Manns, M.P.; Timrott, K.; Kleine, M.; Ramackers, W.; Roessler, S.; Nahnsen, S. Transcriptome profiling identifies TIGIT as a marker of T cell exhaustion in liver cancer. Hepatology 2020, 73, 1399–1418. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Chang, J.J.; Dantanarayana, A.I.; Solomon, A.; Evans, V.A.; Pascoe, R.; Gubser, C.; Trautman, L.; Fromentin, R.; Chomont, N. Combination immune checkpoint blockade enhances IL-2 and CD107a production from HIV-specific T cells ex vivo in people living with HIV on antiretroviral therapy. J. Immunol. 2022, 208, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Masson, J.J.R.; Palmer, C.S. Glucose metabolism in CD4 and CD8 T cells. In Molecular Nutrition: Carbohydrates; Elsevier: Amsterdam, The Netherlands, 2019; pp. 129–147. [Google Scholar]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.-H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, R.P.; Finlay, D.K. Glucose, glycolysis and lymphocyte responses. Mol. Immunol. 2015, 68, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; Macintyre, A.N.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; Van Der Windt, G.J.; Blagih, J.; Qiu, J. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Yang, K.; Li, Y.; Shaw, T.I.; Wang, Y.; Blanco, D.B.; Wang, X.; Cho, J.-H.; Wang, H.; Rankin, S. Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity 2017, 46, 488–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron-Harel, N.; Santos, D.; Ghergurovich, J.M.; Sage, P.T.; Reddy, A.; Lovitch, S.B.; Dephoure, N.; Satterstrom, F.K.; Sheffer, M.; Spinelli, J.B. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab. 2016, 24, 104–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.-R.; Schumacker, P.T.; Licht, J.D.; Perlman, H. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Okoye, I.; Wang, L.; Pallmer, K.; Richter, K.; Ichimura, T.; Haas, R.; Crouse, J.; Choi, O.; Heathcote, D.; Lovo, E. The protein LEM promotes CD8+ T cell immunity through effects on mitochondrial respiration. Science 2015, 348, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Schurich, A.; Pallett, L.J.; Jajbhay, D.; Wijngaarden, J.; Otano, I.; Gill, U.S.; Hansi, N.; Kennedy, P.T.; Nastouli, E.; Gilson, R. Distinct metabolic requirements of exhausted and functional virus-specific CD8 T cells in the same host. Cell Rep. 2016, 16, 1243–1252. [Google Scholar] [CrossRef] [Green Version]
- Valle-Casuso, J.C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M. Cellular metabolism is a major determinant of HIV-1 reservoir seeding in CD4+ T cells and offers an opportunity to tackle infection. Cell Metab. 2019, 29, 611–626. e615. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Baldauf, H.-M.; Keppler, O.T.; Fackler, O.T. Restrictions to HIV-1 replication in resting CD4+ T lymphocytes. Cell Res. 2013, 23, 876–885. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, D.; van der Windt, G.J.; Huang, S.C.; Curtis, J.D.; Chang, C.H.; Buck, M.D.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M.; et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 2014, 41, 75–88. [Google Scholar] [CrossRef] [Green Version]
- van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [Green Version]
- van der Windt, G.J.; O’Sullivan, D.; Everts, B.; Huang, S.C.-C.; Buck, M.D.; Curtis, J.D.; Chang, C.-H.; Smith, A.M.; Ai, T.; Faubert, B. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc. Natl. Acad. Sci. USA 2013, 110, 14336–14341. [Google Scholar] [CrossRef]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.-S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef] [Green Version]
- Burgers, W.A.; Riou, C.; Mlotshwa, M.; Maenetje, P.; de Assis Rosa, D.; Brenchley, J.; Mlisana, K.; Douek, D.C.; Koup, R.; Roederer, M. Association of HIV-specific and total CD8+ T memory phenotypes in subtype C HIV-1 infection with viral set point. J. Immunol. 2009, 182, 4751–4761. [Google Scholar] [CrossRef] [Green Version]
- Masson, J.J.; Murphy, A.J.; Lee, M.K.; Ostrowski, M.; Crowe, S.M.; Palmer, C.S. Assessment of metabolic and mitochondrial dynamics in CD4+ and CD8+ T cells in virologically suppressed HIV-positive individuals on combination antiretroviral therapy. PLoS ONE 2017, 12, e0183931. [Google Scholar]
- Clerc, I.; Abba Moussa, D.; Vahlas, Z.; Tardito, S.; Oburoglu, L.; Hope, T.J.; Sitbon, M.; Dardalhon, V.; Mongellaz, C.; Taylor, N. Entry of glucose-and glutamine-derived carbons into the citric acid cycle supports early steps of HIV-1 infection in CD4 T cells. Nat. Metab. 2019, 1, 717–730. [Google Scholar]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [Green Version]
- Fisicaro, P.; Barili, V.; Montanini, B.; Acerbi, G.; Ferracin, M.; Guerrieri, F.; Salerno, D.; Boni, C.; Massari, M.; Cavallo, M.C. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat. Med. 2017, 23, 327. [Google Scholar] [CrossRef]
- Tarancon-Diez, L.; Rodríguez-Gallego, E.; Rull, A.; Peraire, J.; Viladés, C.; Portilla, I.; Jimenez-Leon, M.R.; Alba, V.; Herrero, P.; Leal, M. Immunometabolism is a key factor for the persistent spontaneous elite control of HIV-1 infection. EBioMedicine 2019, 42, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Vardhana, S.A.; Hwee, M.A.; Berisa, M.; Wells, D.K.; Yost, K.E.; King, B.; Smith, M.; Herrera, P.S.; Chang, H.Y.; Satpathy, A.T. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 2020, 21, 1022–1033. [Google Scholar] [CrossRef]
- Flint, D.H.; Tuminello, J.; Emptage, M. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 1993, 268, 22369–22376. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollizzi, K.N.; Patel, C.H.; Sun, I.-H.; Oh, M.-H.; Waickman, A.T.; Wen, J.; Delgoffe, G.M.; Powell, J.D. mTORC1 and mTORC2 selectively regulate CD8+ T cell differentiation. J. Clin. Investig. 2015, 125, 2090–2108. [Google Scholar] [CrossRef]
- O’Brien, T.F.; Gorentla, B.K.; Xie, D.; Srivatsan, S.; McLeod, I.X.; He, Y.W.; Zhong, X.P. Regulation of T-cell survival and mitochondrial homeostasis by TSC1. Eur. J. Immunol. 2011, 41, 3361–3370. [Google Scholar] [CrossRef] [PubMed]
- Barili, V.; Fisicaro, P.; Montanini, B.; Acerbi, G.; Filippi, A.; Forleo, G.; Romualdi, C.; Ferracin, M.; Guerrieri, F.; Pedrazzi, G. Targeting p53 and histone methyltransferases restores exhausted CD8+ T cells in HCV infection. Nat. Commun. 2020, 11, 604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A.N.-U.; Liu, J.; Mujib, S.; Kidane, S.; Ali, A.; Szep, S.; Han, C.; Bonner, P.; Parsons, M.; Benko, E. Elevated glycolysis imparts functional ability to CD8+ T cells in HIV infection. Life Sci. Alliance 2021, 4, e202101081. [Google Scholar] [CrossRef]
- Kavanagh Williamson, M.; Coombes, N.; Juszczak, F.; Athanasopoulos, M.; Khan, M.B.; Eykyn, T.R.; Srenathan, U.; Taams, L.S.; Dias Zeidler, J.; Da Poian, A.T. Upregulation of glucose uptake and hexokinase activity of primary human CD4+ T cells in response to infection with HIV-1. Viruses 2018, 10, 114. [Google Scholar] [CrossRef] [Green Version]
- Lan, X.; Cheng, K.; Chandel, N.; Lederman, R.; Jhaveri, A.; Husain, M.; Malhotra, A.; Singhal, P.C. High glucose enhances HIV entry into T cells through upregulation of CXCR4. J. Leukoc. Biol. 2013, 94, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Loisel-Meyer, S.; Swainson, L.; Craveiro, M.; Oburoglu, L.; Mongellaz, C.; Costa, C.; Martinez, M.; Cosset, F.-L.; Battini, J.-L.; Herzenberg, L.A. Glut1-mediated glucose transport regulates HIV infection. Proc. Natl. Acad. Sci. USA 2012, 109, 2549–2554. [Google Scholar] [CrossRef] [Green Version]
- Hegedus, A.; Kavanagh Williamson, M.; Huthoff, H. HIV-1 pathogenicity and virion production are dependent on the metabolic phenotype of activated CD4+ T cells. Retrovirology 2014, 11, 98. [Google Scholar] [CrossRef] [Green Version]
- Hollenbaugh, J.A.; Munger, J.; Kim, B. Metabolite profiles of human immunodeficiency virus infected CD4+ T cells and macrophages using LC–MS/MS analysis. Virology 2011, 415, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arunagiri, C.; Macreadie, I.; Hewish, D.; Azad, A. A C-terminal domain of HIV-1 accessory protein Vpr is involved in penetration, mitochondrial dysfunction and apoptosis of human CD4+ lymphocytes. Apoptosis 1997, 2, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Chiang, S.-F.; Lin, T.-Y.; Chiou, S.-H.; Chow, K.-C. HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction. PLoS ONE 2012, 7, e33657. [Google Scholar] [CrossRef] [PubMed]
- Deniaud, A.; Brenner, C.; Kroemer, G. Mitochondrial membrane permeabilization by HIV-1 Vpr. Mitochondrion 2004, 4, 223–233. [Google Scholar] [CrossRef]
- White, A.J. Mitochondrial toxicity and HIV therapy. Sex. Transm. Infect. 2001, 77, 158–173. [Google Scholar] [CrossRef] [Green Version]
- Korencak, M.; Byrne, M.; Richter, E.; Schultz, B.T.; Juszczak, P.; Ake, J.A.; Ganesan, A.; Okulicz, J.F.; Robb, M.L.; de Los Reyes, B. Effect of HIV infection and antiretroviral therapy on immune cellular functions. JCI Insight 2019, 4, e126675. [Google Scholar] [CrossRef] [Green Version]
- Plana, M.; García, F.; Gallart, T.; Tortajada, C.; Soriano, A.; Palou, E.; Maleno, M.J.; Barceló, J.J.; Vidal, C.; Cruceta, A. Immunological benefits of antiretroviral therapy in very early stages of asymptomatic chronic HIV-1 infection. Aids 2000, 14, 1921–1933. [Google Scholar] [CrossRef]
- Calvet-Mirabent, M.; Sánchez-Cerrillo, I.; Martín-Cófreces, N.; Martínez-Fleta, P.; de la Fuente, H.; Tsukalov, I.; Delgado-Arévalo, C.; Calzada, M.J.; Santos, I.; Sanz, J.; et al. Antiretroviral therapy duration and immunometabolic state determine efficacy of ex vivo dendritic cell-based treatment restoring functional HIV-specific CD8+ T cells in people living with HIV. EBioMedicine 2022, 81, 104090. [Google Scholar] [CrossRef]
- Pernas, M.; Tarancon-Diez, L.; Rodriguez-Gallego, E.; Gomez, J.; Prado, J.G.; Casado, C.; Dominguez-Molina, B.; Olivares, I.; Coiras, M.; Leon, A. Factors leading to the loss of natural elite control of HIV-1 infection. J. Virol. 2018, 92, e01805–e01817. [Google Scholar] [CrossRef] [Green Version]
- Akusjärvi, S.S.; Ambikan, A.T.; Krishnan, S.; Gupta, S.; Sperk, M.; Végvári, Á.; Mikaeloff, F.; Healy, K.; Vesterbacka, J.; Nowak, P. Integrative proteo-transcriptomic and immunophenotyping signatures of HIV-1 elite control phenotype: A cross-talk between glycolysis and HIF signaling. Iscience 2022, 25, 103607. [Google Scholar] [CrossRef]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J. The mTOR complex controls HIV latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, F.Z.; Ouyang, Z.; Buzon, M.; Walker, B.D.; Lichterfeld, M.; Yu, X.G. Metabolic pathway activation distinguishes transcriptional signatures of CD8+ T cells from HIV-1 elite controllers. AIDS 2018, 32, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Angin, M.; Volant, S.; Passaes, C.; Lecuroux, C.; Monceaux, V.; Dillies, M.A.; Valle-Casuso, J.C.; Pancino, G.; Vaslin, B.; Le Grand, R.; et al. Metabolic plasticity of HIV-specific CD8(+) T cells is associated with enhanced antiviral potential and natural control of HIV-1 infection. Nat. Metab. 2019, 1, 704–716. [Google Scholar] [CrossRef]
- Taylor, H.E.; Calantone, N.A.; D’Aquila, R.T. mTOR signaling mediates effects of common gamma-chain cytokines on T cell proliferation and exhaustion: Implications for HIV-1 persistence and cure research. AIDS 2018, 32, 2847–2851. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.-J. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [Green Version]
- Dumauthioz, N.; Tschumi, B.; Wenes, M.; Marti, B.; Wang, H.; Franco, F.; Li, W.; Lopez-Mejia, I.C.; Fajas, L.; Ho, P.-C. Enforced PGC-1α expression promotes CD8 T cell fitness, memory formation and antitumor immunity. Cell. Mol. Immunol. 2020, 18, 1761–1771. [Google Scholar] [CrossRef] [Green Version]
- Mylvaganam, G.H.; Chea, L.S.; Tharp, G.K.; Hicks, S.; Velu, V.; Iyer, S.S.; Deleage, C.; Estes, J.D.; Bosinger, S.E.; Freeman, G.J. Combination anti–PD-1 and antiretroviral therapy provides therapeutic benefit against SIV. JCI Insight 2018, 3, e122940. [Google Scholar] [CrossRef]
- Fromentin, R.; DaFonseca, S.; Costiniuk, C.T.; El-Far, M.; Procopio, F.A.; Hecht, F.M.; Hoh, R.; Deeks, S.G.; Hazuda, D.J.; Lewin, S.R. PD-1 blockade potentiates HIV latency reversal ex vivo in CD4+ T cells from ART-suppressed individuals. Nat. Commun. 2019, 10, 814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shikuma, C.M.; Chew, G.M.; Kohorn, L.; Souza, S.A.; Chow, D.; SahBandar, I.N.; Park, E.-Y.; Hanks, N.; Gangcuangco, L.M.A.; Gerschenson, M. metformin reduces CD4 T cell exhaustion in HIV-infected adults on suppressive antiretroviral therapy. AIDS Res. Hum. Retrovir. 2020, 36, 303–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, T.R.; Hanna, D.B.; Kaplan, R.C.; Xue, X.; Kizer, J.R.; Durkin, H.G.; Kassaye, S.G.; Nowicki, M.; Tien, P.C.; Topper, E.T. Elevated CD4+ T-cell glucose metabolism in HIV+ women with diabetes mellitus. AIDS 2022, 36, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Moyo, D.; Tanthuma, G.; Cary, M.; Mushisha, O.; Kwadiba, G.; Chikuse, F.; Steenhoff, A.; Reid, M. Cohort study of diabetes in HIV-infected adult patients: Evaluating the effect of diabetes mellitus on immune reconstitution. Diabetes Res. Clin. Pract. 2014, 103, e34–e36. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, Y.T.; Cheong, H.C.; Tang, T.F.; Rajasuriar, R.; Cheng, K.-K.; Looi, C.Y.; Wong, W.F.; Kamarulzaman, A. Immune Checkpoint Molecules and Glucose Metabolism in HIV-Induced T Cell Exhaustion. Biomedicines 2022, 10, 2809. https://doi.org/10.3390/biomedicines10112809
Chan YT, Cheong HC, Tang TF, Rajasuriar R, Cheng K-K, Looi CY, Wong WF, Kamarulzaman A. Immune Checkpoint Molecules and Glucose Metabolism in HIV-Induced T Cell Exhaustion. Biomedicines. 2022; 10(11):2809. https://doi.org/10.3390/biomedicines10112809
Chicago/Turabian StyleChan, Yee Teng, Heng Choon Cheong, Ting Fang Tang, Reena Rajasuriar, Kian-Kai Cheng, Chung Yeng Looi, Won Fen Wong, and Adeeba Kamarulzaman. 2022. "Immune Checkpoint Molecules and Glucose Metabolism in HIV-Induced T Cell Exhaustion" Biomedicines 10, no. 11: 2809. https://doi.org/10.3390/biomedicines10112809
APA StyleChan, Y. T., Cheong, H. C., Tang, T. F., Rajasuriar, R., Cheng, K.-K., Looi, C. Y., Wong, W. F., & Kamarulzaman, A. (2022). Immune Checkpoint Molecules and Glucose Metabolism in HIV-Induced T Cell Exhaustion. Biomedicines, 10(11), 2809. https://doi.org/10.3390/biomedicines10112809