1. Introduction
The eukaryotic mRNAs with poly(A) tails bind to sequence-specific poly(A)-binding proteins, mediating synthesis of these tails [
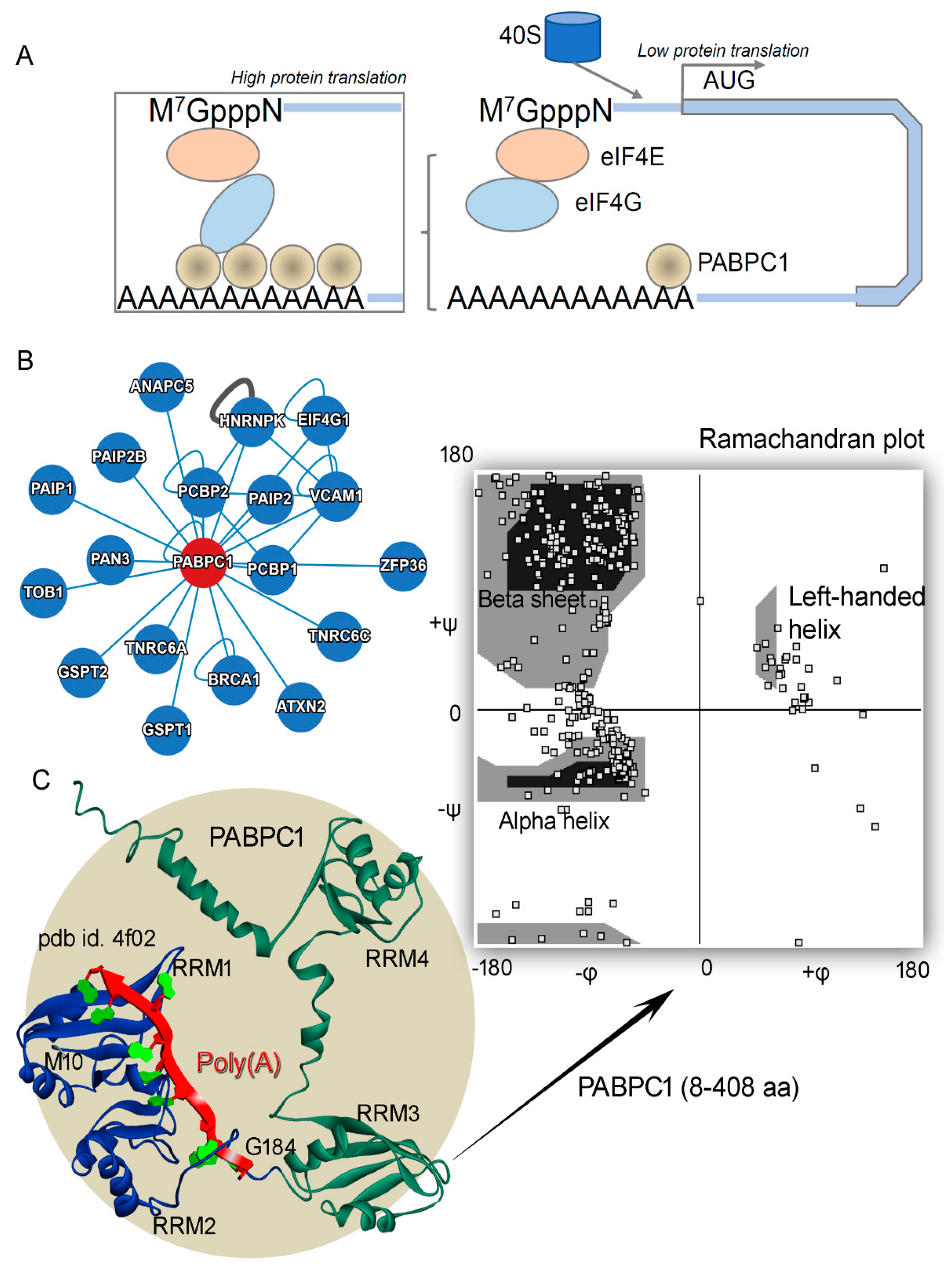
1]. One such protein is poly(A)-binding protein cytoplasmic 1 (PAB1 or PABPC1), associated with the long poly(A) mRNA tails inducing its stability. The PABPC1 protein, with a molecular weight of ~70 kDa, consists of four RNA recognition motifs (RRMs) linked through linker amino acids [
1,
2]. The “faux 3′-UTR” model explains that the proximity of the PABPC1 to the PTC (premature termination codons) is essential for NMD (nonsense-mediated mRNA decay) activation [
2,
3]. For poly(A) shortening, ribosome recruitment, and translation initiation, the PABPC protein plays a crucial role by binding directly to the poly(A) tail of mRNA in cytoplasm [
1]. Along with high specific binding to poly(A) mRNA, the PABPC1 protein is found binding with lower affinity to poly(U) and poly(G) [
4,
5] Among different mechanisms of PABPC is the stimulation of initiation of translation by binding with the eIF4G protein (eukaryotic translation initiation factor 4 G), thus promoting the recruitment of 40S ribosome subunits similar as the eIF4E gene [
6,
7,
8]. Such binding of PABPC1-eIF4G is found to be conserved in different species [
1]. The conformational switch of the eIF4G gene depends on the PAB1 expression; i.e., in normal cells having low PAB1 expression, the binding of eIF4G-PAB1 is lacking (
Figure 1), whereas in cancer cells with high PAB1 expression, binding is induced between these proteins [
9,
10].
Though the PABPC1 protein is proposed to be an antagonist of UPF1 (UP-frameshift 1) from the NMD pathway towards its mRNA activity, it has been found that deletion of either UPF1 or PAB1 significantly increases the production of novel peptide read-through [
11,
12,
13]. Crucial roles of the poly(A)-binding protein for regulation in the translation termination have been investigated [
11], and inefficient termination can result in the activation of the NMD process (initiating mRNA degradation). Different biochemical and structural studies have reported the binding patterns between the RNA recognition motifs with poly(A) mRNA, as well as interaction with different components [
2,
14]. However, detailed insights are still needed for the structural folding and dynamics of the full-length human PABPC1 protein. Due to significant correlation between mRNA degradation and stabilization, herein, we investigated the PABPC1 protein’s dynamics, along with tracing mRNA binding specificity (
Figure 1A). Additionally, considering different cancer-derived mutations for the PABPC1 protein, we presented an overview of different hotspots [
15], along with measuring the change in stability of the structure upon inserting point mutations. Monitoring the mRNA selectivity for PAB1, our findings suggest that aromatic amino acids (Y or W) are associated extensively with the poly(A) mRNA (
Figure 1).
2. Materials and Methods
The crystal structure of PABPC1 containing the RRM1–2 domains with the poly(A) mRNA (pdb id: 4f02 [
2] in blue; M10–G184 aa;
Figure 1) was retrieved from the protein data bank (pdb;
www.rcsb.org accessed on 3 May 2021). The modeled human PABPC1 structure containing all four RRMs was built using the SWISS-MODEL homology modeling approach [
16,
17]. For generating human PABPC1 (RRM1-4), the
Saccharomyces cerevisiae PAB1 cryo-EM (cryogenic electron microscopy) structure from the 90A RNP-Pan2-Pan3 complex (pdb id.: 6r5k [
14]) was considered as a template structure. Superimposing of crystal structure with the modeled PAB1 tertiary structure (
Figure 1) was performed using the BIOVIA Discovery Studio (Dassault Systèmes, BIOVIA Corp., San Diego, CA, USA) pipeline. Different PABPC1 models in the presence or absence of the poly(A) mRNA were energy-minimized via applying CHARMM27 (Chemistry at Harvard Macromolecular Mechanics) forcefield in the Molecular Operating Environment (MOE; Chemical Computing Group Inc., Montreal, QC, Canada) package. Moreover, retrieving coordinates from the PAB1-simulated structure, the active sites were predicted using the “Alpha Shapes” construction geometric method implemented in the MOE modules (Chemical Computing Group Inc., Montreal, QC, Canada) [
18]. The “Alpha Shapes” technique allowed us to construct the binding site of a protein, classified in the alpha sphere form as either “hydrophobic” or “hydrophilic (for lone pair active; LPA)”.
PABPC1 mutations retrieved from the cBioPortal database [
19] were represented over the structure in the BIOVIA Discovery Studio (Dassault Systèmes, BIOVIA Corp., San Diego, CA, USA). Moreover, expression profiles of specific genes within a single dataset (type 1 diabetes (T1D) autoimmune disease), compared with the control samples (GSE60424, Neutrophils; GSE60424, NK; and GSE60424, T cells [
20]), were retrieved from the ADEx database (
https://adex.genyo.es/ accessed on 3 November 2022). The SWISS-MODEL-modeled PABPC1 structure orientation resembles that generated by the Alpha Fold tool [
21] (retrieved from Uniport;
https://www.uniprot.org/ accessed on 3 May 2021). Superimposing both structures (SWISS-MODEL [
16,
17]) and “Alpha Fold” tool [
21]), and tracing stability of individual residues were performed in the MOE (Chemical Computing Group Inc., Montreal, QC, Canada) package.
Frequently occurring mutations for the PABPC1 protein in different cancer types were investigated for the stability change of the protein. Individual variants were inserted in the PABPC1 structure, along with the predicted change in the stability (∆Stability or dStability, kcal/mol) using the “residue scan” module from the MOE (Chemical Computing Group Inc., Montreal, QC, Canada) package. During implementing the “residue scan” over the PABPC1 protein, the “LowModeMD” ensemble was used, and the CHARMM27 forcefield was applied [
22]. In addition, the following parameters were set: 10,000 K search conformations and 50 rounds of iterations. The dStability for a particular mutation is the relative thermostability of the wild-type residue (Boltzmann average).
The PABPC1 protein in apo-form (crystal structure, pdb id: 4f02 [
2]; the modeled system) as well as poly(A) mRNA were investigated further using the molecular dynamics simulation (MDS) technique. MD simulations on each system were performed using the GROMACS 4.6.5 (GROningen MAchine for Chemical Simulations) package [
23] and applying the CHARMM27 forcefield. Individual systems were solvated in water (simple point charge model; SPC) containing N
+Cl
--neutralizing ions, and in a 10 Å-thick dodecahedron simulation box. In this dodecahedron box, the periodic boundary conditions (PBCs) were implemented and systems with the solvent molecules were energy-minimized using the steepest descent algorithm. The Particle Mesh Ewald (PME) method [
24] and the LINCS algorithm [
25] were used to maintain electrostatic interactions (van der Waals and Coulomb interaction cut-off distance was set to 10 Å) and constrain bond lengths, respectively. After equilibrating systems for 1000 ps (in NPT isobaric–isothermal ensemble simulation), the production run was performed for 100 ns using the leapfrog integrator [
26]. The V-rescale thermostat [
25] was used to maintain the temperature of each system at 300 K. In addition, the Parrinello–Rahman barostat [
23] was used to maintain the pressure at 1 bar. The PABPC1-poly(A) mRNA hydrogen bonding interactions were computed using the GROMACS and VMD tools [
15], maintaining the donor–acceptor distance at 3.5 Å and angle cut-off at ≥160°–180°. Protein structure details retrieved from the molecular dynamics or molecular modeling were visualized using the VMD [
15], MOE (Chemical Computing Group Inc., Montreal, QC, Canada), and BIOVIA Discovery Studio (Dassault Systèmes, BIOVIA Corp., San Diego, CA, USA) packages.
3. Results and Discussion
In cancer cells, high expression of the PABPC1 protein can induce binding with different proteins (e.g., eIF4G;
Figure 1B), and several such binding partners were elucidated by the cryo-EM or crystal studies [
1,
2,
14]. However, the dynamics of full-length human PAB1 is the direction that needed further investigation, and therefore, using homology modeling along with molecular dynamics techniques, we measured structural properties of this gene (
Figure 2). The crystal structure of the PAB1 protein consists of only two RNA recognition motifs (RRMs; RRM1–2 domains) with poly(A) mRNA (pdb id: 4f02 [
2]). To implement the homology modeling, we modeled the possible full-length structure of all four RNA recognition motifs (RRM1–4;
Figure 1C and
Figure 2). The modeled PAB1-mRNA and apo systems were optimized or energy-minimized by applying the CHARMM27 forcefield in the MOE package (Chemical Computing Group Inc., Montreal, QC, Canada) [
22]. The “Ramachandran plot” demonstrated in
Figure 1C highlights the well-defined secondary structures that were modeled for the protein. Moreover, this modeled PABPC1 structure orientation resembled that generated by the “Alpha Fold” tool [
21] (retrieved from Uniport;
www.uniprot.org accessed on 3 May 2021). Superimposing both structures (SWISS-MODEL [
16,
17]) and “Alpha Fold” tool [
21]) showed that when the RRM4 domain was excluded, the majority of the structure had less flexibility (
Figure 3). These structures were further investigated using the MD simulation approach, along with considering different systems with the poly(A) mRNA.
Measuring the stability based on the RMSFs (root-mean-square fluctuation) or RMSDs (root-mean-square deviation, excluding hydrogen atoms) for the PABPC1 structure demonstrated that the presence of poly(A) mRNA induced stability within the residues (
Figure 2A,B). In addition, the modeled PAB1 structure (8–408 aa; RRM1-4 domains) obtained a pattern of fluctuations similar to that of other simulated crystal structures consisting of RRM1–2 domains (10–184 aa; pdb id.: 4f02 [
2];
Figure 2A and
Videos S1–S2). In particular, the poly(A) mRNA was found to be changing its conformations, which stabilized by the end of the MD simulation time (
Figure 2B). For the modeled apo-form of PAB1 (
Model S1 and Video S3), the R176-Y408 residues formed a folded structure after a large displacement, as shown in
Figure 2C and
Video S3 (or Model S1), whereas the RRM1–2 domains were found to be conserved and less flexible, almost positioned like those in the crystal structure [
2]. In comparing different conformations from MD simulation for PAB1 in the presence or absence of the mRNA motif, it was observed that the region binding with the poly(A) mRNA was more stable (
Figure 2D) and gained a slightly different conformation compared to the apo-form.
Visualizing significant conformational folding in the RRM3–4 domains, we further investigated changes in the structural properties of these domains (RRM1–4;
Figure 3). Extracting RMSDs from only the RRM3–4 domains (
Figure 3B), it was observed that the majority of the protein flexibility emerged from these two RRMs within the PABPC1 structure. In addition, the intramolecular interactions among the RRM3–4 (180–408 aa) domains significantly increased over time (
Figure 3B). RRM4 domain residues were mainly involved in high-occupancy intramolecular interactions (
Figure 3C), despite their high flexibility (
Figure 3D). In addition to conformational change, the surface hydrophobicity (mostly being hydrophilic) showed fluctuations (
Figure 3D) in the PABPC1 apo-form.
The intermolecular interactions between poly(A) mRNA-PABPC1 suggest that ~8 H-bonds were formed every nanosecond (ns), and the PAB1 amino acids were found to be distributed over the mRNA (
Figure 4A). The PAB1 residues D45, Y54, Y56, N58, Q88, and N100 formed long-lasting interactions having an occupancy ≥40% with the mRNA (
Figure 4B). In particular, the poly(A) mRNA showed a unique pattern in the presence of PAB1. Every second nucleotide from both 5′ and 3′ ends was in an “inward-position” facing towards PAB1, whereas every third nucleotide had an “outward-position” conformation. The centered three nucleotides were found facing towards the region between RRM1–2 domains of the PABPC1 protein (
Figure 2A,B). Specifically, aromatic amino acids from the PABPC1 protein (Y14, Y54, Y56, W86, and Y140) were found binding with poly(A) mRNA. These findings correlate with previously reported R94 and R179 residues involved in hydrogen bond interaction with the mRNA [
9].
PABPC1 has been shown to perform crucial roles in cancer and autoimmune diseases (ADs). Particularly, induced neoantigen production may lead to immune cell infiltration in the ADs, whereas in cancer cells, there is a lack of immune cell activation [
11,
27]. Deletion of PABPC1 protein in cancer cells could significantly increase the production of novel peptide read-through [
11,
12,
13]. Moreover, the weighted gene co-expression network analysis (WGCNA) over microarray samples collected from ischemic stroke (IS) females revealed the expression profile of different genes, including the PABPC1 protein [
27]. It has been proposed that in IS patients, a set of genes are linked to T cells, macrophages, NK cells, and neutrophils. In the samples studied by Haipeng et al. [
27], it was demonstrated that the primary immune-infiltrating cells were neutrophils, resting NK cells, macrophages, and CD8 T cells. In particular, NK cells (large granular lymphocytes needed for IS immunosurveillance) blocking CD8 T cell activation were found regulating cellular immune response. Considering such involvement of PABPC1 in cell infiltration, we retrieved protein expression data for autoimmune diseases from the ADEx database (Autoimmune Diseases Explorer;
https://adex.genyo.es/ accessed on 3 November 2022) [
20]. This database covers common organ-specific autoimmune diseases (from >5000 samples), type 1 diabetes (T1D), rheumatoid arthritis (RA), etc. In particular, the expression profiles of PABPC1 in T1D from (GSE60424; Neutrophils, GSE60424; NK, and GSE60424, T cells [
20]) different datasets are presented in
Figure 5.
Moreover, the dataset describing or containing mutation frequency of individual amino acids from different cancer types was retrieved from the cBioPortal (
Figure 5A) [
19]. The majority of the PABPC1 point mutations in cancer reside within the RRM domains, and in particular, the regions within 300350 aa (RRM4 domain), have high mutation frequency (
Figure 5A). Comparing these data with the hydrogen-binding residues of the poly(A) mRNA, it was observed that amino acids engaging in long-lasting interactions were found to be preserved in different cancer types (
Figure 5A); i.e., none of them were found to be mutated in the retrieved cancer dataset. Furthermore, mutations with high frequency (>4) were investigated to trace changes in the stability of the protein structure (
Figure 6). However, in the G123C variant, the majority of the cancer-derived mutants reduced the stability of the protein (
Figure 6B). In particular, both R331H and R331C mutations (
Figure 6B) significantly disrupted the PABPC1 structure.
In addition, one of the active sites (from three active sites built using the “Alpha Shapes” approach) originated in the region (300–350 aa; RRM4 domain) that is highly mutated in different cancer types (
Figure 7A and
Table 1). For most of the eukaryotic mRNAs, stability depends on a ribonucleoprotein, and PAB1 is known to be bound with the poly(A) tail [
14]. This PAB1 protein at the poly(A) tail can bind with different proteins, gaining slight structural conformations or movements. Cryo-EM data have shown multiple binding conformations or structural folding of the PABPC1 protein in the 90A RNP-Pan2-Pan3 complex [
14]. Different structural movements observed in our MD simulations can be directly correlated with such dynamics traced in the cryo-EM studies [
14]. Our initial conformation from beginning of MD simulation was almost identical to the second PAB1 (as shown in
Figure 7B), and structure from end of MD simulation correlates with the first PAB1 (
Figure 7B) molecule. Moreover, it is known that in eukaryotic cells, diverse stresses can enhance coalescence of RNA-binding proteins, making them stress granules. Similarly, the PABPC1 protein has been suggested as a defined marker of stress granules under physiological stress conditions [
28]. Since multiple conformations were observed in our MD simulations, it could be hypothesized that in cellular stress, PABPC1 could represent distinct folding, affecting the predicted active sites (
Figure 7A and
Table 1). However, we believe that in non-stress conditions, PABPC1 may form the active sites presented in
Figure 7.
4. Conclusions
The PAB1 or PABPC1 protein is proposed to induce mRNA stability; hence, it is suggested as an antagonist of the NMD factors when targeting mRNA Despite this, it has been found that deletion of either PAB1 or UPF1 (NMD) significantly increases the production of novel peptide read-through. This could result in the increased production of mutant peptides that can be presented over the HLA molecules and could trigger the immune response in cancer. Hence, applying different target inhibition strategies to completely or partially block the activity of PAB1 may result in induced production of peptide read-through over mRNA, which eventually produces mutant peptide or neoantigens in cancer cells. A detailed understanding of the PAB1 protein structure can guide such experimental designs to change the activity of this protein. Herein, we investigated the structural folding or dynamics (correlating with the cryo-EM) of the human PAB1 protein, and proposed several key residues involved in mRNA binding, as well as highlighted different active sites.
Applying the homology modeling techniques, we modeled and optimized the possible full-length structure of all four RRMs for the PAB1 protein. For this PAB1 model, it has been observed that the residue range R176-Y408 initiated a folded structure after a large displacement. The D45, Y54, Y56, N58, Q88, and N100 residues formed long-lasting high-occupancy interactions with the poly(A) mRNA. Monitoring the specific mRNA selectivity for PAB1, we reviewed aromatic amino acids (Y or W) associated extensively with the poly(A) mRNA. On the other hand, the poly(A) mRNA has shown a unique pattern towards the PAB1 protein. Every second nucleotide from both the 5′ or 3′ ends was in an “inward-position” facing towards protein, whereas every third nucleotide had an “outward-position” conformation, and the center three nucleotides faced towards the region between RRM1–2 domains. Moreover, residues from the RRM4 domain were mainly involved in high-occupancy intramolecular interactions, despite high flexibility within its residue. The majority of the high-frequency cancer mutations in PAB1 reside within the RRM domains, and in particular, the regions within 300–350 aa (RRM4) have high mutation frequency. Amino acids engaging in long-lasting interactions with poly(A) mRNA were found to be preserved in different cancer types. However, in the G123C variant, the majority of the cancer derived mutants reduced the stability of the protein. We believe that the molecular details from this study provide a detailed understanding of the PABPC1 structure, and can guide future in vitro or in vivo experiments to modulate the activity of this gene.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}