
Growth Arrest of Alveolar Cells in Response to Cytokines from Spike S1-Activated Macrophages: Role of IFN-γ
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Models
2.2. Experimental Treatments
2.3. Cell Proliferation and Viability Assay
2.4. Cell Cycle Analysis
2.5. Autophagy Detection
2.6. Determination of the Intracellular Amino Acid Content
2.7. Amino Acid Uptake
2.8. RT-qPCR Analysis
2.9. Cytokine Analysis
2.10. Western Blot Analysis
2.11. Statistical Analysis
2.12. Materials
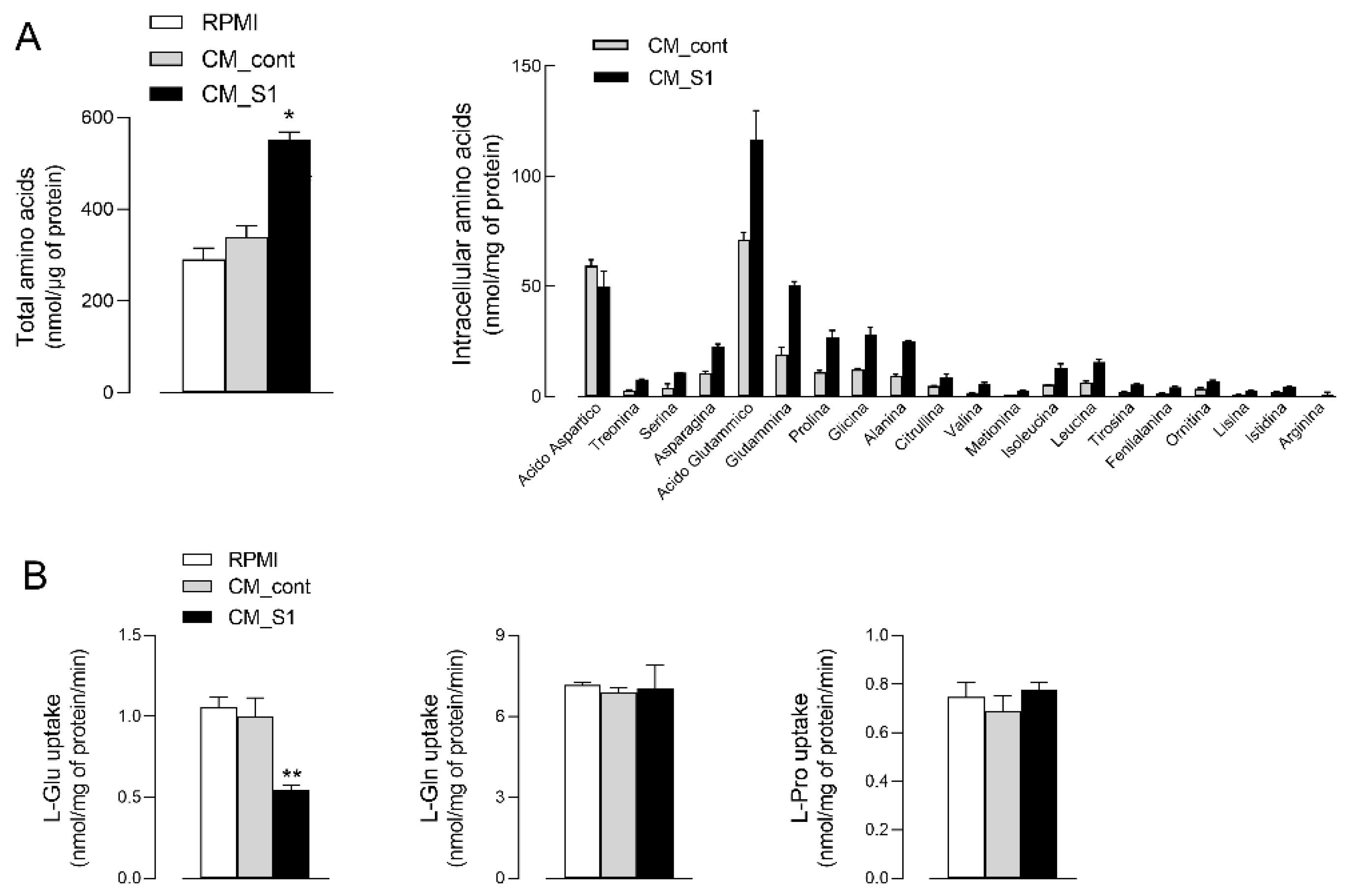
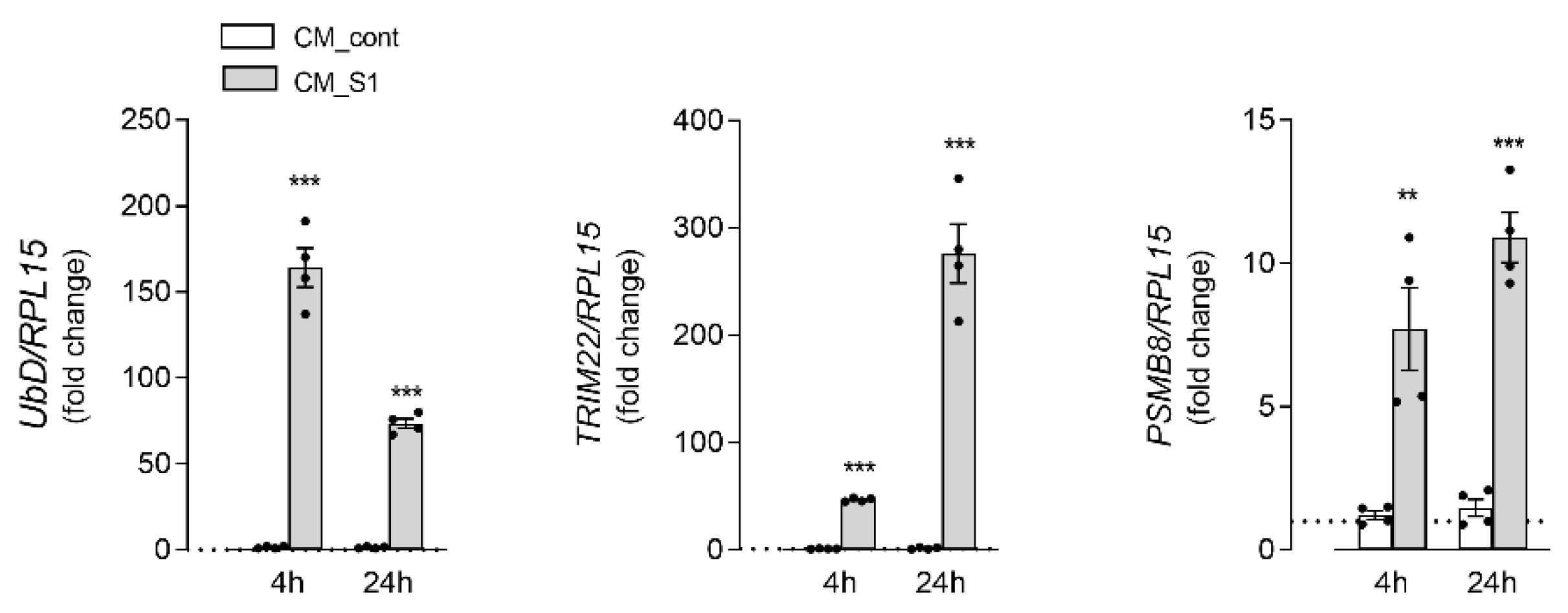
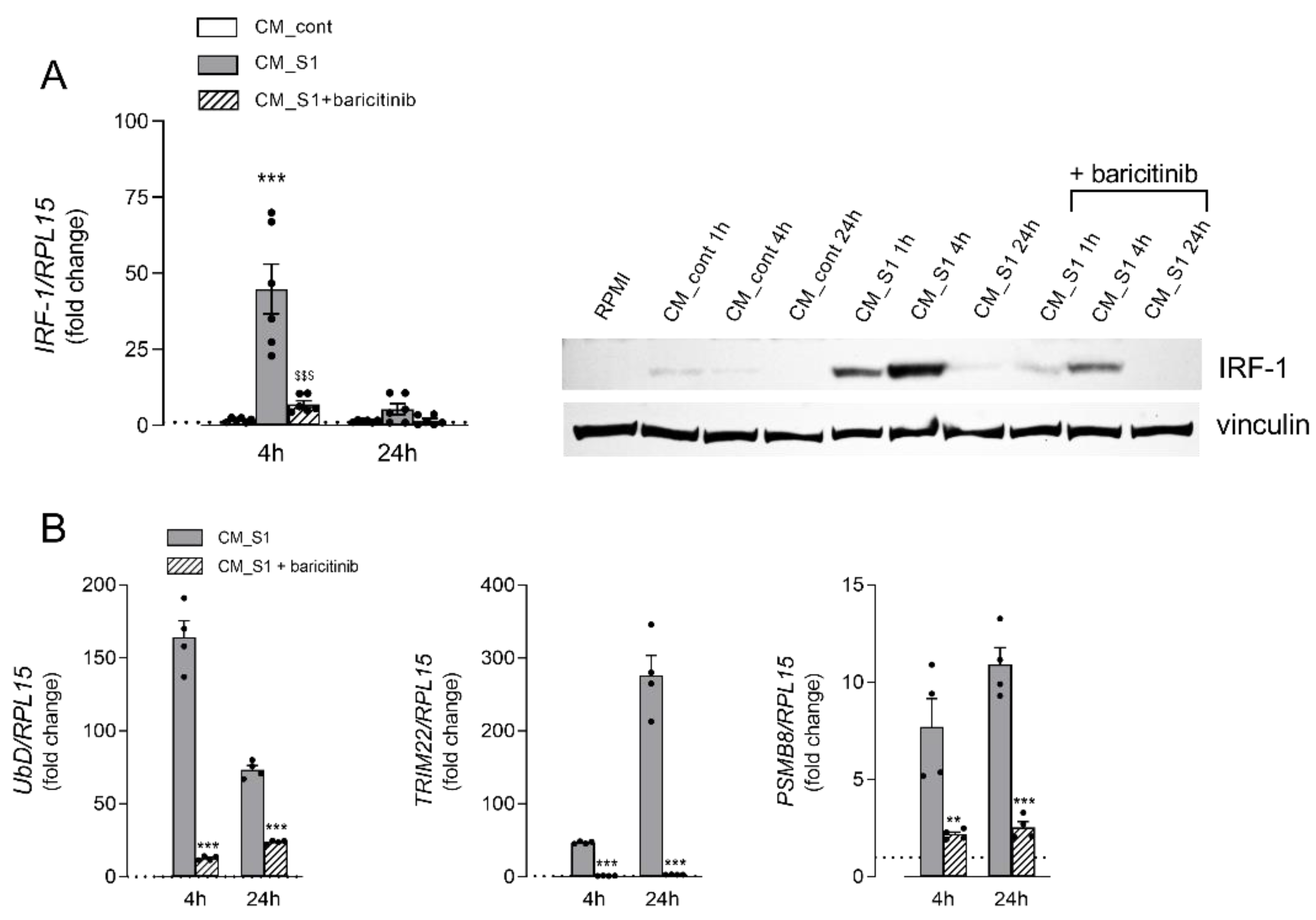
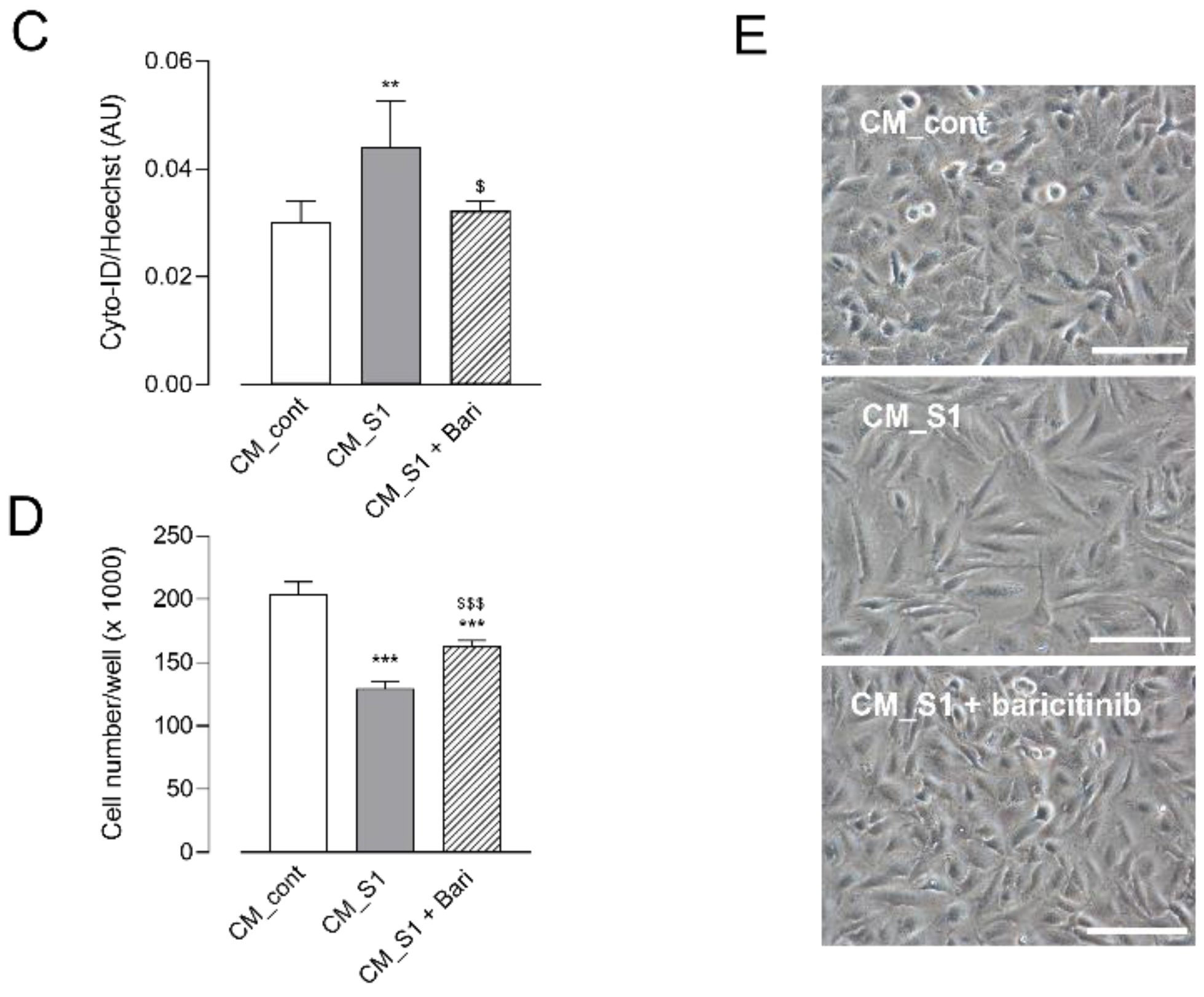
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aslan, A.; Aslan, C.; Zolbanin, N.M.; Jafari, R. Acute respiratory distress syndrome in COVID-19: Possible mechanisms and therapeutic management. Pneumonia 2021, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in wuhan, china. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, N.J.; Gattinoni, L.; Calfee, C.S. Acute respiratory distress syndrome. Lancet 2021, 398, 622–637. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Prim. 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Bradley, B.T.; Maioli, H.; Johnston, R.; Chaudhry, I.; Fink, S.L.; Xu, H.; Najafian, B.; Deutsch, G.; Lacy, J.M.; Williams, T.; et al. Histopathology and ultrastructural findings of fatal COVID-19 infections in washington state: A case series. Lancet 2020, 396, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Gandet, X.D.; et al. High risk of thrombosis in patients with severe Sars-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Pu, D.; Zhai, X.; Zhou, Y.; Xie, Y.; Tang, L.; Yin, L.; Liu, H.; Li, L. A narrative review of COVID-19-related acute respiratory distress syndrome (cards): “Typical” or “atypical” ards? Ann. Transl. Med. 2022, 10, 908. [Google Scholar] [CrossRef]
- Batah, S.S.; Fabro, A.T. Pulmonary pathology of ards in COVID-19: A pathological review for clinicians. Respir. Med. 2021, 176, 106239. [Google Scholar] [CrossRef]
- Ragab, D.; Eldin, H.S.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 cytokine storm; what we know so far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Kosyreva, A.; Dzhalilova, D.; Lokhonina, A.; Vishnyakova, P.; Fatkhudinov, T. The role of macrophages in the pathogenesis of SARS-CoV-2-associated acute respiratory distress syndrome. Front. Immunol. 2021, 12, 682871. [Google Scholar] [CrossRef]
- Wang, J.; Yang, X.; Li, Y.; Huang, J.-A.; Jiang, J.; Su, N. Specific cytokines in the inflammatory cytokine storm of patients with COVID-19-associated acute respiratory distress syndrome and extrapulmonary multiple-organ dysfunction. Virol. J. 2021, 18, 117. [Google Scholar] [CrossRef] [PubMed]
- Boroujeni, M.E.; Sekrecka, A.; Antonczyk, A.; Hassani, S.; Sekrecki, M.; Nowicka, H.; Lopacinska, N.; Olya, A.; Kluzek, K.; Wesoly, J.; et al. Dysregulated interferon response and immune hyperactivation in severe COVID-19: Targeting stats as a novel therapeutic strategy. Front. Immunol. 2022, 13, 888897. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Ashkar, A.A. The dual nature of type i and type ii interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.E.; Kessler, D.S.; Pine, R.; Darnell, J.E., Jr. Cytoplasmic activation of isgf3, the positive regulator of interferon-alpha-stimulated transcription, reconstituted in vitro. Genes Dev. 1989, 3, 1362–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [Green Version]
- Kroger, A.; Koster, M.; Schroeder, K.; Hauser, H.; Mueller, P.P. Activities of irf-1. J. Interferon Cytokine Res. 2002, 22, 5–14. [Google Scholar] [CrossRef]
- Feng, H.; Zhang, Y.B.; Gui, J.F.; Lemon, S.M.; Yamane, D. Interferon regulatory factor 1 (irf1) and anti-pathogen innate immune responses. PLoS Pathog. 2021, 17, e1009220. [Google Scholar] [CrossRef]
- Barilli, A.; Visigalli, R.; Ferrari, F.; Bianchi, M.G.; Dall’Asta, V.; Rotoli, B.M. Immune-mediated inflammatory responses of alveolar epithelial cells: Implications for covid-19 lung pathology. Biomedicines 2022, 10, 618. [Google Scholar] [CrossRef]
- Rotoli, B.M.; Barilli, A.; Visigalli, R.; Ferrari, F.; Dall’Asta, V. Endothelial cell activation by sars-cov-2 spike s1 protein: A crosstalk between endothelium and innate immune cells. Biomedicines 2021, 9, 1220. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the alamar blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Barilli, A.; Visigalli, R.; Ferrari, F.; Borsani, G.; Dall’Asta, V.; Rotoli, B.M. Flagellin from pseudomonas aeruginosa stimulates atb(0,+) transporter for arginine and neutral amino acids in human airway epithelial cells. Front. Immunol. 2021, 12, 641563. [Google Scholar] [CrossRef] [PubMed]
- Ingoglia, F.; Visigalli, R.; Rotoli, B.M.; Barilli, A.; Riccardi, B.; Puccini, P.; Dall’Asta, V. Functional activity of l-carnitine transporters in human airway epithelial cells. Biochim. Biophys. Acta Biomembr. 2016, 1858, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Rotoli, B.M.; Barilli, A.; Visigalli, R.; Ferrari, F.; Dall’Asta, V. Y+lat1 and y+lat2 contribution to arginine uptake in different human cell models: Implications in the pathophysiology of lysinuric protein intolerance. J. Cell. Mol. Med. 2020, 24, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barilli, A.; Visigalli, R.; Ferrari, F.; Luciani, G.R.; Soli, M.; Dall’Asta, V.; Rotoli, B.M. The jak1/2 inhibitor baricitinib mitigates the spike-induced inflammatory response of immune and endothelial cells in vitro. Biomedicines 2022, 10, 2324. [Google Scholar] [CrossRef]
- Jorgensen, S.C.J.; Tse, C.L.Y.; Burry, L.; Dresser, L.D. Baricitinib: A review of pharmacology, safety, and emerging clinical experience in COVID-19. Pharmacotherapy 2020, 40, 843–856. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk between mammalian autophagy and the ubiquitin-proteasome system. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef] [Green Version]
- Buchsbaum, S.; Bercovich, B.; Ciechanover, A. Fat10 is a proteasomal degradation signal that is itself regulated by ubiquitination. Mol. Biol. Cell 2012, 23, 225–232. [Google Scholar] [CrossRef]
- Lou, J.; Wang, Y.; Zheng, X.; Qiu, W. Trim22 regulates macrophage autophagy and enhances mycobacterium tuberculosis clearance by targeting the nuclear factor-multiplicity kappab/beclin 1 pathway. J. Cell. Biochem. 2018, 119, 8971–8980. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, H.X.; Zhang, C.M.; Zou, M.; Zou, B.B.; Wei, W.; Hu, W. Foxo3/trim22 axis abated the antitumor effect of gemcitabine in non-small cell lung cancer via autophagy induction. Transl. Cancer Res. 2020, 9, 937–948. [Google Scholar] [CrossRef]
- Li, F.; Li, J.; Wang, P.H.; Yang, N.; Huang, J.; Ou, J.; Xu, T.; Zhao, X.; Liu, T.; Huang, X.; et al. SARS-CoV-2 spike promotes inflammation and apoptosis through autophagy by ros-suppressed pi3k/akt/mtor signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166260. [Google Scholar] [CrossRef]
- Ge, Y.; Huang, M.; Yao, Y.M. Autophagy and proinflammatory cytokines: Interactions and clinical implications. Cytokine Growth Factor Rev. 2018, 43, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Forero, A.; Ozarkar, S.; Li, H.; Lee, C.H.; Hemann, E.A.; Nadjsombati, M.S.; Hendricks, M.R.; So, L.; Green, R.; Roy, C.N.; et al. Differential activation of the transcription factor irf1 underlies the distinct immune responses elicited by type i and type iii interferons. Immunity 2019, 51, 451–464.e6. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Du, Q.; Cao, Z.; Guo, Z.; Evankovich, J.; Yan, W.; Chang, Y.; Shao, L.; Stolz, D.B.; Tsung, A.; et al. Interferon-gamma induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (hcc) cells through interferon-regulatory factor-1 (irf-1). Cancer Lett. 2012, 314, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.; Toyoda, S.; Nishitani, S.; Onodera, T.; Fukuda, S.; Kita, S.; Fukuhara, A.; Shimomura, I. SARS-CoV-2 infection impairs the insulin/igf signaling pathway in the lung, liver, adipose tissue, and pancreatic cells via irf1. Metabolism 2022, 133, 155236. [Google Scholar] [CrossRef]
- Farahani, M.; Niknam, Z.; Amirabad, L.M.; Amiri-Dashatan, N.; Koushki, M.; Nemati, M.; Pouya, F.D.; Rezaei-Tavirani, M.; Rasmi, Y.; Tayebi, L.; et al. Molecular pathways involved in covid-19 and potential pathway-based therapeutic targets. Biomed. Pharm. 2022, 145, 112420. [Google Scholar] [CrossRef]
- Luo, W.; Li, Y.X.; Jiang, L.J.; Chen, Q.; Wang, T.; Ye, D.W. Targeting jak-stat signaling to control cytokine release syndrome in covid-19. Trends Pharm. Sci. 2020, 41, 531–543. [Google Scholar] [CrossRef]
- Levy, G.; Guglielmelli, P.; Langmuir, P.; Constantinescu, S.N. Jak inhibitors and COVID-19. J. Immunother. Cancer 2022, 10, e002838. [Google Scholar] [CrossRef]
- Bronte, V.; Ugel, S.; Tinazzi, E.; Vella, A.; De Sanctis, F.; Cane, S.; Batani, V.; Trovato, R.; Fiore, A.; Petrova, V.; et al. Baricitinib restrains the immune dysregulation in patients with severe COVID-19. J. Clin. Investig. 2020, 130, 6409–6416. [Google Scholar] [CrossRef]
- Marconi, V.C.; Ramanan, A.V.; de Bono, S.; Kartman, C.E.; Krishnan, V.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Alatorre-Alexander, J.; Pellegrini, R.D.C.; et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (cov-barrier): A randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 2021, 9, 1407–1418. [Google Scholar] [CrossRef]
- Titanji, B.K.; Farley, M.M.; Mehta, A.; Connor-Schuler, R.; Moanna, A.; Cribbs, S.K.; O’Shea, J.; DeSilva, K.; Chan, B.; Edwards, A.; et al. Use of baricitinib in patients with moderate to severe coronavirus disease 2019. Clin. Infect. Dis. 2021, 72, 1247–1250. [Google Scholar]
- Kalil, A.C.; Stebbing, J. Baricitinib: The first immunomodulatory treatment to reduce COVID-19 mortality in a placebo-controlled trial. Lancet Respir. Med. 2021, 9, 1349–1351. [Google Scholar] [CrossRef] [PubMed]
- Ely, E.W.; Ramanan, A.V.; Kartman, C.E.; de Bono, S.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Saraiva, J.F.K.; Chakladar, S.; Marconi, V.C.; et al. Efficacy and safety of baricitinib plus standard of care for the treatment of critically ill hospitalised adults with COVID-19 on invasive mechanical ventilation or extracorporeal membrane oxygenation: An exploratory, randomised, placebo-controlled trial. Lancet Respir. Med. 2022, 10, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, A.; Ueki, I.; Min-Oo, G.; Ballon-Landa, E.; Knoff, D.; Galen, B.; Lanier, L.L.; Nadel, J.A.; Koff, J.L. Egfr activation suppresses respiratory virus-induced irf1-dependent cxcl10 production. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L186–L196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaheer, R.S.; Proud, D. Human rhinovirus-induced epithelial production of cxcl10 is dependent upon ifn regulatory factor-1. Am. J. Respir. Cell Mol. Biol. 2010, 43, 413–421. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (Gene ID) | Forward Primer | Reverse Primer |
|---|---|---|
| RPL15 (6138) | GCAGCCATCAGGTAAGCCAAG | AGCGGACCCTCAGAAGAAAGC |
| IRF1 (3659) | CTGTGCGAGTGTACCGGATG | ATCCCCACATGACTTCCTCTT |
| PSMB8 (5696) | CACGCTCGCCTTCAAGTTC | AGGCACTAATGTAGGACCCAG |
| TRIM22 (10346) | ACCAAACATTCCGCATAAACGA | AGGCGGTTCTCTCTTGTCTGA |
| UBD (10537) | GAAGCCTCTCATCTTATGGCATT | CCTCATCACCTGACTCCACAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barilli, A.; Visigalli, R.; Ferrari, F.; Recchia Luciani, G.; Soli, M.; Dall'Asta, V.; Rotoli, B.M. Growth Arrest of Alveolar Cells in Response to Cytokines from Spike S1-Activated Macrophages: Role of IFN-γ. Biomedicines 2022, 10, 3085. https://doi.org/10.3390/biomedicines10123085
Barilli A, Visigalli R, Ferrari F, Recchia Luciani G, Soli M, Dall'Asta V, Rotoli BM. Growth Arrest of Alveolar Cells in Response to Cytokines from Spike S1-Activated Macrophages: Role of IFN-γ. Biomedicines. 2022; 10(12):3085. https://doi.org/10.3390/biomedicines10123085
Chicago/Turabian StyleBarilli, Amelia, Rossana Visigalli, Francesca Ferrari, Giulia Recchia Luciani, Maurizio Soli, Valeria Dall'Asta, and Bianca Maria Rotoli. 2022. "Growth Arrest of Alveolar Cells in Response to Cytokines from Spike S1-Activated Macrophages: Role of IFN-γ" Biomedicines 10, no. 12: 3085. https://doi.org/10.3390/biomedicines10123085