
Relationship between Cognitive Dysfunction and Age-Related Variability in Oxidative Markers in Isolated Mitochondria of Alzheimer’s Disease Transgenic Mouse Brains


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Behavioral Assessment
2.2.1. Morris Water Maze
2.2.2. Rota-Rod Test
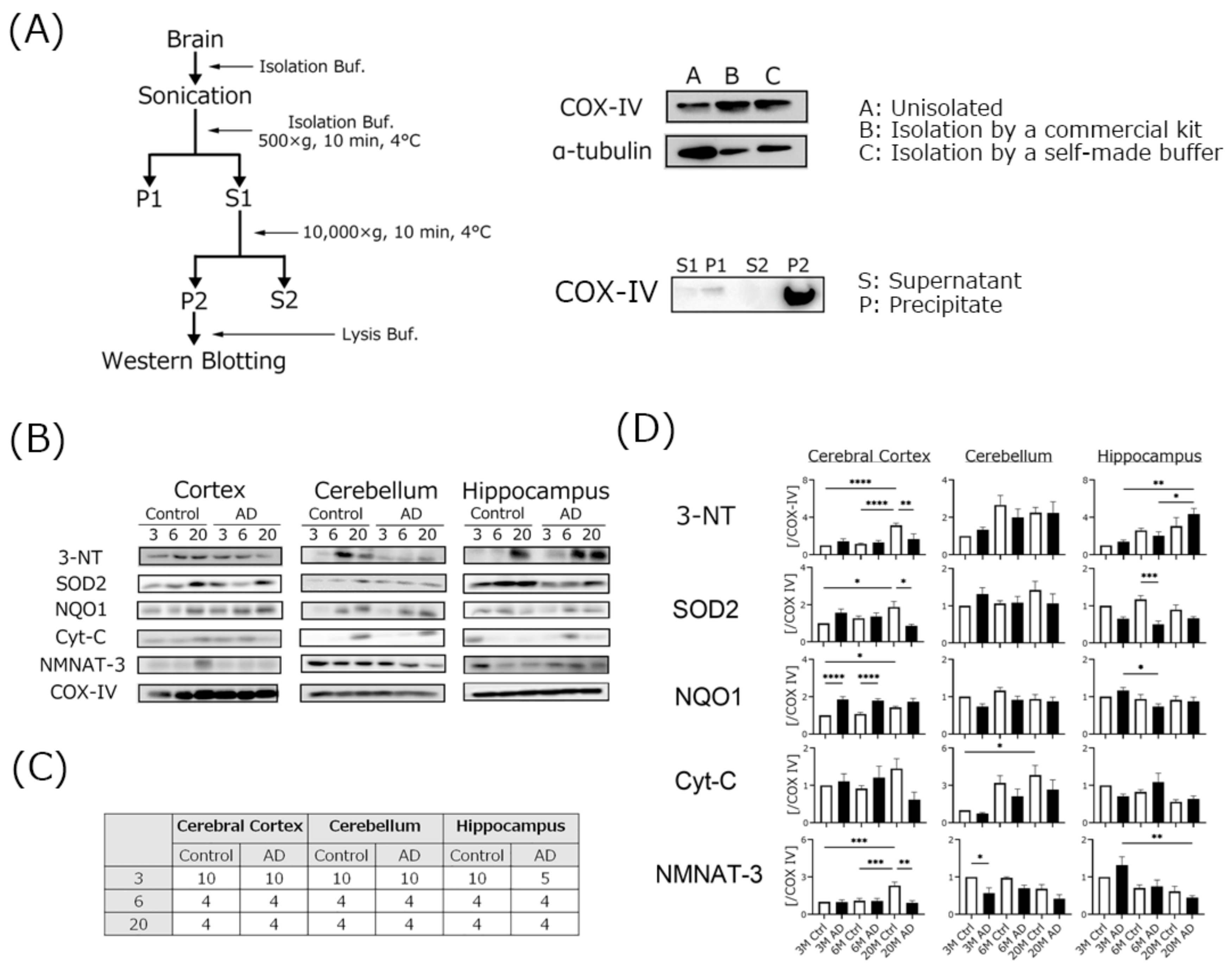
2.3. Mitochondria Isolation
2.4. Western Blotting
2.5. Statistical Analysis
3. Results
4. Discussion
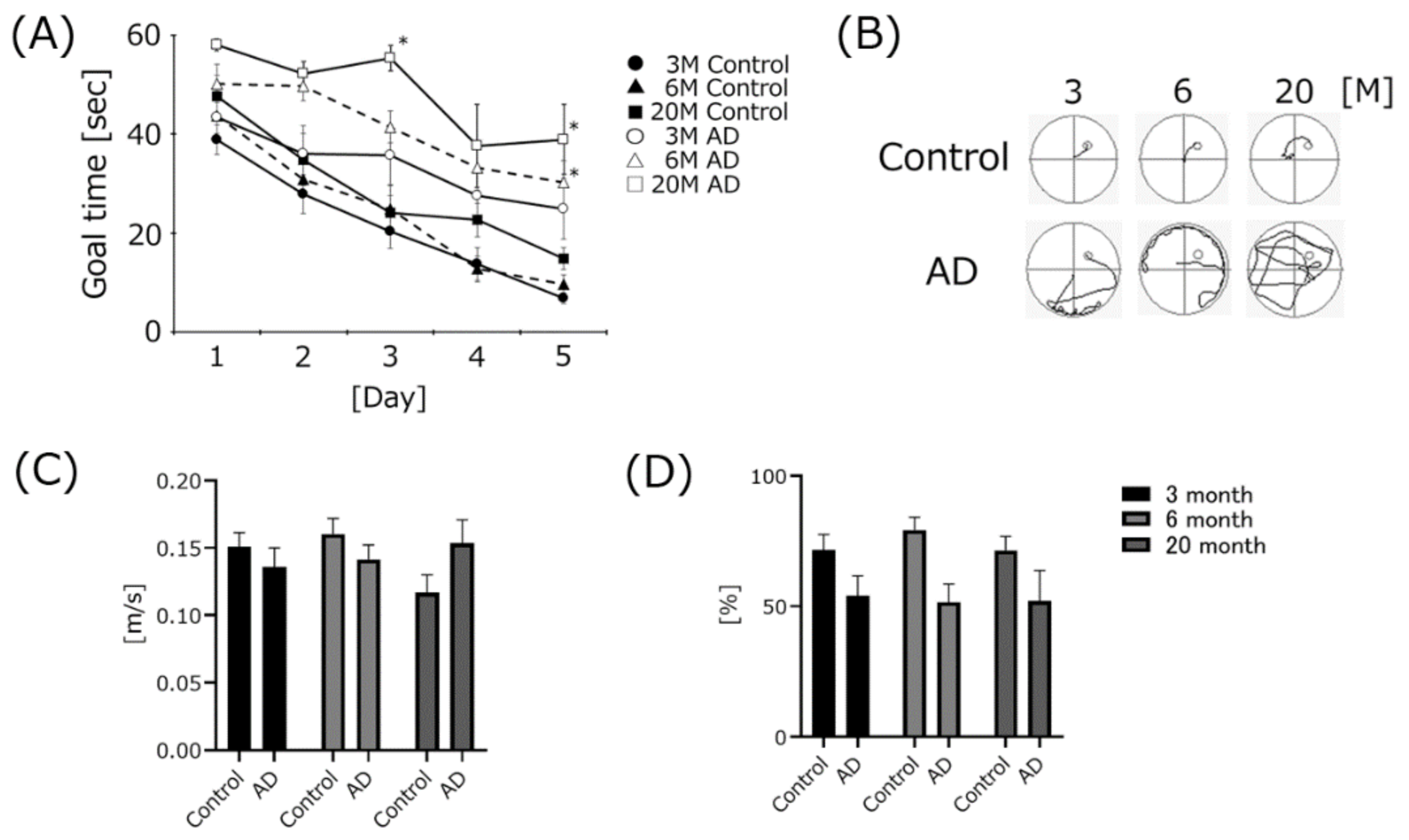
4.1. AD Transgenic Mice Developed Cognitive Dysfunction, but Swimming Speed Did Not Change
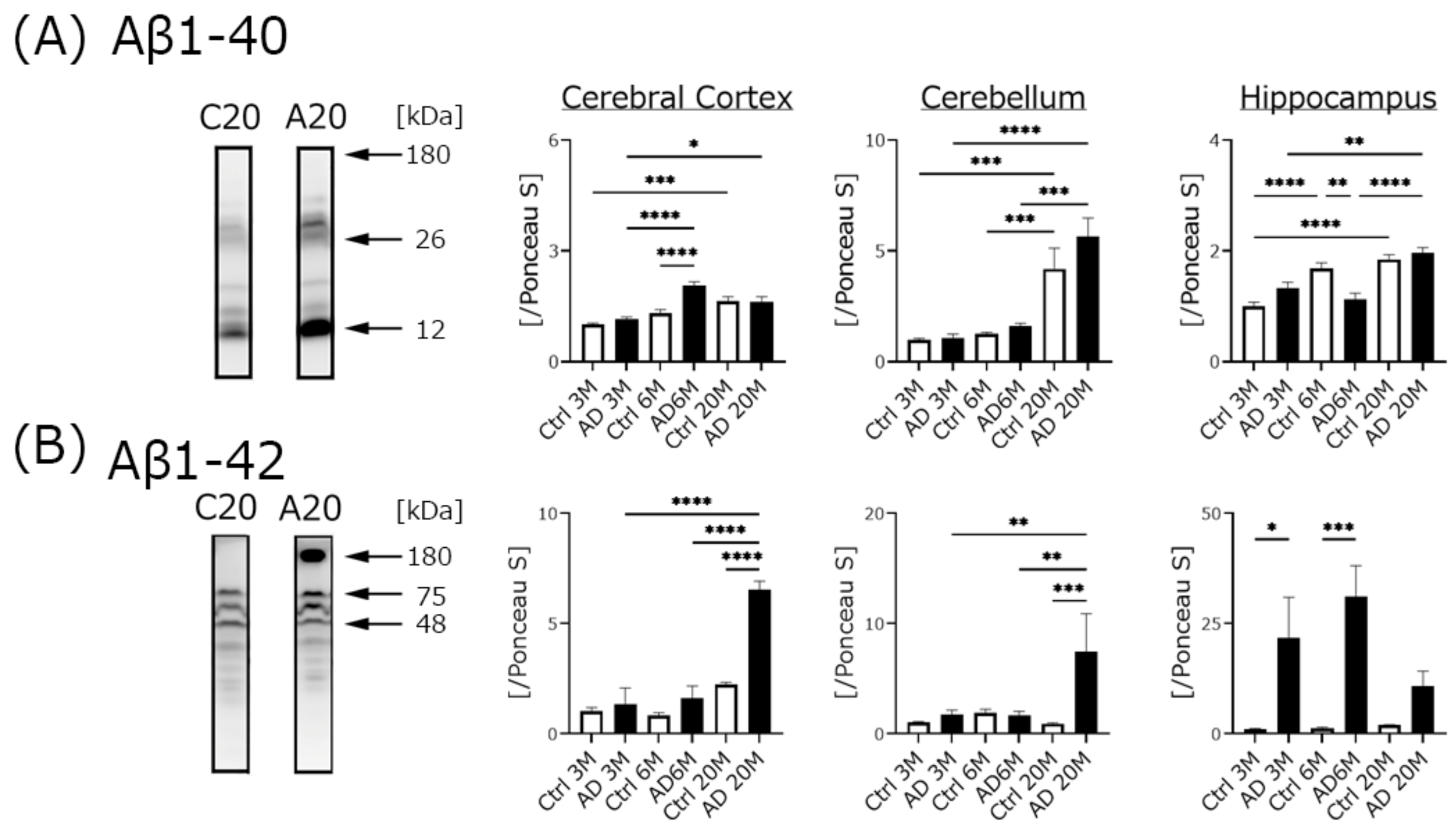
4.2. Aβ Expression, but Not Phospho-Tau Was Significantly Increased in AD Transgenic Mice
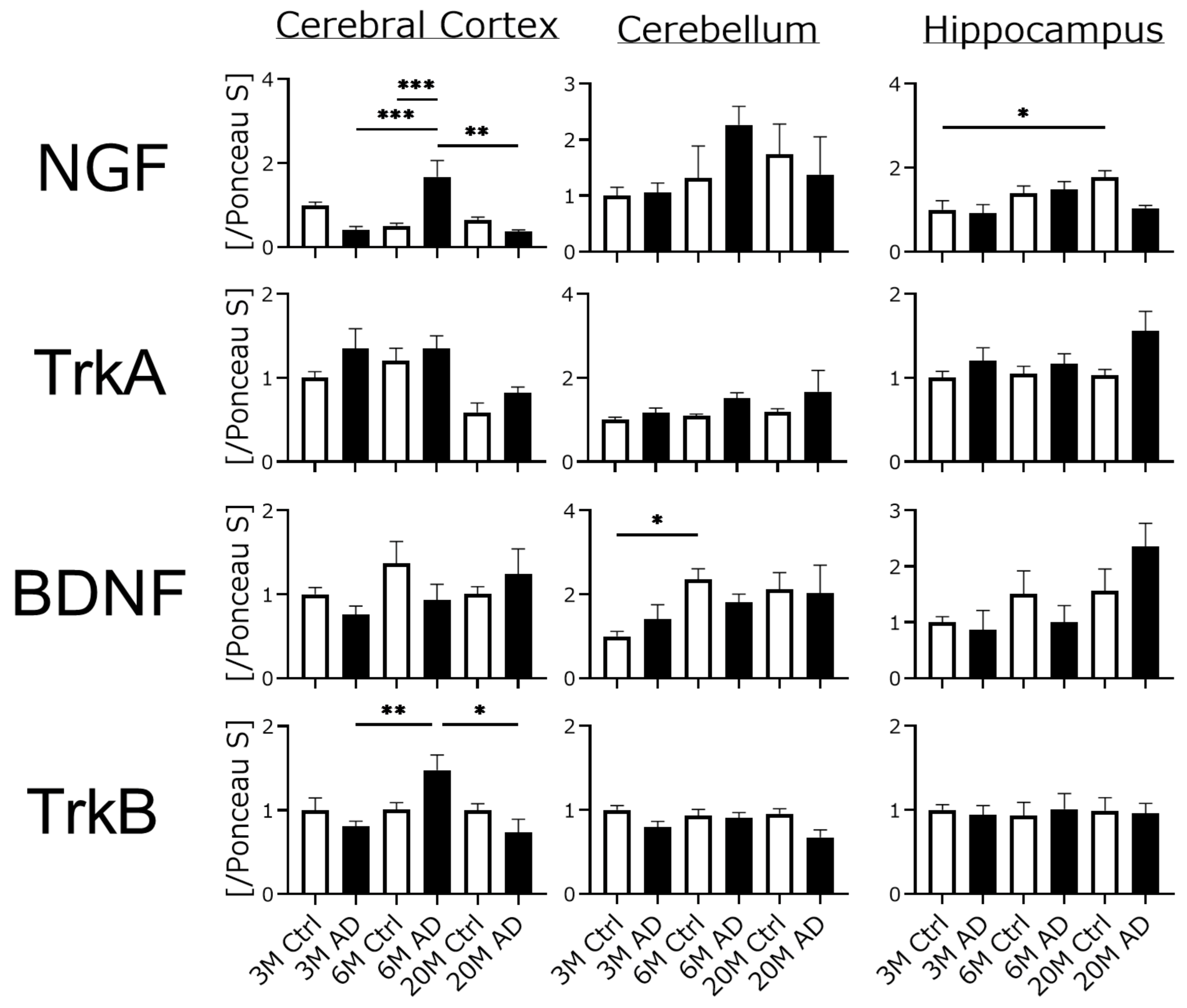
4.3. Neurotrophic Factors and Their Receptor Expression Were Unchanged in AD Transgenic Mouse Brains
4.4. Mitochondrial Oxidative Damage Was Increased in AD Transgenic Mouse Brains
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ohno, S.; Chen, Y.; Sakamaki, H.; Matsumaru, N.; Yoshino, M.; Tsukamoto, K. Humanistic burden among caregivers of patients with Alzheimer’s disease or dementia in Japan: A large-scale cross-sectional survey. J. Med. Econ. 2021, 24, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Cause and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, A.K.; Walker, A.C.; Moudgal, R.A.; Myrick, D.A.; Kyle, S.M.; Bai, Y.; Rowley, M.J.; Katz, D.J. The inhibition of LSD1 via sequestration contributes to tau-mediated neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 29133–29143. [Google Scholar] [CrossRef] [PubMed]
- Vaz Miguel Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar]
- Alzheimer’s Association. 2020 Alzheimer’s diseases facts and figures. Alzheimers Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Wang, X.; Davis, R.L. Early mitochondrial fragmentation and dysfunction in a drosophila model for Alzheimer’s disease. Mol. Neurobiol. 2021, 58, 143–155. [Google Scholar] [CrossRef]
- Malar, D.S.; Prasanth, M.I.; Jeyakumar, M.; Balamurugan, K.; Devi, K.P. Vitexin prevents Aβ proteotoxicity in transgenic Caenorhabditis elegans model of Alzheimer’s disease by modulating unfolded protein response. J. Biochem. Mol. Toxicol. 2021, 35, e22632. [Google Scholar] [CrossRef]
- Beckman, D.; Chakrabarty, P.; Ott, S.; Dao, A.; Zhou, E.; Janssen, W.G.; Donis-Cox, K.; Muller, S.; Kordower, J.H.; Morrision, J.H. A novel tau-based rhesus monkey model of Alzheimer’s pathogenesis. Alzheimers Dement. 2021, 17, 933–945. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neurpeptides 2015, 52, 1–18. [Google Scholar] [CrossRef]
- Wisniewski, T.; Boutajangout, A. Tau-based therapeutic approaches for Alzheimer’s disease—A mini-review. Gerontology 2014, 60, 381–385. [Google Scholar]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [Green Version]
- Alfadda, A.A.; Sallam, R.M. Reactive oxygen species in health and disease. J. Biomed. Biotechnol. 2012, 2012, 936486. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K.; Takatsu, H.; Shinkai, T.; Suzuki, S.; Abe, K.; Urano, S. Appearance of amyloid β-like substances and delayed-type apoptosis in rat hippocampus CA1 region through aging and oxidative stress. J. Alzheimers Dis. 2005, 8, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davis, P.; Masliah, E.; Williams, D.S. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Krstic, D.; Knuesel, I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat. Rev. Neurol. 2013, 9, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, S.H.; Yu, Z.X.; Shelbourne, P.; Li, X.J. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J. Neurosci. 2001, 21, 8473–8481. [Google Scholar] [CrossRef] [Green Version]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mörk, S.; Bö, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef]
- Fukui, K.; Ushiki, K.; Takatsu, H.; Koike, T.; Urano, S. Tocotrienols prevent hydrogen peroxide-induced axon and dendrite degeneration in cerebellar granule cells. Free Radic. Res. 2012, 46, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Nakanishi, A.; Okihiro, S.; Urano, S.; Fukui, K. Ionomycin-induced calcium influx induces neurite degeneration in mouse neuroblastoma cells: Analysis of a time-lapse live cell imaging system. Free Radic. Res. 2016, 50, 1214–1225. [Google Scholar] [CrossRef]
- Fukui, K.; Onodera, K.; Shinkai, T.; Suzuki, S.; Urano, S. Impairment of learning and memory in rats caused by oxidative stress and aging and changes in antioxidative defense systems. Ann. N. Y. Acad. Sci. 2001, 928, 168–175. [Google Scholar] [CrossRef]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Kato, Y.; Aoki, Y.; Fukui, K. Tocotrienols influence body weight gain and brain protein expression in long-term high-fat diet-treated mice. Int. J. Mol. Sci. 2020, 21, 4533. [Google Scholar] [CrossRef]
- Fukui, K.; Okihiro, S.; Ohfuchi, Y.; Hashimoto, M.; Kato, Y.; Yoshida, N.; Mochizuki, K.; Tsumoto, H.; Miura, Y. Proteomic study on neurite responses to oxidative stress: Search for differentially expressed proteins in isolated neurites of N1E-115 cells. J. Clin. Biochem. Nutr. 2019, 64, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Uchiumi, H.; Usami, R.; Takatsu, H.; Aoki, Y.; Yanai, S.; Endo, S.; Fukui, K. Tocotrienols reach the brain and play roles in the attenuation of body weight gain and improvement of cognitive function in high-fat diet-treated mice. J. Clin. Biochem. Nutr. 2021, 69, 256–264. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Richard, B.C.; Kurdakova, A.; Baches, S.; Bayer, T.A.; Weggen, S.; Wirths, O. Gene Dosage Dependent Aggravation of the Neurological Phenotype in the 5XFAD Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2015, 45, 1223–1236. [Google Scholar] [CrossRef]
- Sanderberg, A.; Luheshi, L.M.; Söllvander, S.; Pereira de Barros, T.; Macao, B.; Knowles, T.P.; Biverstål, H.; Lendel, C.; Ekholm-Petterson, F.; Dubnovitsky, A.; et al. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. Proc. Natl. Acad. Sci. USA 2010, 107, 15595–15600. [Google Scholar] [CrossRef] [Green Version]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T., Jr. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef] [PubMed]
- Kuperstein, I.; Broersen, K.; Benilova, I.; Rozenski, J.; Jonckheere, W.; Debulpaep, M.; Vandersteen, A.; Segers-Nolten, I.; van der Werf, K.; Subramaniam, V.; et al. Neurotoxicity of Alzheimer’s disease Aβ peptides id induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 2010, 29, 3408–3420. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Yeh, M.L.; Cuzon Carlson, V.C.; Johnson-Venkatesh, E.M.; Yeh, H.H. Nerve growth factor in the hippocamposeptal system: Evidence for activity-dependent anterograde delivery and modulation of synaptic activity. J. Neurosci. 2012, 32, 7701–7710. [Google Scholar] [CrossRef] [Green Version]
- Lärkfors, L.; Ebendal, T.; Whittemore, S.R.; Persson, H.; Hoffer, B.; Olson, L. Decreased level of nerve growth factor (NGF) and its messenger RNA in the aged rat brain. Brain Res. 1987, 427, 55–60. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Pao, P.C.; Patnaik, D.; Watson, L.A.; Gao, F.; Pan, L.; Wang, J.; Adaikkan, C.; Penney, J.; Cam, H.P.; Huang, W.C.; et al. HDAC1 modulates OGC1-initiated oxidative DNA damage repair in the aging brain and Alzheimer’s disease. Nat. Commun. 2020, 11, 2484. [Google Scholar] [CrossRef]
- Annesley, S.; Fisher, P.R. Mitochondria in health and disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s disease: Cellular and molecular mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Fukui, K.; Masuda, A.; Hosono, A.; SUwabe, R.; Yamashita, K.; Shinkai, T.; Urano, S. Changes in microtubule-related proteins and authohagy in long-term vitamin E-deficient mice. Free Radic. Res. 2014, 48, 649–658. [Google Scholar] [CrossRef]
- Flynn, J.M.; Melov, S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radic. Biol. Med. 2013, 62, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahsan, H. 3-Nitrotyrosine: A biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum. Immunol. 2013, 74, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Thomson, L. 3-Nitrotyrosine modified proteins in arteriosclerosis. Dis. Markers 2015, 2015, 708282. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Reed, T.T.; Perluigi, M.; de Marco, C.; Coccia, R.; Keller, J.N.; Markesbery, W.R.; Sultana, R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: Implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res. 2007, 1148, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Cambronne, X.A.; Stewart, M.L.; Kim, D.H.; Jones-Brunette, A.M.; Morgan, R.K.; Farrens, D.L.; Cohen, M.S.; Goodman, R.H. NAD+ biosensor reveals multiple sources for mitochondrial NAD+. Science 2016, 352, 1474–1477. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Gulshan, M.; Yaku, K.; Okabe, K.; Mohamood, A.; Sasaki, T.; Yamamoto, M.; Hikosaka, K.; Usui, I.; Kitamura, T.; Tobe, K.; et al. Overexpression of Nmnat3 efficiently increases NAD and NGD levels and ameliorates age-associated insulin resistance. Aging Cell 2018, 17, e12798. [Google Scholar] [CrossRef]
- Chhetri, J.; King, A.E.; Gueven, N. Alzheimer’s disease and NQO-1: Is there a link? Curr. Alzheimer Res. 2018, 15, 56–66. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free. Radc. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.; Talalay, P. NAD(P)H: Quinone acceptor oxidoreductase 1 (NQO) 1, a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, S.M.; Proença, M.T.; Santos, S.; Santana, I.; Oliveira, C.R. Cytochrome c oxidase is decreased in Alzheimer’s disease platelet. Neurobiol. Aging 2004, 25, 105–110. [Google Scholar] [CrossRef]
- Maurer, I.; Zierz, S.; Möller, H.J. A selective defect of cytochrome c oxidaseis present in brain of Alzheimer disease patients. Neurobiol. Aging 2000, 21, 455–462. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshida, N.; Kato, Y.; Takatsu, H.; Fukui, K. Relationship between Cognitive Dysfunction and Age-Related Variability in Oxidative Markers in Isolated Mitochondria of Alzheimer’s Disease Transgenic Mouse Brains. Biomedicines 2022, 10, 281. https://doi.org/10.3390/biomedicines10020281
Yoshida N, Kato Y, Takatsu H, Fukui K. Relationship between Cognitive Dysfunction and Age-Related Variability in Oxidative Markers in Isolated Mitochondria of Alzheimer’s Disease Transgenic Mouse Brains. Biomedicines. 2022; 10(2):281. https://doi.org/10.3390/biomedicines10020281
Chicago/Turabian StyleYoshida, Naoki, Yugo Kato, Hirokatsu Takatsu, and Koji Fukui. 2022. "Relationship between Cognitive Dysfunction and Age-Related Variability in Oxidative Markers in Isolated Mitochondria of Alzheimer’s Disease Transgenic Mouse Brains" Biomedicines 10, no. 2: 281. https://doi.org/10.3390/biomedicines10020281
APA StyleYoshida, N., Kato, Y., Takatsu, H., & Fukui, K. (2022). Relationship between Cognitive Dysfunction and Age-Related Variability in Oxidative Markers in Isolated Mitochondria of Alzheimer’s Disease Transgenic Mouse Brains. Biomedicines, 10(2), 281. https://doi.org/10.3390/biomedicines10020281