
Metformin Inhibits ROS Production by Human M2 Macrophages via the Activation of AMPK

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Chemicals and Reagents
2.2. Ethics Statement and Monocytes Isolation
2.3. Human Monocyte Differentiation and Polarization
2.4. Monocyte and Macrophage Cytotoxicity Tests
2.5. Reactive Oxygen Species (ROS) Measurement by the NBT Reduction Assay
2.6. Luminol-Amplified Chemiluminescence Assay
2.7. Animal Experiments
2.8. Western Blot Analysis
2.9. Statistical Analysis
3. Results
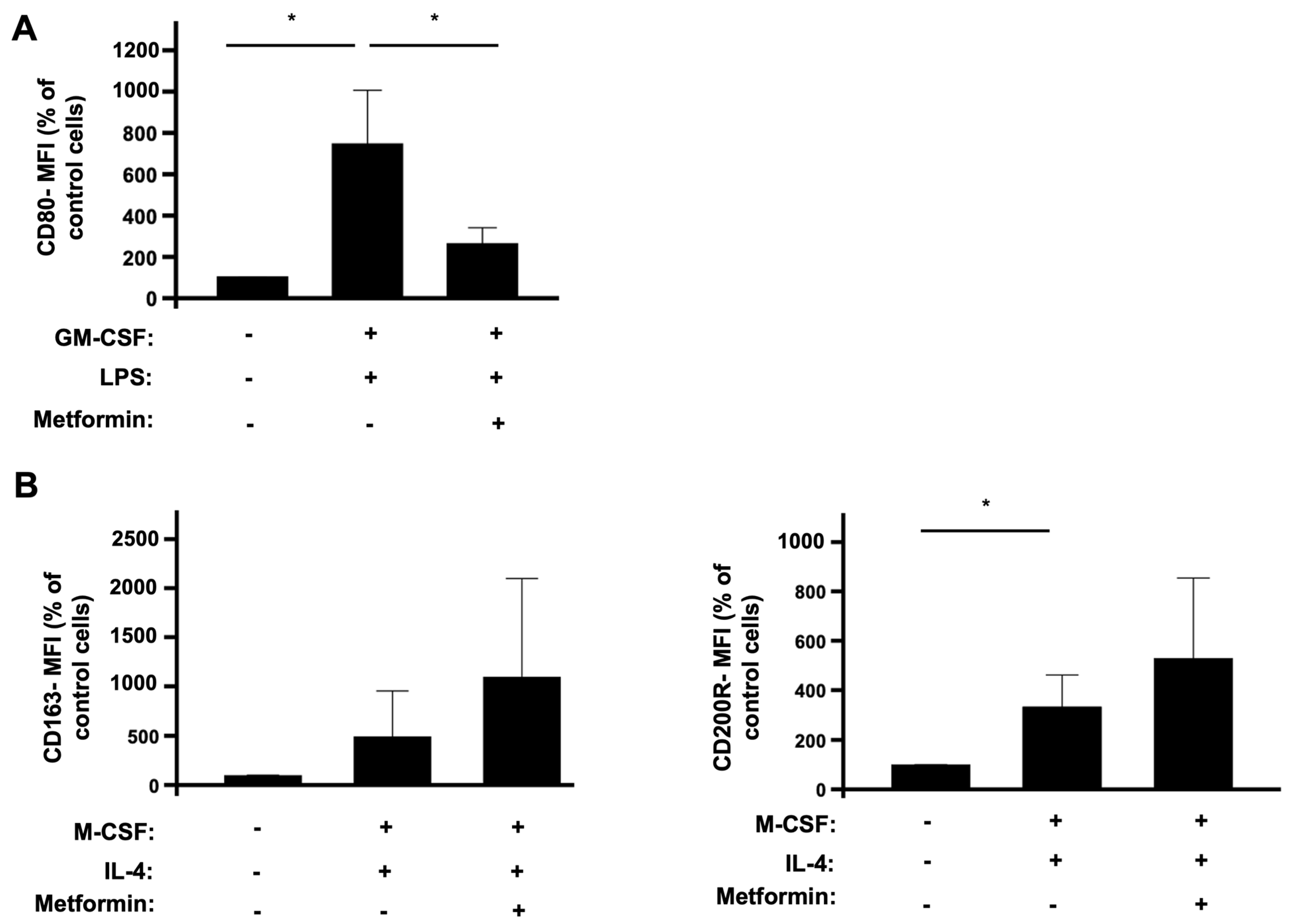
3.1. The Effects of Metformin on the Polarization of Human Monocytes into M1 and M2 Macrophages
3.2. Metformin Inhibits Reactive Oxygen Species (ROS) Production in M2 Macrophages
3.3. Metformin Does Not Affect Cell Viability
3.4. Metformin Does Not Affect the Expression of the NADPH Oxidase/NOX2 Components nor the Phosphorylation of p47phox on Ser345 and Its Upstream Kinases p38 and ERK1/2
3.5. Metformin and LPS Enhanced AMPK Phosphorylation in Human M2 Macrophages
3.6. AMPK Plays the Role of a Brake of ROS Production by Human and Mice Phagocytes
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AICAR | 5-Aminoimidazole-4-carboxamide ribonucleotide |
| AMPK | adenosine monophosphate activated protein kinase |
| CD | cluster of differentiation |
| fMLF | N-formyl-methionyl-leucyl-phenylalanine |
| GM-CSF | granulocyte macrophages colony stimulating factor |
| LPS | lipopolysaccharide |
| M-CSF | colony-stimulating factor |
| NBT | nitro blue tetrazolium |
| NOX | nicotinamide adenine dinucleotide phosphate oxidase |
| PBMC | peripheral blood mononuclear cells |
| ROS | reactive oxygen species |
References
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernicova, I.; Korbonits, M. Metforminmode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2004, 10, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Zi, F.; Zi, H.; Li, Y.; He, J.; Shi, Q.; Cai, Z. Metformin and cancer: An existing drug for cancer prevention and therapy. Oncol. Lett. 2017, 15, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; He, S.; Feng, X.; Cheng, J.; Luo, Y.; Tian, L.; Huang, Q. Metformin: A review of its potential indications. Drug Des. Devel. Ther. 2017, 11, 2421–2429. [Google Scholar] [CrossRef] [Green Version]
- Horakova, O.; Kroupova, P.; Bardova, K.; Buresova, J.; Janovska, P.; Kopecky, J.; Rossmeisl, M. Metformin acutely lowers blood glucose levels by inhibition of intestinal glucose transport. Sci. Rep. 2019, 9, 6156. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Yan, Y.; Zhou, X.E.; Xu, H.E.; Melcher, K. Structure and Physiological Regulation of AMPK. Int. J. Mol. Sci. 2018, 19, 3534. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef] [Green Version]
- Cusi, K.; Consoli, A.; DeFronzo, R.A. Metabolic effects of metformin on glucose and lactate metabolism in noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1996, 81, 4059–4067. [Google Scholar] [PubMed] [Green Version]
- He, L.; Wondisford, F.E. Metformin Action: Concentrations Matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loubière, C.; Dirat, B.; Tanti, J.; Bost, F. Metformine et cancer: De nouvelles perspectives pour un ancien médicament. Ann. Endocrinol. 2013, 74, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.S.N.; Masharani, U. Pancreatic Hormones & Antidiabetic Drugs. In Basic & Clinical Pharmacology; Katzung, B.G., Ed.; McGraw-Hill Education: New York, NY, USA, 2017. [Google Scholar]
- Faubert, B.; Vincent, E.E.; Poffenberger, M.C.; Jones, R.G. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer Lett. 2014, 356, 165–170. [Google Scholar] [CrossRef]
- Tamargo-Gómez, I.; Mariño, G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galic, S.; Fullerton, M.D.; Schertzer, J.D.; Sikkema, S.; Marcinko, K.; Walkley, C.R.; Izon, D.; Honeyman, J.; Chen, Z.; van Denderen, B.J.; et al. Steinberg, G.R. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Investig. 2011, 121, 4903–4915. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, M.; O’Brien, E.L.; Bigornia, S.; Mott, M.; Cacicedo, J.M.; Xu, X.J.; Gokce, N.; Apovian, C.; Ruderman, N. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem. Biophys. Res. Commun. 2011, 404, 382–387. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef]
- Xie, J.; Xia, L.; Xiang, W.; He, W.; Yin, H.; Wang, F.; Gao, T.; Qi, W.; Yang, Z.; Yang, X.; et al. Metformin selectively inhibits metastatic colorectal cancer with the KRAS mutation by intracellular accumulation through silencing MATE1. Proc. Natl. Acad. Sci. USA 2020, 117, 13012–13022. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Chatterjee, A.; Kogan, D.; Patel, D.; Foster, D.A. 5-Aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) enhances the efficacy of rapamycin in human cancer cells. Cell Cycle 2015, 14, 3331–33319. [Google Scholar] [CrossRef] [Green Version]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood Monocytes Consist of Two Principal Subsets with Distinct Migratory Properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Orekhov, A.N.; Orekhova, V.A.; Nikiforov, N.G.; Myasoedova, V.A.; Grechko, A.V.; Romanenko, E.B.; Zhang, D.; Chistiakov, D.A. Monocyte differentiation and macrophage polarization. Vessel Plus 2019, 3, 10. [Google Scholar] [CrossRef]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Investig. 1973, 52, 741–744. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.; Wang, H. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Immunological Genome Consortium, the Immunological Genome Consortium. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [Green Version]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.; Elbim, C. Phagocyte NADPH oxidase: A multicomponent enzyme essential for host defenses. Arch. Immunol. Ther. Exp. 2005, 53, 199–206. [Google Scholar]
- Migdal, C.; Serres, M. Espèces réactives de l’oxygène et stress oxydant. Med. Sci. 2011, 27, 405–412. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef]
- Roos, D. Chronic granulomatous disease. Br. Med. Bull. 2016, 118, 50–63. [Google Scholar] [CrossRef] [Green Version]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M. Role of the NADPH oxidase systems Nox and Duox in host defense and inflammation. Exp. Mol. Med. 2007, 3, 111–115. [Google Scholar] [CrossRef]
- Singel, K.L.; Segal, B.H. NOX2-dependent regulation of inflammation. Clin. Sci. 2016, 130, 479–490. [Google Scholar] [CrossRef] [Green Version]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.; Marie, J.; Braut-Boucher, F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Cross, A.R.; Segal, A.W. The NADPH oxidase of professional phagocytes—prototype of the NOX electron transport chain systems. Biochim. Biophys. Acta 2004, 1657, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Paik, Y.H.; Brenner, D.A. NADPH oxidase mediated oxidative stress in hepatic fibrogenesis. Korean J. Hepatol. 2011, 17, 251–257. [Google Scholar] [CrossRef]
- Dang, P.M.; Dewas, C.; Gaudry, M.; Fay, M.; Pedruzzi, E.; Gougerot-Pocidalo, M.A.; El Benna, J. Priming of human neutrophil respiratory burst by granulocyte/macrophage colony-stimulating factor (GM-CSF) involves partial phosphorylation of p47(phox). J. Biol. Chem. 1999, 274, 20704–20708. [Google Scholar] [CrossRef] [Green Version]
- Dang, P.M.; Stensballe, A.; Boussetta, T.; Raad, H.; Dewas, C.; Kroviarski, Y.; Hayem, G.; Jensen, O.N.; Gougerot-Pocidalo, M.; El-Benna, J. A specific p47phox -serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J. Clin. Investig. 2006, 116, 2033–2043. [Google Scholar] [CrossRef] [Green Version]
- Dewas, C.; Dang, P.M.; Gougerot-Pocidalo, M.; El-Benna, J. TNF-alpha induces phosphorylation of p47(phox) in human neutrophils: Partial phosphorylation of p47phox is a common event of priming of human neutrophils by TNF-alpha and granulocyte-macrophage colony-stimulating factor. J. Immunol. 2003, 171, 4392–4398. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.L.; Park, J.; Benna, J.E.; Faust, L.P.; Inanami, O.; Babior, B.M. Activation of p47PHOX, a Cytosolic Subunit of the Leukocyte NADPH Oxidase: Phosphorylation of ser-359 or ser-370 precedes phosphorylation at other sites and is required for activity. J. Biol. Chem. 1998, 273, 35147–35152. [Google Scholar] [CrossRef] [Green Version]
- Harrison, I.P.; Selemidis, S. Understanding the biology of reactive oxygen species and their link to cancer: NADPH oxidases as novel pharmacological targets. Clin. Exp. Pharmacol. Physiol. 2014, 41, 533–542. [Google Scholar] [CrossRef]
- Tan, H.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The reactive oxygen species in macrophage polarization: Reflecting its dual role in progression and treatment of human diseases. Oxid. Med. Cell. Longev. 2016, 2016, 2795090. [Google Scholar] [CrossRef] [Green Version]
- Kamata, T. Roles of Nox1 and other Nox isoforms in cancer development. Cancer Sci. 2009, 100, 1382–1388. [Google Scholar] [CrossRef]
- Viollet, B.; Andreelli, F.; Jørgensen, S.B.; Perrin, C.; Geloen, A.; Flamez, D.; Mu, J.; Lenzner, C.; Baud, O.; Bennoun, M.; et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J. Clin. Investig. 2003, 111, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Benna, J.E.; Dang, P.M.; Gaudry, M.; Fay, M.; Morel, F.; Hakim, J.; Gougerot-Pocidalo, M.A. Phosphorylation of the respiratory burst oxidase subunit p67(phox) during human neutrophil activation. Regulation by protein kinase C-dependent and independent pathways. J. Biol. Chem. 1997, 272, 17204. [Google Scholar] [CrossRef] [Green Version]
- Ziegler-Heitbrock, L. The CD14+ CD16+ blood monocytes: Their role in infection and inflammation. J. Leukoc. Biol. 2007, 81, 584–592. [Google Scholar] [CrossRef]
- Jessen, N.; Pold, R.; Buhl, E.S.; Jensen, L.S.; Schmitz, O.; Lund, S. Effects of AICAR and exercise on insulin-stimulated glucose uptake, signaling, and GLUT-4 content in rat muscles. J. Appl. Physiol. 2003, 94, 1373–1379. [Google Scholar] [CrossRef] [Green Version]
- Dolinar, K.; Jan, V.; Pavlin, M.; Chibalin, A.V.; Pirkmajer, S. Nucleosides block AICAR-stimulated activation of AMPK in skeletal muscle and cancer cells. Am. J. Physiol. Cell. Physiol. 2018, 315, C803–C817. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Wu, F.; Li, D.; Yang, L.; Li, Q.; Li, R. Metformin improves obesity-associated inflammation by altering macrophages polarization. Mol. Cell. Endocrinol. 2018, 461, 256–264. [Google Scholar] [CrossRef]
- Martin, L.M.; Möller, M.; Weiss, U.; Russe, O.Q.; Scholich, K.; Pierre, S.; Geisslinger, G.; Niederberger, E. 5-Amino-1-β-D-Ribofuranosyl-Imidazole-4-Carboxamide (AICAR) Reduces Peripheral Inflammation by Macrophage Phenotype Shift. Int. J. Mol. Sci. 2019, 20, 3255. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Tian, X.; Zhu, J.; Lu, Z.; Yu, L.; Zhang, D.; Liu, Y.; Yang, C.; Zhu, Q.; Cao, X. Metformin suppresses UHMWPE particle-induced osteolysis in the mouse calvaria by promoting polarization of macrophages to an anti-inflammatory phenotype. Mol. Med. 2018, 24, 20. [Google Scholar] [CrossRef]
- Qing, L.; Fu, J.; Wu, P.; Zhou, Z.; Yu, F.; Tang, J. Metformin induces the M2 macrophage polarization to accelerate the wound healing via regulating AMPK/mTOR/NLRP3 inflammasome singling pathway. Am. J. Transl. Res. 2019, 11, 655. [Google Scholar]
- Wang, J.; Sun, X.; Ma, Q.; Fu, G.; Cong, L.; Zhang, H.; Fan, D.; Feng, J.; Lu, S.; Liu, J.; et al. Metformin’s antitumour and anti-angiogenic activities are mediated by skewing macrophage polarization. J. Cell. Mol. Med. 2018, 22, 3825–3836. [Google Scholar] [CrossRef]
- Sag, D.; Carling, D.; Stout, R.D.; Suttles, J. AMP-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype 1. J. Immunol. 2008, 181, 8633–8641. [Google Scholar] [CrossRef]
- Vasamsetti, S.B.; Karnewar, S.; Kanugula, A.K.; Thatipalli, A.R.; Kumar, J.M.; Kotamraju, S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: Potential role in atherosclerosis. Diabetes 2015, 64, 2028–2041. [Google Scholar] [CrossRef] [Green Version]
- Chedid, P.; Hurtado-Nedelec, M.; Marion-Gaber, B.; Bournier, O.; Hayem, G.; Gougerot-Pocidalo, M.; Frystyk, J.; Flyvbjerg, A.; El Benna, J.; Marie, J. Adiponectin and Its Globular Fragment Differentially Modulate the Oxidative Burst of Primary Human Phagocytes. Am. J. Pathol. 2012, 180, 682–692. [Google Scholar] [CrossRef]
- Towler, M.C.; Hardie, D.G. AMP-Activated Protein Kinase in Metabolic Control and Insulin Signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef]
- Shrivastava, A. Activation of macrophages with N-formyl-methionyl-leucyl-phenylalanine: Involvement of protein kinase C and tyrosine kinase. Indian J. Exp. Biol. 2007, 45, 755–763. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nassif, R.M.; Chalhoub, E.; Chedid, P.; Hurtado-Nedelec, M.; Raya, E.; Dang, P.M.-C.; Marie, J.-C.; El-Benna, J. Metformin Inhibits ROS Production by Human M2 Macrophages via the Activation of AMPK. Biomedicines 2022, 10, 319. https://doi.org/10.3390/biomedicines10020319
Nassif RM, Chalhoub E, Chedid P, Hurtado-Nedelec M, Raya E, Dang PM-C, Marie J-C, El-Benna J. Metformin Inhibits ROS Production by Human M2 Macrophages via the Activation of AMPK. Biomedicines. 2022; 10(2):319. https://doi.org/10.3390/biomedicines10020319
Chicago/Turabian StyleNassif, Rana M., Elias Chalhoub, Pia Chedid, Margarita Hurtado-Nedelec, Elia Raya, Pham My-Chan Dang, Jean-Claude Marie, and Jamel El-Benna. 2022. "Metformin Inhibits ROS Production by Human M2 Macrophages via the Activation of AMPK" Biomedicines 10, no. 2: 319. https://doi.org/10.3390/biomedicines10020319