
Identification of Novel Mitochondrial Pyruvate Carrier Inhibitors by Homology Modeling and Pharmacophore-Based Virtual Screening


 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General Procedure for the Synthesis of 3-Substituted-2-Cyano-2-Propenoic Acids
2.1.2. (E)-3-[5-(1,3-Benzothiazol-2-yl)-2-Furyl]-2-Cyanoacrylic Acid (BE1976)
2.1.3. (E)-2-Cyano-3-(1-Phenyl-4-Pyrazolyl)acrylic Acid (BE1978)
2.1.4. (E)-2-Cyano-3-(3,5-Dimethyl-1-Phenyl-4-Pyrazolyl)acrylic Acid (BE1980)
2.1.5. (E)-2-Cyano-3-(p-Tolyl)acrylic Acid (BE1984)
2.1.6. (E)-2-Cyano-3-(4-Phenyl-3-Pyrazolyl)acrylic Acid (BE 2617)
2.1.7. (E)-2-Cyano-3-[4-(p-Tolyl)-3-Pyrazolyl]acrylic Acid (BE 2623)
2.2. Homology Modeling
2.3. Pharmacophore Modeling, Database Preparation, and Pharmacophore-Based Virtual Screening
2.4. Bioluminescent Resonance Energy Transfer (BRET)-Based Assays for Inhibitor Binding
2.5. Experimental Animals
2.6. Mitochondrial Isolation and Respiration
2.7. Primary Murine Hepatocyte Isolation and Culture
2.8. Western Blotting Procedures
2.9. Statistical Analyses
3. Results
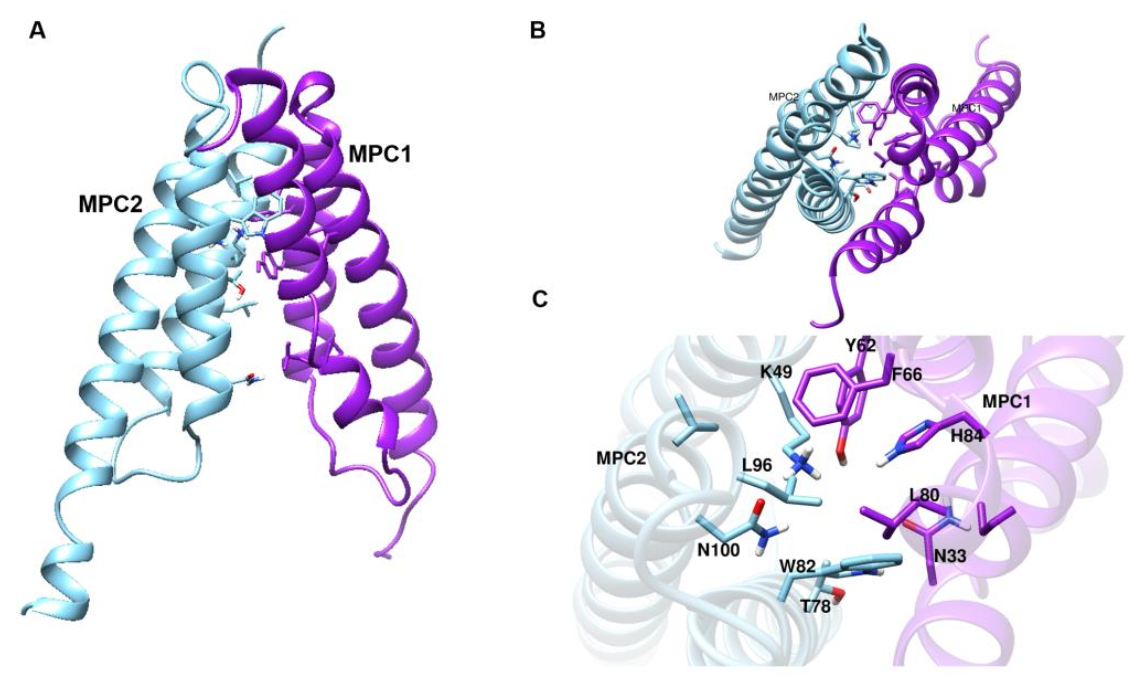
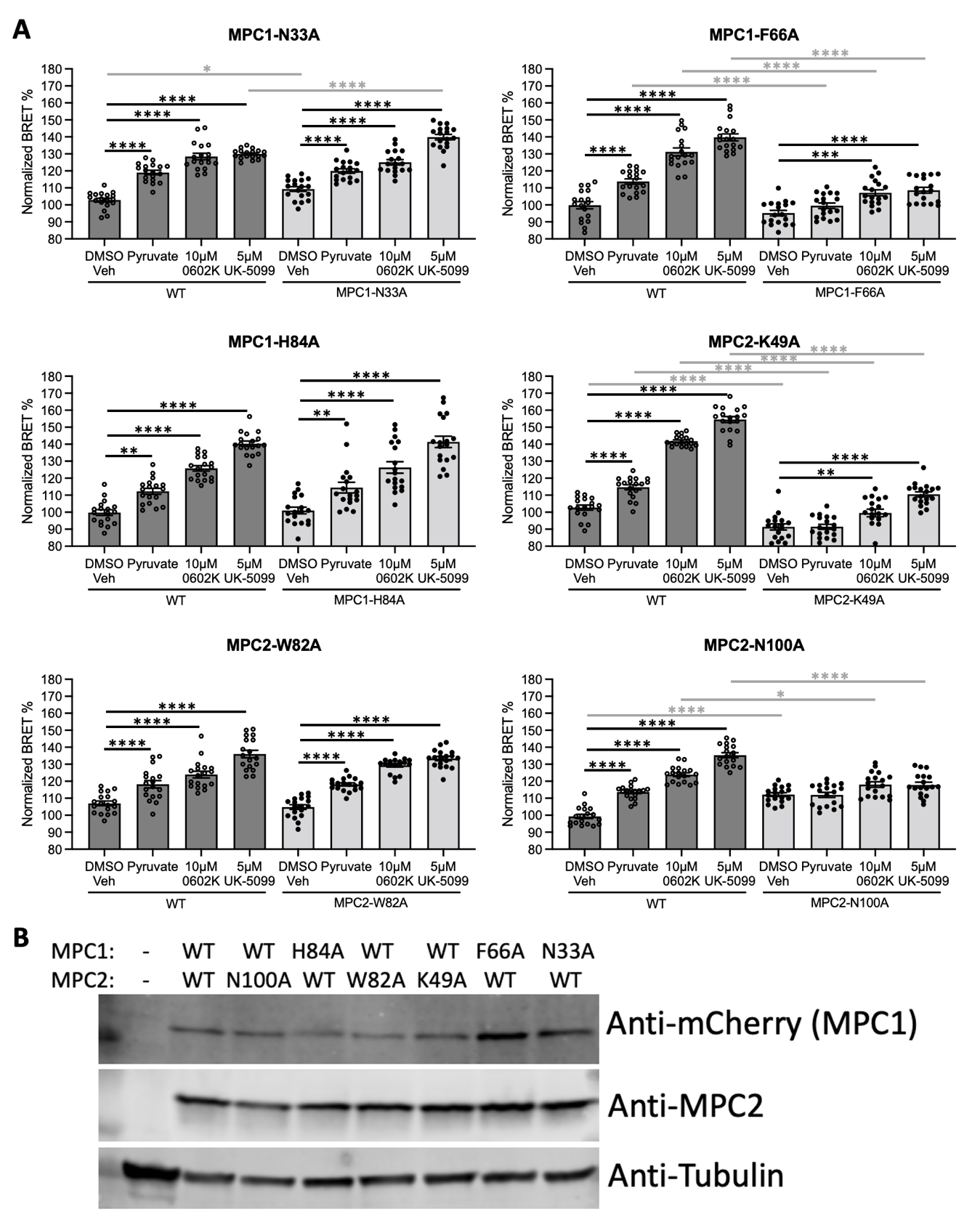
3.1. Homology Modeling of the MPC and Mutagenesis Studies
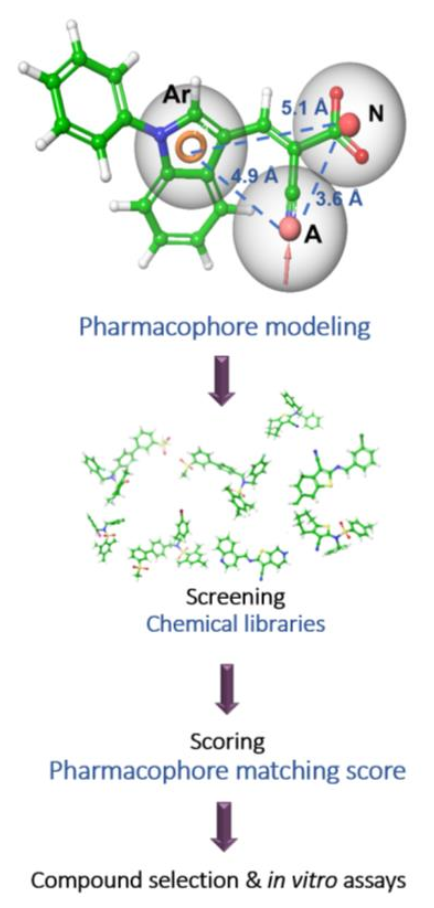
3.2. Pharmacophore Modeling
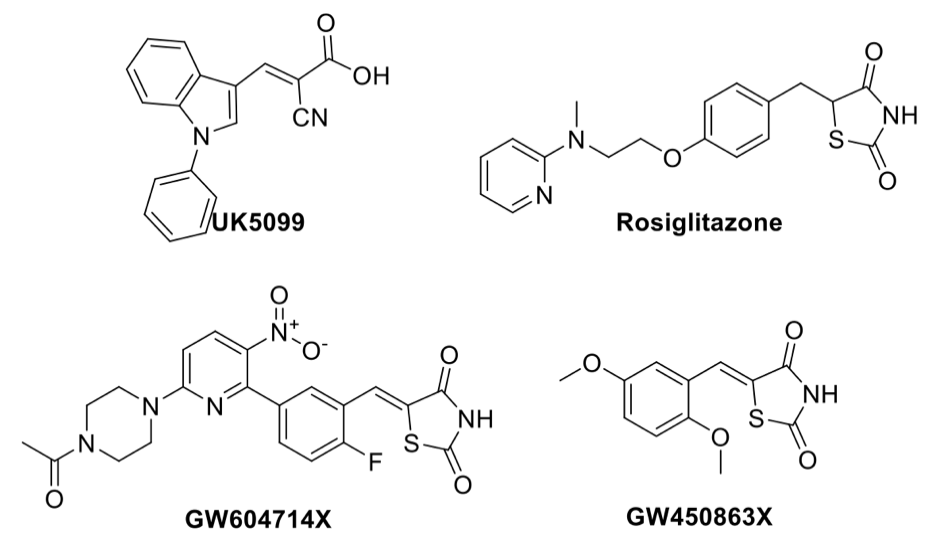
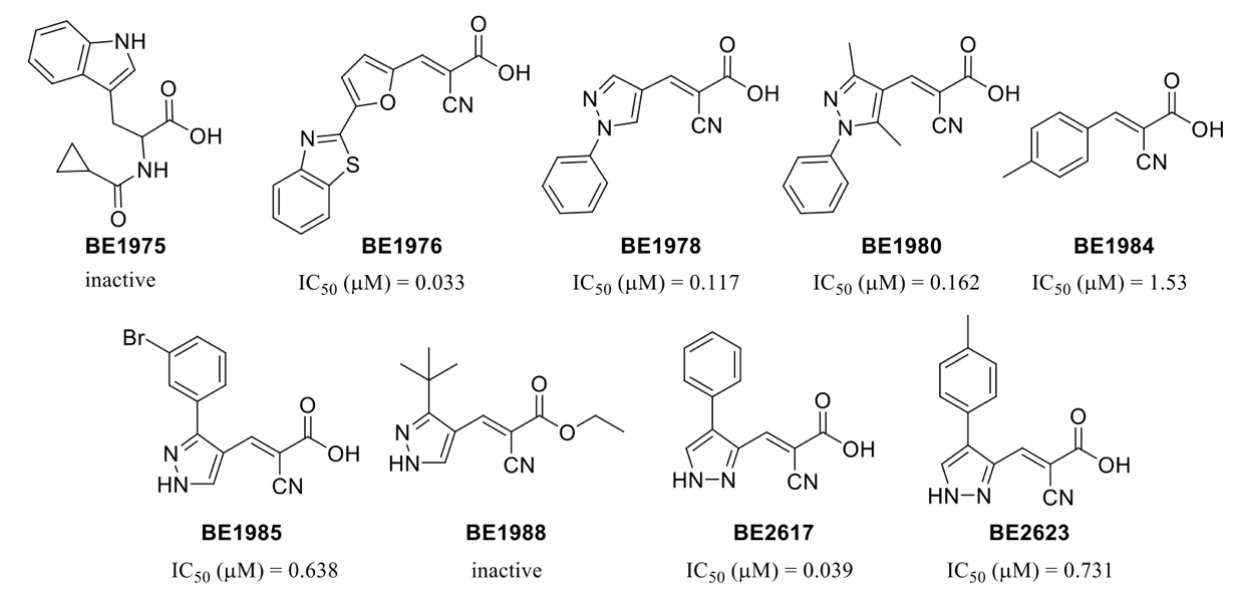
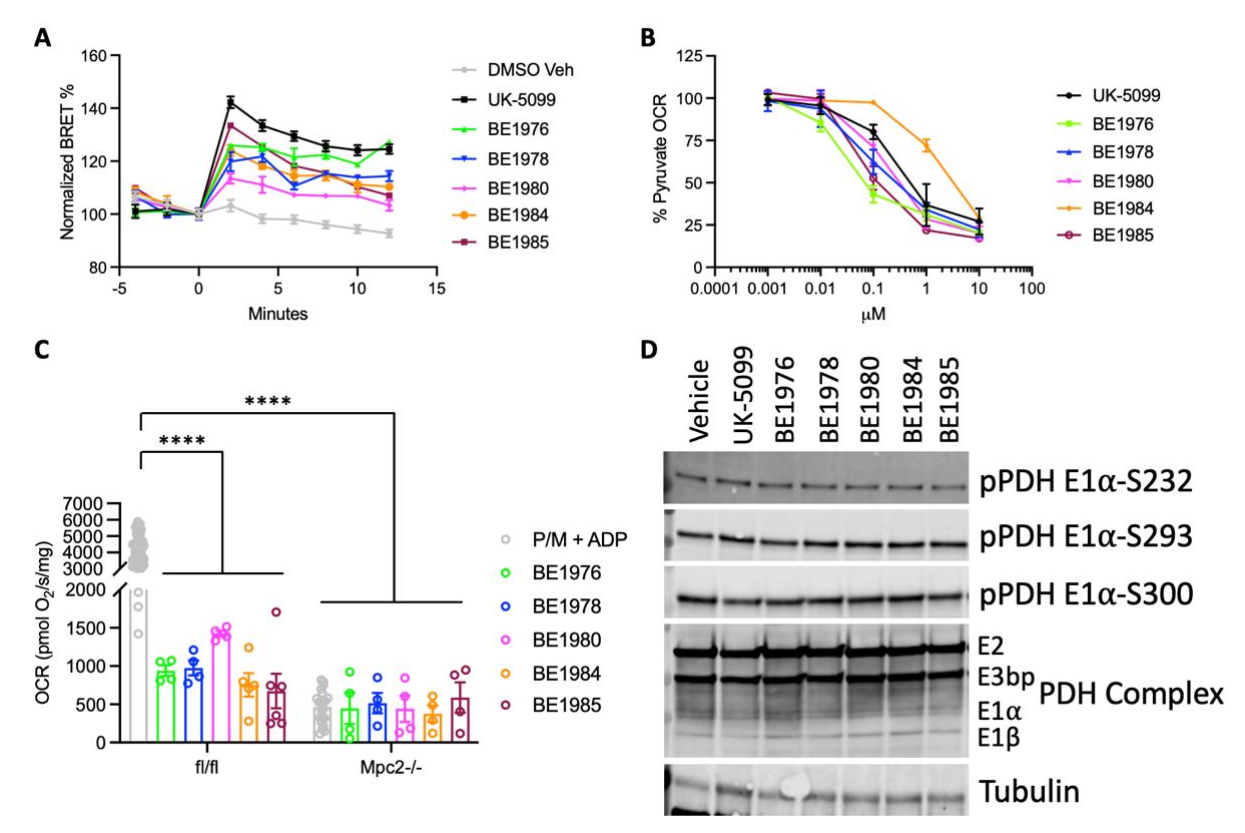
3.3. Identification and Validation of Novel MPC Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herzig, S.; Raemy, E.; Montessuit, S.; Veuthey, J.-L.; Zamboni, N.; Westermann, B.; Kunji, E.R.S.; Martinou, J.-C. Identification and Functional Expression of the Mitochondrial Pyruvate Carrier. Science 2012, 337, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Bricker, D.K.; Taylor, E.B.; Schell, J.C.; Orsak, T.; Boutron, A.; Chen, Y.-C.; Cox, J.E.; Cardon, C.M.; Van Vranken, J.G.; Dephoure, N.; et al. A Mitochondrial Pyruvate Carrier Required for Pyruvate Uptake in Yeast, Drosophila, and Humans. Science 2012, 337, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigueira, P.A.; McCommis, K.S.; Schweitzer, G.G.; Remedi, M.S.; Chambers, K.T.; Fu, X.; McDonald, W.G.; Cole, S.L.; Colca, J.R.; Kletzien, R.F.; et al. Mitochondrial pyruvate carrier 2 hypomorphism in mice leads to defects in glucose-stimulated insulin secretion. Cell Rep. 2014, 7, 2042–2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, C.E.; Zhao, L.; Hartung, T.; Wolfgang, M.J. Requirement for the Mitochondrial Pyruvate Carrier in Mammalian Development Revealed by a Hypomorphic Allelic Series. Mol. Cell. Biol. 2016, 36, 2089–2104. [Google Scholar] [CrossRef] [Green Version]
- McCommis, K.S.; Chen, Z.; Fu, X.; McDonald, W.G.; Colca, J.R.; Kletzien, R.F.; Burgess, S.C.; Finck, B.N. Loss of Mitochondrial Pyruvate Carrier 2 in the Liver Leads to Defects in Gluconeogenesis and Compensation via Pyruvate-Alanine Cycling. Cell Metab. 2015, 22, 682–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, L.R.; Sultana, M.R.; Rauckhorst, A.J.; Oonthonpan, L.; Tompkins, S.C.; Sharma, A.; Fu, X.; Miao, R.; Pewa, A.D.; Brown, K.S.; et al. Hepatic Mitochondrial Pyruvate Carrier 1 Is Required for Efficient Regulation of Gluconeogenesis and Whole-Body Glucose Homeostasis. Cell Metab. 2015, 22, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Rauckhorst, A.J.; Gray, L.R.; Sheldon, R.D.; Fu, X.; Pewa, A.D.; Feddersen, C.R.; Dupuy, A.J.; Gibson-Corley, K.N.; Cox, J.E.; Burgess, S.C.; et al. The mitochondrial pyruvate carrier mediates high fat diet-induced increases in hepatic TCA cycle capacity. Mol. Metab. 2017, 6, 1468–1479. [Google Scholar] [CrossRef] [PubMed]
- Veliova, M.; Ferreira, C.M.; Benador, I.Y.; Jones, A.E.; Mahdaviani, K.; Brownstein, A.J.; Desousa, B.R.; Acín-Pérez, R.; Petcherski, A.; Assali, E.A.; et al. Blocking mitochondrial pyruvate import in brown adipocytes induces energy wasting via lipid cycling. EMBO Rep. 2020, 21, e49634. [Google Scholar] [CrossRef]
- Panic, V.; Pearson, S.; Banks, J.; Tippetts, T.S.; Velasco-Silva, J.N.; Lee, S.; Simcox, J.; Geoghegan, G.; Bensard, C.L.; van Ry, T.; et al. Mitochondrial pyruvate carrier is required for optimal brown fat thermogenesis. eLife 2020, 9, e52558. [Google Scholar] [CrossRef]
- Sharma, A.; Oonthonpan, L.; Sheldon, R.D.; Rauckhorst, A.J.; Zhu, Z.; Tompkins, S.C.; Cho, K.; Grzesik, W.J.; Gray, L.R.; Scerbo, D.A.; et al. Impaired skeletal muscle mitochondrial pyruvate uptake rewires glucose metabolism to drive whole-body leanness. eLife 2019, 8, e45873. [Google Scholar] [CrossRef]
- Liu, X.; Flores, A.A.; Situ, L.; Gu, W.; Ding, H.; Christofk, H.R.; Lowry, W.E.; Jung, M.E. Development of Novel Mitochondrial Pyruvate Carrier Inhibitors to Treat Hair Loss. J. Med. Chem. 2021, 64, 2046–2063. [Google Scholar] [CrossRef]
- Halestrap, A.P. The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors. Biochem. J. 1975, 148, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Divakaruni, A.S.; Wiley, S.E.; Rogers, G.W.; Andreyev, A.Y.; Petrosyan, S.; Loviscach, M.; Wall, E.A.; Yadava, N.; Heuck, A.P.; Ferrick, D.A.; et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc. Natl. Acad. Sci. USA 2013, 110, 5422–5427. [Google Scholar] [CrossRef] [Green Version]
- Hildyard, J.C.W.; Ämmälä, C.; Dukes, I.D.; Thomson, S.A.; Halestrap, A.P. Identification and characterisation of a new class of highly specific and potent inhibitors of the mitochondrial pyruvate carrier. Biochim. Biophys. Acta Bioenerg. 2005, 1707, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Zhu, Y.; Liu, M.; Zhou, Y.; Lu, G.; Lan, L.; Wang, X.; Zhao, Y.; Zhang, X.C. Molecular mechanism of substrate recognition and transport by the AtSWEET13 sugar transporter. Proc. Natl. Acad. Sci. USA 2017, 114, 10089–10094. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Nishizawa, T.; Yamashita, K.; Ishitani, R.; Nureki, O. Structural basis for the facilitative diffusion mechanism by SemiSWEET transporter. Nat. Commun. 2015, 6, 6112. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Tao, Y.; Cheung, L.S.; Fan, C.; Chen, L.-Q.; Xu, S.; Perry, K.; Frommer, W.B.; Feng, L. Structures of bacterial homologues of SWEET transporters in two distinct conformations. Nature 2014, 515, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided. Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-1: QikProp; Schrödinger, LLC: New York, NY, USA, 2020.
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Compan, V.; Pierredon, S.; Vanderperre, B.; Krznar, P.; Marchiq, I.; Zamboni, N.; Pouyssegur, J.; Martinou, J.C. Monitoring Mitochondrial Pyruvate Carrier Activity in Real Time Using a BRET-Based Biosensor: Investigation of the Warburg Effect. Mol. Cell 2015, 59, 491–501. [Google Scholar] [CrossRef] [Green Version]
- McCommis, K.S.; Kovacs, A.; Weinheimer, C.J.; Shew, T.M.; Koves, T.R.; Ilkayeva, O.R.; Kamm, D.R.; Pyles, K.D.; King, M.T.; Veech, R.L.; et al. Nutritional modulation of heart failure in mitochondrial pyruvate carrier–deficient mice. Nat. Metab. 2020, 2, 1232–1247. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Phelix, C.F.; Chen, L.Y. Structural Insights into the Human Mitochondrial Pyruvate Carrier Complexes. J. Chem. Inf. Model. 2021, 61, 5614–5625. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Samanta, I.; Mondal, A.; Liu, W.R. Covalent Inhibition in Drug Discovery. ChemMedChem 2019, 14, 889–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nancolas, B.; Guo, L.; Zhou, R.; Nath, K.; Nelson, D.S.; Leeper, D.B.; Blair, I.A.; Glickson, J.D.; Halestrap, A.P. The anti-tumour agent lonidamine is a potent inhibitor of the mitochondrial pyruvate carrier and plasma membrane monocarboxylate transporters. Biochem. J. 2016, 473, 929–936. [Google Scholar] [CrossRef] [Green Version]
- Divakaruni, A.S.; Wallace, M.; Buren, C.; Martyniuk, K.; Andreyev, A.Y.; Li, E.; Fields, J.A.; Cordes, T.; Reynolds, I.J.; Bloodgood, B.L.; et al. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J. Cell Biol. 2017, 216, 1091–1105. [Google Scholar] [CrossRef]
- Harrison, S.A.; Alkhouri, N.; Davison, B.A.; Sanyal, A.; Edwards, C.; Colca, J.R.; Lee, B.H.; Loomba, R.; Cusi, K.; Kolterman, O.; et al. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study. J. Hepatol. 2020, 72, 613–626. [Google Scholar] [CrossRef]
- McCommis, K.S.; Hodges, W.T.; Brunt, E.M.; Nalbantoglu, I.; McDonald, W.G.; Holley, C.; Fujiwara, H.; Schaffer, J.E.; Colca, J.R.; Finck, B.N. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology 2017, 65, 1543–1556. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | PhaseScreenScore | IC50 (µM) |
|---|---|---|
| UK-5099 | 2.66 | 0.241 |
| BE1975 | 2.61 | inactive |
| BE1976 | 2.61 | 0.033 |
| BE1978 | 2.57 | 0.117 |
| BE1980 | 2.62 | 0.162 |
| BE1984 | 2.60 | 1.53 |
| BE1985 | 2.58 | 0.638 |
| BE1988 | 2.39 | inactive |
| BE2617 | N.D. | 0.039 |
| BE2623 | N.D. | 0.731 |
| Compound | IC50 (µM) | cLogP | HERG Log IC50 | %Human Oral Absorption | LogSwat (m/L) (Solubility) |
|---|---|---|---|---|---|
| Range for 95% of Known Drugs | −2.0–6.5 | >−5 | >25% | −6.5–0.5 | |
| BE1976 | 0.033 | 2.37 | −3.879 | 70.347 | −4.728 |
| BE1978 | 0.117 | 2.07 | −3.305 | 69.044 | −3.838 |
| BE1980 | 0.162 | 3.05 | −3.209 | 78.955 | −5.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hegazy, L.; Gill, L.E.; Pyles, K.D.; Kaiho, C.; Kchouk, S.; Finck, B.N.; McCommis, K.S.; Elgendy, B. Identification of Novel Mitochondrial Pyruvate Carrier Inhibitors by Homology Modeling and Pharmacophore-Based Virtual Screening. Biomedicines 2022, 10, 365. https://doi.org/10.3390/biomedicines10020365
Hegazy L, Gill LE, Pyles KD, Kaiho C, Kchouk S, Finck BN, McCommis KS, Elgendy B. Identification of Novel Mitochondrial Pyruvate Carrier Inhibitors by Homology Modeling and Pharmacophore-Based Virtual Screening. Biomedicines. 2022; 10(2):365. https://doi.org/10.3390/biomedicines10020365
Chicago/Turabian StyleHegazy, Lamees, Lauren E. Gill, Kelly D. Pyles, Christopher Kaiho, Sophia Kchouk, Brian N. Finck, Kyle S. McCommis, and Bahaa Elgendy. 2022. "Identification of Novel Mitochondrial Pyruvate Carrier Inhibitors by Homology Modeling and Pharmacophore-Based Virtual Screening" Biomedicines 10, no. 2: 365. https://doi.org/10.3390/biomedicines10020365