
Role of Base Excision Repair in Innate Immune Cells and Its Relevance for Cancer Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Base Excision Repair in Innate Immune Cells
3. BER Modulates DNA Damage Induced Innate Immune Inflammatory Response
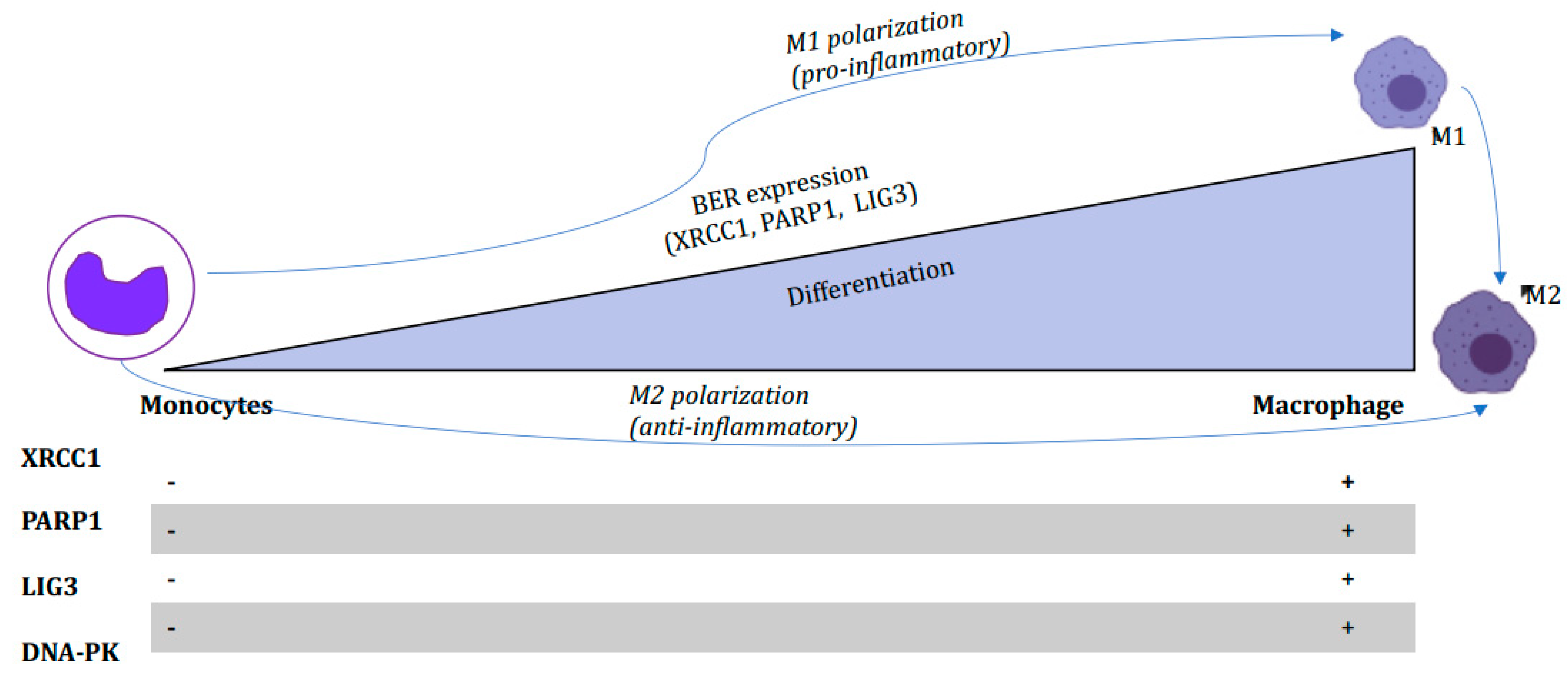
4. Role of BER in Macrophage Plasticity
5. Targeting PARP1 Modulate Macrophage Mediated Tumor Inflammatory Response
6. Targeting APE1 Modulate Macrophage Mediated Tumor Inflammatory Response
7. Exploiting BER as Innate Immune Modulator for Cancer Therapy
8. Conclusions and Future Direction
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C., Jr. Innate immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar] [CrossRef]
- Koenderman, L.; Buurman, W.; Daha, M.R. The innate immune response. Immunol. Lett. 2014, 162, 95–102. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R.; Janeway, C., Jr. Innate immune recognition: Mechanisms and pathways. Immunol. Rev. 2000, 173, 89–97. [Google Scholar] [CrossRef]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, G.M.; Kagan, J.C. A cell biological view of Toll-like receptor function: Regulation through compartmentalization. Nat. Rev. Immunol. 2009, 9, 535–542. [Google Scholar] [CrossRef]
- Chuenchor, W.; Jin, T.; Ravilious, G.; Xiao, T.S. Structures of pattern recognition receptors reveal molecular mechanisms of autoinhibition, ligand recognition and oligomerization. Curr. Opin. Immunol. 2014, 26, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, A.; Mikkelsen, H.; Jungersen, G. Intracellular Pathogens: Host Immunity and Microbial Persistence Strategies. J. Immunol. Res. 2019, 2019, 1356540. [Google Scholar] [CrossRef]
- Gause, W.C.; Wynn, T.A.; Allen, J.E. Type 2 immunity and wound healing: Evolutionary refinement of adaptive immunity by helminths. Nat. Rev. Immunol. 2013, 13, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coquel, F.; Silva, M.J.; Techer, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.L.; et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef]
- Briegert, M.; Kaina, B. Human monocytes, but not dendritic cells derived from them, are defective in base excision repair and hypersensitive to methylating agents. Cancer Res. 2007, 67, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auffray, C.; Sieweke, M.H.; Geissmann, F. Blood monocytes: Development, heterogeneity, and relationship with dendritic cells. Annu. Rev. Immunol. 2009, 27, 669–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S. Macrophage neutral proteinases and chronic inflammation. Ann. N. Y. Acad. Sci. 1976, 278, 176–189. [Google Scholar] [CrossRef]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Raggatt, L.J.; Wullschleger, M.E.; Alexander, K.A.; Wu, A.C.; Millard, S.M.; Kaur, S.; Maugham, M.L.; Gregory, L.S.; Steck, R.; Pettit, A.R. Fracture healing via periosteal callus formation requires macrophages for both initiation and progression of early endochondral ossification. Am. J. Pathol. 2014, 184, 3192–3204. [Google Scholar] [CrossRef] [PubMed]
- Minutti, C.M.; Knipper, J.A.; Allen, J.E.; Zaiss, D.M. Tissue-specific contribution of macrophages to wound healing. Semin. Cell Dev. Biol. 2017, 61, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Schraml, B.U.; Reis e Sousa, C. Defining dendritic cells. Curr. Opin. Immunol. 2015, 32, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.F.; Leong, C.F.; Cheong, S.K. Observation of dendritic cell morphology under light, phase-contrast or confocal laser scanning microscopy. Malays. J. Pathol. 2010, 32, 97–102. [Google Scholar] [PubMed]
- Bauer, M.; Goldstein, M.; Christmann, M.; Becker, H.; Heylmann, D.; Kaina, B. Human monocytes are severely impaired in base and DNA double-strand break repair that renders them vulnerable to oxidative stress. Proc. Natl. Acad. Sci. USA 2011, 108, 21105–21110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef] [Green Version]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Ueha, S.; Shand, F.H.; Matsushima, K. Myeloid cell population dynamics in healthy and tumor-bearing mice. Int. Immunopharmacol. 2011, 11, 783–788. [Google Scholar] [CrossRef]
- Durackova, Z. Some current insights into oxidative stress. Physiol. Res. 2010, 59, 459–469. [Google Scholar] [CrossRef]
- Nathan, C.; Shiloh, M.U. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. USA 2000, 97, 8841–8848. [Google Scholar] [CrossRef] [Green Version]
- Kawanishi, S.; Hiraku, Y.; Pinlaor, S.; Ma, N. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol. Chem. 2006, 387, 365–372. [Google Scholar] [CrossRef]
- Ischiropoulos, H.; Zhu, L.; Beckman, J.S. Peroxynitrite formation from macrophage-derived nitric oxide. Arch. Biochem. Biophys. 1992, 298, 446–451. [Google Scholar] [CrossRef]
- Zingarelli, B.; O’Connor, M.; Wong, H.; Salzman, A.L.; Szabo, C. Peroxynitrite-mediated DNA strand breakage activates poly-adenosine diphosphate ribosyl synthetase and causes cellular energy depletion in macrophages stimulated with bacterial lipopolysaccharide. J. Immunol. 1996, 156, 350–358. [Google Scholar]
- Xia, Y.; Zweier, J.L. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc. Natl. Acad. Sci. USA 1997, 94, 6954–6958. [Google Scholar] [CrossRef] [Green Version]
- Ponath, V.; Kaina, B. Death of Monocytes through Oxidative Burst of Macrophages and Neutrophils: Killing in Trans. PLoS ONE 2017, 12, e0170347. [Google Scholar] [CrossRef]
- Ohshima, H.; Sawa, T.; Akaike, T. 8-nitroguanine, a product of nitrative DNA damage caused by reactive nitrogen species: Formation, occurrence, and implications in inflammation and carcinogenesis. Antioxid. Redox Signal. 2006, 8, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Klattenhoff, A.W.; Thakur, M.; Sebastian, M.; Kidane, D. Mutation in DNA Polymerase Beta Causes Spontaneous Chromosomal Instability and Inflammation-Associated Carcinogenesis in Mice. Cancers 2019, 11, 1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Chaudhry, M.A.; Wallace, S.S. Base excision repair by hNTH1 and hOGG1: A two edged sword in the processing of DNA damage in gamma-irradiated human cells. DNA Repair 2006, 5, 43–51. [Google Scholar] [CrossRef]
- Eccles, L.J.; O’Neill, P.; Lomax, M.E. Delayed repair of radiation induced clustered DNA damage: Friend or foe? Mutat. Res. 2011, 711, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Sage, E.; Harrison, L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutat. Res. 2011, 711, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehrke, N.; Mertens, C.; Zillinger, T.; Wenzel, J.; Bald, T.; Zahn, S.; Tuting, T.; Hartmann, G.; Barchet, W. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 2013, 39, 482–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, Y.; Nash, R.A.; Klungland, A.; Schar, P.; Barnes, D.E.; Lindahl, T. Reconstitution of DNA base excision-repair with purified human proteins: Interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996, 15, 6662–6670. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.F.; Robins, P.; Carter, K.; Caldecott, K.; Pappin, D.J.; Yu, G.L.; Wang, R.P.; Shell, B.K.; Nash, R.A.; Schar, P.; et al. Molecular cloning and expression of human cDNAs encoding a novel DNA ligase IV and DNA ligase III, an enzyme active in DNA repair and recombination. Mol. Cell. Biol. 1995, 15, 3206–3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Geng, J.; Gao, J.; Zhao, H.; Li, J.; Shi, Y.; Yang, B.; Xiao, C.; Linghu, Y.; Sun, X.; et al. Macrophage achieves self-protection against oxidative stress-induced ageing through the Mst-Nrf2 axis. Nat. Commun. 2019, 10, 755. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid. Med. Cell Longev. 2016, 2016, 2795090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallucci, S.; Matzinger, P. Danger signals: SOS to the immune system. Curr. Opin. Immunol. 2001, 13, 114–119. [Google Scholar] [CrossRef]
- Krokan, H.E.; Nilsen, H.; Skorpen, F.; Otterlei, M.; Slupphaug, G. Base excision repair of DNA in mammalian cells. FEBS Lett. 2000, 476, 73–77. [Google Scholar] [CrossRef]
- Bauer, M.; Goldstein, M.; Heylmann, D.; Kaina, B. Human monocytes undergo excessive apoptosis following temozolomide activating the ATM/ATR pathway while dendritic cells and macrophages are resistant. PLoS ONE 2012, 7, e39956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N. Dietary carcinogens and anticarcinogens. Oxygen radicals and degenerative diseases. Science 1983, 221, 1256–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, D.E.; Lindahl, T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu. Rev. Genet. 2004, 38, 445–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.L.; Souza-Pinto, N.; Nyaga, S.G.; Thybo, T.; Stevnsner, T.; Bohr, V.A. Base excision repair in nuclear and mitochondrial DNA. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 285–297. [Google Scholar] [CrossRef]
- Wilson, S.H. Mammalian base excision repair and DNA polymerase beta. Mutat. Res. 1998, 407, 203–215. [Google Scholar] [CrossRef]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Fortini, P.; Dogliotti, E. Base damage and single-strand break repair: Mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair 2007, 6, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, A.E.; Chen, L.; Dong, Z.; Leppard, J.B.; Levin, D.S.; Mackey, Z.B.; Motycka, T.A. Completion of base excision repair by mammalian DNA ligases. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.S.; Balakrishnan, L.; Buncher, N.A.; Opresko, P.L.; Bambara, R.A. Telomere proteins POT1, TRF1 and TRF2 augment long-patch base excision repair in vitro. Cell Cycle 2012, 11, 998–1007. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-J.; Wilson, D.M., 3rd. Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 2012, 5, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Salati, S.; Bianchi, E.; Zini, R.; Tenedini, E.; Quaglino, D.; Manfredini, R.; Ferrari, S. Eosinophils, but not neutrophils, exhibit an efficient DNA repair machinery and high nucleolar activity. Haematologica 2007, 92, 1311–1318. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Lin, P.; Wu, Q.; Pu, Q.; Zhou, C.; Wang, B.; Gao, P.; Wang, Z.; Gao, A.; Overby, M.; et al. Small-Molecule Inhibitor of 8-Oxoguanine DNA Glycosylase 1 Regulates Inflammatory Responses during Pseudomonas aeruginosa Infection. J. Immunol. 2020, 205, 2231–2242. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yuan, K.; Yan, C.; Fox, J., 3rd; Gaid, M.; Breitwieser, W.; Bansal, A.K.; Zeng, H.; Gao, H.; Wu, M. 8-Oxoguanine-DNA glycosylase 1 deficiency modifies allergic airway inflammation by regulating STAT6 and IL-4 in cells and in mice. Free Radic. Biol. Med. 2012, 52, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Zhu, B.; Hao, W.; Zeng, X.; Vlahopoulos, S.A.; Hazra, T.K.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Brasier, A.R.; et al. Oxidized Guanine Base Lesions Function in 8-Oxoguanine DNA Glycosylase-1-mediated Epigenetic Regulation of Nuclear Factor kappaB-driven Gene Expression. J. Biol. Chem. 2016, 291, 25553–25566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, A.; Wakamiya, M.; Venkova-Canova, T.; Pandita, R.K.; Aguilera-Aguirre, L.; Sarker, A.H.; Singh, D.K.; Hosoki, K.; Wood, T.G.; Sharma, G.; et al. Neil2-null Mice Accumulate Oxidized DNA Bases in the Transcriptionally Active Sequences of the Genome and Are Susceptible to Innate Inflammation. J. Biol. Chem. 2015, 290, 24636–24648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontes, F.L.; Pinheiro, D.M.L.; Oliveira, A.H.S.d.; Oliveira, R.K.d.M.; Lajus, T.B.P.; Agnez-Lima, L.F. Role of DNA repair in host immune response and inflammation. Mutat. Res.-Rev. Mutat. Res. 2015, 763, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Yamtich, J.; Nemec, A.A.; Keh, A.; Sweasy, J.B. A germline polymorphism of DNA polymerase beta induces genomic instability and cellular transformation. PLoS Genet. 2012, 8, e1003052. [Google Scholar] [CrossRef] [PubMed]
- Ting, L.; Lingyun, Z.; Donghyun, J.; Shao-Cong, S. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Kidane, D.; Chae, W.J.; Czochor, J.; Eckert, K.A.; Glazer, P.M.; Bothwell, A.L.M.; Sweasy, J.B. Interplay between DNA repair and inflammation, and the link to cancer. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 116–139. [Google Scholar] [CrossRef]
- Li, N.; Banin, S.; Ouyang, H.; Li, G.C.; Courtois, G.; Shiloh, Y.; Karin, M.; Rotman, G. ATM is required for IkappaB kinase (IKKk) activation in response to DNA double strand breaks. J. Biol. Chem. 2001, 276, 8898–8903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ba, X.; Bacsi, A.; Luo, J.; Aguilera-Aguirre, L.; Zeng, X.; Radak, Z.; Brasier, A.R.; Boldogh, I. 8-oxoguanine DNA glycosylase-1 augments proinflammatory gene expression by facilitating the recruitment of site-specific transcription factors. J. Immunol. 2014, 192, 2384–2394. [Google Scholar] [CrossRef] [PubMed]
- Seifermann, M.; Ulges, A.; Bopp, T.; Melcea, S.; Schafer, A.; Oka, S.; Nakabeppu, Y.; Klungland, A.; Niehrs, C.; Epe, B. Role of the DNA repair glycosylase OGG1 in the activation of murine splenocytes. DNA Repair 2017, 58, 13–20. [Google Scholar] [CrossRef]
- Visnes, T.; Cazares-Korner, A.; Hao, W.; Wallner, O.; Masuyer, G.; Loseva, O.; Mortusewicz, O.; Wiita, E.; Sarno, A.; Manoilov, A.; et al. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 2018, 362, 834–839. [Google Scholar] [CrossRef] [Green Version]
- Mabley, J.G.; Pacher, P.; Deb, A.; Wallace, R.; Elder, R.H.; Szabo, C. Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. FASEB J. 2005, 19, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017, 31, 353–369. [Google Scholar] [CrossRef] [Green Version]
- Soulat, D.; Bauch, A.; Stockinger, S.; Superti-Furga, G.; Decker, T. Cytoplasmic Listeria monocytogenes stimulates IFN-beta synthesis without requiring the adapter protein MAVS. FEBS Lett. 2006, 580, 2341–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, S.; Reutterer, B.; Schaljo, B.; Schellack, C.; Brunner, S.; Materna, T.; Yamamoto, M.; Akira, S.; Taniguchi, T.; Murray, P.J.; et al. IFN regulatory factor 3-dependent induction of type I IFNs by intracellular bacteria is mediated by a TLR- and Nod2-independent mechanism. J. Immunol. 2004, 173, 7416–7425. [Google Scholar] [CrossRef] [Green Version]
- Nociari, M.; Ocheretina, O.; Schoggins, J.W.; Falck-Pedersen, E. Sensing infection by adenovirus: Toll-like receptor-independent viral DNA recognition signals activation of the interferon regulatory factor 3 master regulator. J. Virol. 2007, 81, 4145–4157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.B.; Sorensen, L.N.; Malmgaard, L.; Ank, N.; Baines, J.D.; Chen, Z.J.; Paludan, S.R. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J. Virol. 2007, 81, 13315–13324. [Google Scholar] [CrossRef] [Green Version]
- Stetson, D.B.; Medzhitov, R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 2006, 24, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klattenhoff, A.W.; Thakur, M.; Chu, C.S.; Ray, D.; Habib, S.L.; Kidane, D. Loss of NEIL3 DNA glycosylase markedly increases replication associated double strand breaks and enhances sensitivity to ATR inhibitor in glioblastoma cells. Oncotarget 2017, 8, 112942–112958. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83. [Google Scholar] [CrossRef]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA damage response. Cold Spring Harb. Perspect. Biol. 2011, 3, a000745. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Z.J. Innate Immune Sensing and Signaling of Cytosolic Nucleic Acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef]
- Dhanwani, R.; Takahashi, M.; Sharma, S. Cytosolic sensing of immuno-stimulatory DNA, the enemy within. Curr. Opin. Immunol. 2018, 50, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Doublié, S.; Wallace, S.S. Neil3, the final frontier for the DNA glycosylases that recognize oxidative damage. Mutat. Res. 2013, 743–744, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, M.; Fleming, A.M.; Burrows, C.J.; Wallace, S.S. Neil3 and NEIL1 DNA glycosylases remove oxidative damages from quadruplex DNA and exhibit preferences for lesions in the telomeric sequence context. J. Biol. Chem. 2013, 288, 27263–27272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massaad, M.J.; Zhou, J.; Tsuchimoto, D.; Chou, J.; Jabara, H.; Janssen, E.; Glauzy, S.; Olson, B.G.; Morbach, H.; Ohsumi, T.K.; et al. Deficiency of base excision repair enzyme NEIL3 drives increased predisposition to autoimmunity. J. Clin. Investig. 2016, 126, 4219–4236. [Google Scholar] [CrossRef]
- Sakumi, K.; Tominaga, Y.; Furuichi, M.; Xu, P.; Tsuzuki, T.; Sekiguchi, M.; Nakabeppu, Y. Ogg1 knockout-associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Res. 2003, 63, 902–905. [Google Scholar]
- Casorelli, I.; Pannellini, T.; De Luca, G.; Degan, P.; Chiera, F.; Iavarone, I.; Giuliani, A.; Butera, A.; Boirivant, M.; Musiani, P.; et al. The Mutyh base excision repair gene influences the inflammatory response in a mouse model of ulcerative colitis. PLoS ONE 2010, 5, e12070. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Dai, H.; Zhou, M.; Li, M.; Singh, P.; Qiu, J.; Tsark, W.; Huang, Q.; Kernstine, K.; Zhang, X.; et al. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat. Med. 2007, 13, 812–819. [Google Scholar] [CrossRef]
- He, L.; Zhang, Y.; Sun, H.; Jiang, F.; Yang, H.; Wu, H.; Zhou, T.; Hu, S.; Kathera, C.S.; Wang, X.; et al. Targeting DNA Flap Endonuclease 1 to Impede Breast Cancer Progression. EBioMedicine 2016, 14, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Schaar, B.; Tallapragada, S.; Dorigo, O. Tumor associated macrophages in gynecologic cancers. Gynecol. Oncol. 2018, 149, 205–213. [Google Scholar] [CrossRef]
- Gupta, V.; Yull, F.; Khabele, D. Bipolar Tumor-Associated Macrophages in Ovarian Cancer as Targets for Therapy. Cancers 2018, 10, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Smallwood, H.S.; Lopez-Ferrer, D.; Eberlein, P.E.; Watson, D.J.; Squier, T.C. Calmodulin mediates DNA repair pathways involving H2AX in response to low-dose radiation exposure of RAW 264.7 macrophages. Chem. Res. Toxicol. 2009, 22, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Paludan, S.R.; Bowie, A.G. Immune sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Wassermann, R.; Gulen, M.F.; Sala, C.; Perin, S.G.; Lou, Y.; Rybniker, J.; Schmid-Burgk, J.L.; Schmidt, T.; Hornung, V.; Cole, S.T.; et al. Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host Microbe 2015, 17, 799–810. [Google Scholar] [CrossRef] [Green Version]
- Rahman, I.; Marwick, J.; Kirkham, P. Redox modulation of chromatin remodeling: Impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem. Pharmacol. 2004, 68, 1255–1267. [Google Scholar] [CrossRef]
- Roux, C.; Jafari, S.M.; Shinde, R.; Duncan, G.; Cescon, D.W.; Silvester, J.; Chu, M.F.; Hodgson, K.; Berger, T.; Wakeham, A.; et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc. Natl. Acad. Sci. USA 2019, 116, 4326–4335. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [Green Version]
- Dale, D.C.; Boxer, L.; Liles, W.C. The phagocytes: Neutrophils and monocytes. Blood 2008, 112, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.S.; Chen, F.H.; Wang, C.C.; Huang, H.L.; Jung, S.M.; Wu, C.J.; Lee, C.C.; McBride, W.H.; Chiang, C.S.; Hong, J.H. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 499–507. [Google Scholar] [CrossRef]
- van Ravenswaay Claasen, H.H.; Kluin, P.M.; Fleuren, G.J. Tumor infiltrating cells in human cancer. On the possible role of CD16+ macrophages in antitumor cytotoxicity. Lab. Investig. 1992, 67, 166–174. [Google Scholar] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Denton, A.E.; Roberts, E.W.; Fearon, D.T. Stromal Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2018, 1060, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.N.; Watari, K.; Fotovati, A.; Hosoi, F.; Yasumoto, K.; Izumi, H.; Kohno, K.; Umezawa, K.; Iguchi, H.; Shirouzu, K.; et al. Inflammatory stimuli from macrophages and cancer cells synergistically promote tumor growth and angiogenesis. Cancer Sci. 2007, 98, 2009–2018. [Google Scholar] [CrossRef]
- Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol. Rev. 2008, 222, 155–161. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, J.; Xiao, Z.; Guo, C.; Pu, Q.; Ma, L.; Liu, C.; Lin, F.; Liao, H.; You, Z.; Liu, L. Prognostic impact of tumor-associated macrophage infiltration in non-small cell lung cancer: A systemic review and meta-analysis. Oncotarget 2016, 7, 34217–34228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, S.; Huang, J.; Li, Z.; Zhang, J.; Luo, J.; Lu, C.; Xu, H.; Xu, H. The Prognostic and Clinicopathological Significance of Tumor-Associated Macrophages in Patients with Gastric Cancer: A Meta-Analysis. PLoS ONE 2017, 12, e0170042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Zajac, E.; Schweighofer, B.; Kupriyanova, T.A.; Juncker-Jensen, A.; Minder, P.; Quigley, J.P.; Deryugina, E.I. Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood 2013, 122, 4054–4067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Sayaman, R.W.; Saad, M.; Thorsson, V.; Hu, D.; Hendrickx, W.; Roelands, J.; Porta-Pardo, E.; Mokrab, Y.; Farshidfar, F.; Kirchhoff, T.; et al. Germline genetic contribution to the immune landscape of cancer. Immunity 2021, 54, 367–386.e8. [Google Scholar] [CrossRef]
- Zhu, J.; Jia, W.; Wu, C.; Fu, W.; Xia, H.; Liu, G.; He, J. Base Excision Repair Gene Polymorphisms and Wilms Tumor Susceptibility. EBioMedicine 2018, 33, 88–93. [Google Scholar] [CrossRef]
- Mattar, M.M.; Zekri, A.N.; Hussein, N.; Morsy, H.; Esmat, G.; Amin, M.A. Polymorphisms of base-excision repair genes and the hepatocarcinogenesis. Gene 2018, 675, 62–68. [Google Scholar] [CrossRef]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef] [Green Version]
- Rozacky, J.; Nemec, A.A.; Sweasy, J.B.; Kidane, D. Gastric cancer associated variant of DNA polymerase beta (Leu22Pro) promotes DNA replication associated double strand breaks. Oncotarget 2015, 6, 24474–24487. [Google Scholar] [CrossRef] [Green Version]
- Popanda, O.; Seibold, P.; Nikolov, I.; Oakes, C.C.; Burwinkel, B.; Hausmann, S.; Flesch-Janys, D.; Plass, C.; Chang-Claude, J.; Schmezer, P. Germline variants of base excision repair genes and breast cancer: A polymorphism in DNA polymerase gamma modifies gene expression and breast cancer risk. Int. J. Cancer 2013, 132, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Carter, H.; Marty, R.; Hofree, M.; Gross, A.M.; Jensen, J.; Fisch, K.M.; Wu, X.; DeBoever, C.; Van Nostrand, E.L.; Song, Y.; et al. Interaction Landscape of Inherited Polymorphisms with Somatic Events in Cancer. Cancer Discov. 2017, 7, 410–423. [Google Scholar] [CrossRef] [Green Version]
- Consortium, I.T.P.-C.A.o.W.G. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Goulielmaki, E.; Ioannidou, A.; Tsekrekou, M.; Stratigi, K.; Poutakidou, I.K.; Gkirtzimanaki, K.; Aivaliotis, M.; Evangelou, K.; Topalis, P.; Altmuller, J.; et al. Tissue-infiltrating macrophages mediate an exosome-based metabolic reprogramming upon DNA damage. Nat. Commun. 2020, 11, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Macrophages define the invasive microenvironment in breast cancer. J. Leukoc. Biol. 2008, 84, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Ohtaki, Y.; Ishii, G.; Nagai, K.; Ashimine, S.; Kuwata, T.; Hishida, T.; Nishimura, M.; Yoshida, J.; Takeyoshi, I.; Ochiai, A. Stromal macrophage expressing CD204 is associated with tumor aggressiveness in lung adenocarcinoma. J. Thorac. Oncol. 2010, 5, 1507–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, L.A.; Rafat, M.; Colomer, M.V.; Giaccia, A.; Graves, E. Tumor-associated macrophages enhance DNA damage repair and improve survival of murine breast cancers after irradiation. Cancer Res. 2016, 76, 1645. [Google Scholar]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Mitri, D.; Mirenda, M.; Vasilevska, J.; Calcinotto, A.; Delaleu, N.; Revandkar, A.; Gil, V.; Boysen, G.; Losa, M.; Mosole, S.; et al. Re-education of Tumor-Associated Macrophages by CXCR2 Blockade Drives Senescence and Tumor Inhibition in Advanced Prostate Cancer. Cell Rep. 2019, 28, 2156–2168.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Bottazzi, B.; Colotta, F.; Sozzani, S.; Ruco, L. The origin and function of tumor-associated macrophages. Immunol. Today 1992, 13, 265–270. [Google Scholar] [CrossRef]
- Sun, Y.; Nelson, P.S. Molecular pathways: Involving microenvironment damage responses in cancer therapy resistance. Clin. Cancer Res. 2012, 18, 4019–4025. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.A.; Yang, B.; Tang, T.; Yang, Y.; Zhang, D.; Xiao, H.; Xu, J.; Wang, L.; Lin, L.; Jiang, J. Correlation of APE1 with VEGFA and CD163(+) macrophage infiltration in bladder cancer and their prognostic significance. Oncol. Lett. 2020, 20, 2881–2887. [Google Scholar] [CrossRef]
- Mehta, A.K.; Cheney, E.M.; Hartl, C.A.; Pantelidou, C.; Oliwa, M.; Castrillon, J.A.; Lin, J.R.; Hurst, K.E.; de Oliveira Taveira, M.; Johnson, N.T.; et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat. Cancer 2021, 2, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Mouchemore, K.A.; Pixley, F.J. CSF-1 signaling in macrophages: Pleiotrophy through phosphotyrosine-based signaling pathways. Crit. Rev. Clin. Lab. Sci. 2012, 49, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Gouon-Evans, V.; Nguyen, A.V.; Pollard, J.W. The macrophage growth factor CSF-1 in mammary gland development and tumor progression. J. Mammary Gland. Biol. Neoplasia 2002, 7, 147–162. [Google Scholar] [CrossRef]
- Azzam, G.; Wang, X.; Bell, D.; Murphy, M.E. CSF1 is a novel p53 target gene whose protein product functions in a feed-forward manner to suppress apoptosis and enhance p53-mediated growth arrest. PLoS ONE 2013, 8, e74297. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, M.B.; Danielsen, A.V.; Hamilton-Dutoit, S.J.; Bendix, K.; Norgaard, P.; Moller, M.B.; Steiniche, T.; d’Amore, F. High intratumoral macrophage content is an adverse prognostic feature in anaplastic large cell lymphoma. Histopathology 2014, 65, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Nguewa, P.A.; Fuertes, M.A.; Valladares, B.; Alonso, C.; Perez, J.M. Poly(ADP-ribose) polymerases: Homology, structural domains and functions. Novel therapeutical applications. Prog. Biophys. Mol. Biol. 2005, 88, 143–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, A. PARP inhibitors: Its role in treatment of cancer. Chin. J. Cancer 2011, 30, 463–471. [Google Scholar] [CrossRef]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef] [Green Version]
- Sobczak, M.; Zyma, M.; Robaszkiewicz, A. The Role of PARP1 in Monocyte and Macrophage Commitment and Specification: Future Perspectives and Limitations for the Treatment of Monocyte and Macrophage Relevant Diseases with PARP Inhibitors. Cells 2020, 9, 2040. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, P.; Płoszaj, T.; Regdon, Z.; Virag, L.; Robaszkiewicz, A. PARP1-LSD1 functional interplay controls transcription of SOD2 that protects human pro-inflammatory macrophages from death under an oxidative condition. Free Radic. Biol. Med. 2019, 131, 218–224. [Google Scholar] [CrossRef]
- Lee, E.K.; Konstantinopoulos, P.A. PARP inhibition and immune modulation: Scientific rationale and perspectives for the treatment of gynecologic cancers. Ther. Adv. Med. Oncol. 2020, 12, 1758835920944116. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Scaffidi, P.; Degryse, B.; Bonaldi, T.; Ronfani, L.; Agresti, A.; Beltrame, M.; Bianchi, M.E. New EMBO members’ review: The double life of HMGB1 chromatin protein: Architectural factor and extracellular signal. EMBO J. 2001, 20, 4337–4340. [Google Scholar] [CrossRef]
- Falciola, L.; Spada, F.; Calogero, S.; Langst, G.; Voit, R.; Grummt, I.; Bianchi, M.E. High mobility group 1 protein is not stably associated with the chromosomes of somatic cells. J. Cell Biol. 1997, 137, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.E. HMGB1 loves company. J. Leukoc. Biol. 2009, 86, 573–576. [Google Scholar] [CrossRef]
- Gorgulho, C.M.; Romagnoli, G.G.; Bharthi, R.; Lotze, M.T. Johnny on the Spot-Chronic Inflammation Is Driven by HMGB1. Front. Immunol. 2019, 10, 1561. [Google Scholar] [CrossRef]
- Rojas, A.; Delgado-Lopez, F.; Perez-Castro, R.; Gonzalez, I.; Romero, J.; Rojas, I.; Araya, P.; Anazco, C.; Morales, E.; Llanos, J. HMGB1 enhances the protumoral activities of M2 macrophages by a RAGE-dependent mechanism. Tumor Biol. 2016, 37, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Xiang, A.; Peng, T.; Doran, A.C.; Tracey, K.J.; Barnes, B.J.; Tabas, I.; Son, M.; Diamond, B. HMGB1-C1q complexes regulate macrophage function by switching between leukotriene and specialized proresolving mediator biosynthesis. Proc. Natl. Acad. Sci. USA 2019, 116, 23254–23263. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Henon, C.; Dorvault, N.; Rouanne, M.; et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Investig. 2019, 129, 1211–1228. [Google Scholar] [CrossRef]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jonsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappaB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almine, J.F.; O’Hare, C.A.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef]
- Jonsson, K.L.; Laustsen, A.; Krapp, C.; Skipper, K.A.; Thavachelvam, K.; Hotter, D.; Egedal, J.H.; Kjolby, M.; Mohammadi, P.; Prabakaran, T.; et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat. Commun. 2017, 8, 14391. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Curran, T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992, 11, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Demple, B.; Herman, T.; Chen, D.S. Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: Definition of a family of DNA repair enzymes. Proc. Natl. Acad. Sci. USA 1991, 88, 11450–11454. [Google Scholar] [CrossRef] [Green Version]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antioxid. Redox Signal. 2009, 11, 601–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, J.; Park, H.; Sung, S.H.; Moon, B.I.; Suh, H.; Lim, W. Prognostic value of human apurinic/apyrimidinic endonuclease 1 (APE1) expression in breast cancer. PLoS ONE 2014, 9, e99528. [Google Scholar] [CrossRef]
- Yoo, D.G.; Song, Y.J.; Cho, E.J.; Lee, S.K.; Park, J.B.; Yu, J.H.; Lim, S.P.; Kim, J.M.; Jeon, B.H. Alteration of APE1/ref-1 expression in non-small cell lung cancer: The implications of impaired extracellular superoxide dismutase and catalase antioxidant systems. Lung Cancer 2008, 60, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.L.; He, F.; Ye, J.Z.; Wu, H.N.; Zhang, J.Y.; Liu, Z.H.; Li, Y.Q.; Luo, X.L.; Lin, Y.; Liang, R. APE1 overexpression is associated with poor survival in patients with solid tumors: A meta-analysis. Oncotarget 2017, 8, 59720–59728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedinak, A.; Dudhgaonkar, S.; Kelley, M.R.; Sliva, D. Apurinic/Apyrimidinic endonuclease 1 regulates inflammatory response in macrophages. Anticancer Res. 2011, 31, 379–385. [Google Scholar] [PubMed]
- Qing, Y.; Li, Q.; Ren, T.; Xia, W.; Peng, Y.; Liu, G.L.; Luo, H.; Yang, Y.X.; Dai, X.Y.; Zhou, S.F.; et al. Upregulation of PD-L1 and APE1 is associated with tumorigenesis and poor prognosis of gastric cancer. Drug Des. Dev. Ther. 2015, 9, 901–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.; Charles, K.A.; Mantovani, A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005, 7, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T.; Willoughby, D.A.; Gilroy, D.W. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat. Rev. Immunol. 2002, 2, 787–795. [Google Scholar] [CrossRef]
- Kaplanski, G.; Marin, V.; Montero-Julian, F.; Mantovani, A.; Farnarier, C. IL-6: A regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003, 24, 25–29. [Google Scholar] [CrossRef]
- Bosca, L.; Zeini, M.; Traves, P.G.; Hortelano, S. Nitric oxide and cell viability in inflammatory cells: A role for NO in macrophage function and fate. Toxicology 2005, 208, 249–258. [Google Scholar] [CrossRef]
- Aguilera-Aguirre, L.; Bacsi, A.; Radak, Z.; Hazra, T.K.; Mitra, S.; Sur, S.; Brasier, A.R.; Ba, X.; Boldogh, I. Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1-KRAS-NF-kappaB pathway. J. Immunol. 2014, 193, 4643–4653. [Google Scholar] [CrossRef] [Green Version]
- Arneth, B. Systemic Lupus Erythematosus and DNA Degradation and Elimination Defects. Front Immunol. 2019, 10, 1697. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.; Kinsella, T.J. Essential role of DNA base excision repair on survival in an acidic tumor microenvironment. Cancer Res. 2009, 69, 7285–7293. [Google Scholar] [CrossRef] [Green Version]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016, 14, 282–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.S.; Sansanaphongpricha, K.; Xie, Y.; Donnelly, C.R.; Luo, X.; Heath, B.R.; Zhao, X.; Bellile, E.; Hu, H.; Chen, H.; et al. Mitigating SOX2-potentiated Immune Escape of Head and Neck Squamous Cell Carcinoma with a STING-inducing Nanosatellite Vaccine. Clin. Cancer Res. 2018, 24, 4242–4255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Wang, X.D.; Yu, Y. PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. eLife 2020, 9, e60637. [Google Scholar] [CrossRef]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef]
- Wang, C.; Guan, Y.; Lv, M.; Zhang, R.; Guo, Z.; Wei, X.; Du, X.; Yang, J.; Li, T.; Wan, Y.; et al. Manganese Increases the Sensitivity of the cGAS-STING Pathway for Double-Stranded DNA and Is Required for the Host Defense against DNA Viruses. Immunity 2018, 48, 675–687.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Ma, Z.; Wang, B.; Guan, Y.; Su, X.D.; Jiang, Z. Mn(2+) Directly Activates cGAS and Structural Analysis Suggests Mn(2+) Induces a Noncanonical Catalytic Synthesis of 2′3′-cGAMP. Cell Rep. 2020, 32, 108053. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Donigan, K.A.; Hile, S.E.; Eckert, K.A.; Sweasy, J.B. The human gastric cancer-associated DNA polymerase beta variant D160N is a mutator that induces cellular transformation. DNA Repair 2012, 11, 381–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, B.; Castellsague, E.; Bah, I.; van Kempen, L.C.; Foulkes, W.D. Biallelic NTHL1 Mutations in a Woman with Multiple Primary Tumors. N. Engl. J. Med. 2015, 373, 1985–1986. [Google Scholar] [CrossRef] [PubMed]
- Donigan, K.A.; Sun, K.W.; Nemec, A.A.; Murphy, D.L.; Cong, X.; Northrup, V.; Zelterman, D.; Sweasy, J.B. Human POLB gene is mutated in high percentage of colorectal tumors. J. Biol. Chem. 2012, 287, 23830–23839. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.S.; Kudchadkar, R.R.; Yu, B.; Gallenstein, D.; Horak, C.E.; Inzunza, H.D.; Zhao, X.; Martinez, A.J.; Wang, W.; Gibney, G.; et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J. Clin. Oncol. 2013, 31, 4311–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Shan, B.; Zhang, Y.; Ren, Y.; Liang, S.; Zhao, J.; Zhao, Z.; Wang, G.; Zhao, X.; Peng, D.; et al. Association between DNA damage repair gene somatic mutations and immune-related gene expression in ovarian cancer. Cancer Med. 2020, 9, 2190–2200. [Google Scholar] [CrossRef] [Green Version]
- Permata, T.B.M.; Hagiwara, Y.; Sato, H.; Yasuhara, T.; Oike, T.; Gondhowiardjo, S.; Held, K.D.; Nakano, T.; Shibata, A. Base excision repair regulates PD-L1 expression in cancer cells. Oncogene 2019, 38, 4452–4466. [Google Scholar] [CrossRef] [PubMed]
- Colebatch, A.; Hitchins, M.; Williams, R.; Meagher, A.; Hawkins, N.J.; Ward, R.L. The role of MYH and microsatellite instability in the development of sporadic colorectal cancer. Br. J. Cancer 2006, 95, 1239–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizotte, P.H.; Baird, J.R.; Stevens, C.A.; Lauer, P.; Green, W.R.; Brockstedt, D.G.; Fiering, S.N. Attenuated Listeria monocytogenes reprograms M2-polarized tumor-associated macrophages in ovarian cancer leading to iNOS-mediated tumor cell lysis. Oncoimmunology 2014, 3, e28926. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, S.; Habib, S.L.; Senejani, A.G.; Sebastian, M.; Kidane, D. Role of Base Excision Repair in Innate Immune Cells and Its Relevance for Cancer Therapy. Biomedicines 2022, 10, 557. https://doi.org/10.3390/biomedicines10030557
Zhao S, Habib SL, Senejani AG, Sebastian M, Kidane D. Role of Base Excision Repair in Innate Immune Cells and Its Relevance for Cancer Therapy. Biomedicines. 2022; 10(3):557. https://doi.org/10.3390/biomedicines10030557
Chicago/Turabian StyleZhao, Shengyuan, Samy L. Habib, Alireza G. Senejani, Manu Sebastian, and Dawit Kidane. 2022. "Role of Base Excision Repair in Innate Immune Cells and Its Relevance for Cancer Therapy" Biomedicines 10, no. 3: 557. https://doi.org/10.3390/biomedicines10030557
APA StyleZhao, S., Habib, S. L., Senejani, A. G., Sebastian, M., & Kidane, D. (2022). Role of Base Excision Repair in Innate Immune Cells and Its Relevance for Cancer Therapy. Biomedicines, 10(3), 557. https://doi.org/10.3390/biomedicines10030557