
Immune-Mediated Inflammatory Responses of Alveolar Epithelial Cells: Implications for COVID-19 Lung Pathology

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Models
2.2. Experimental Treatments
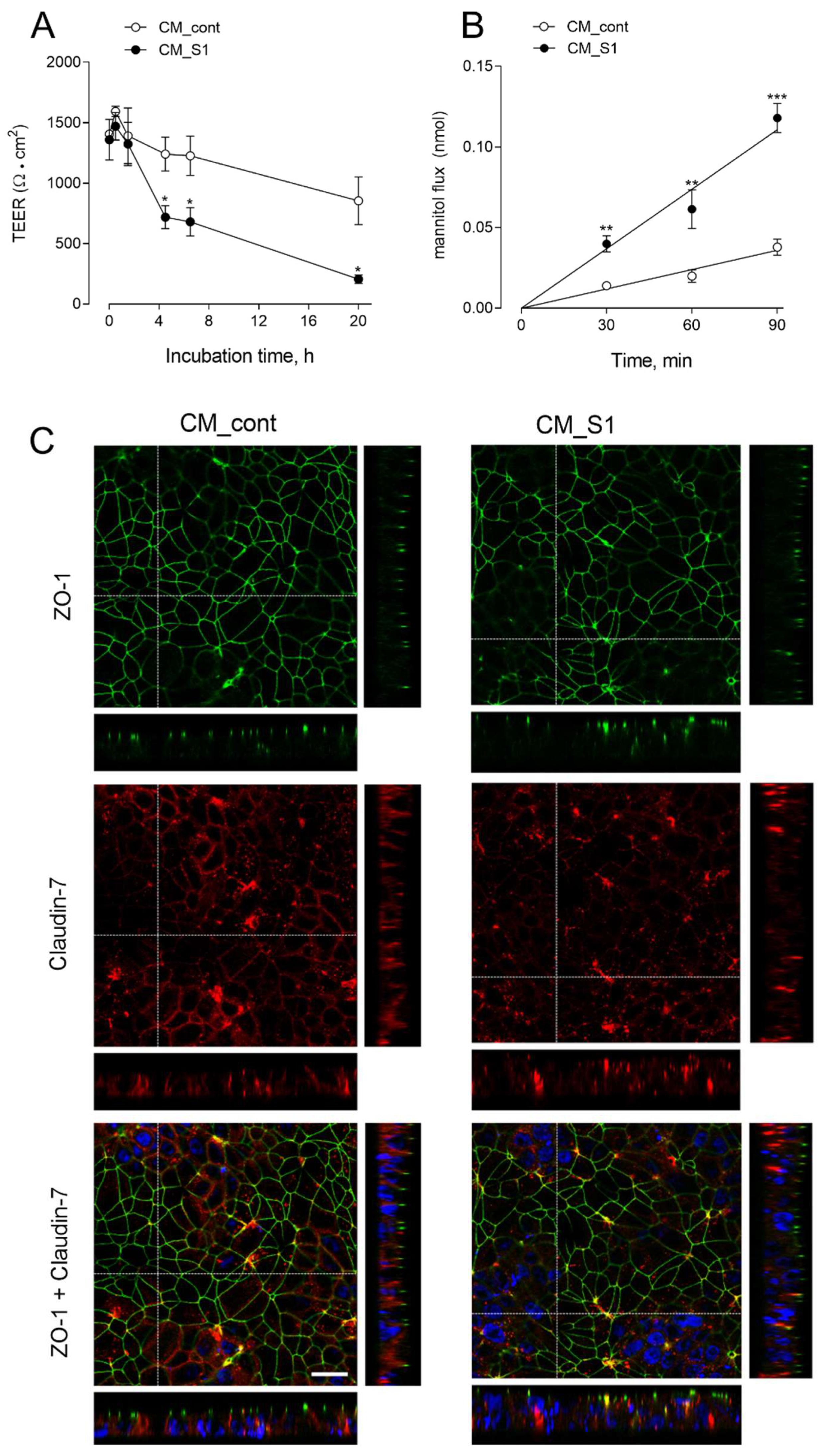
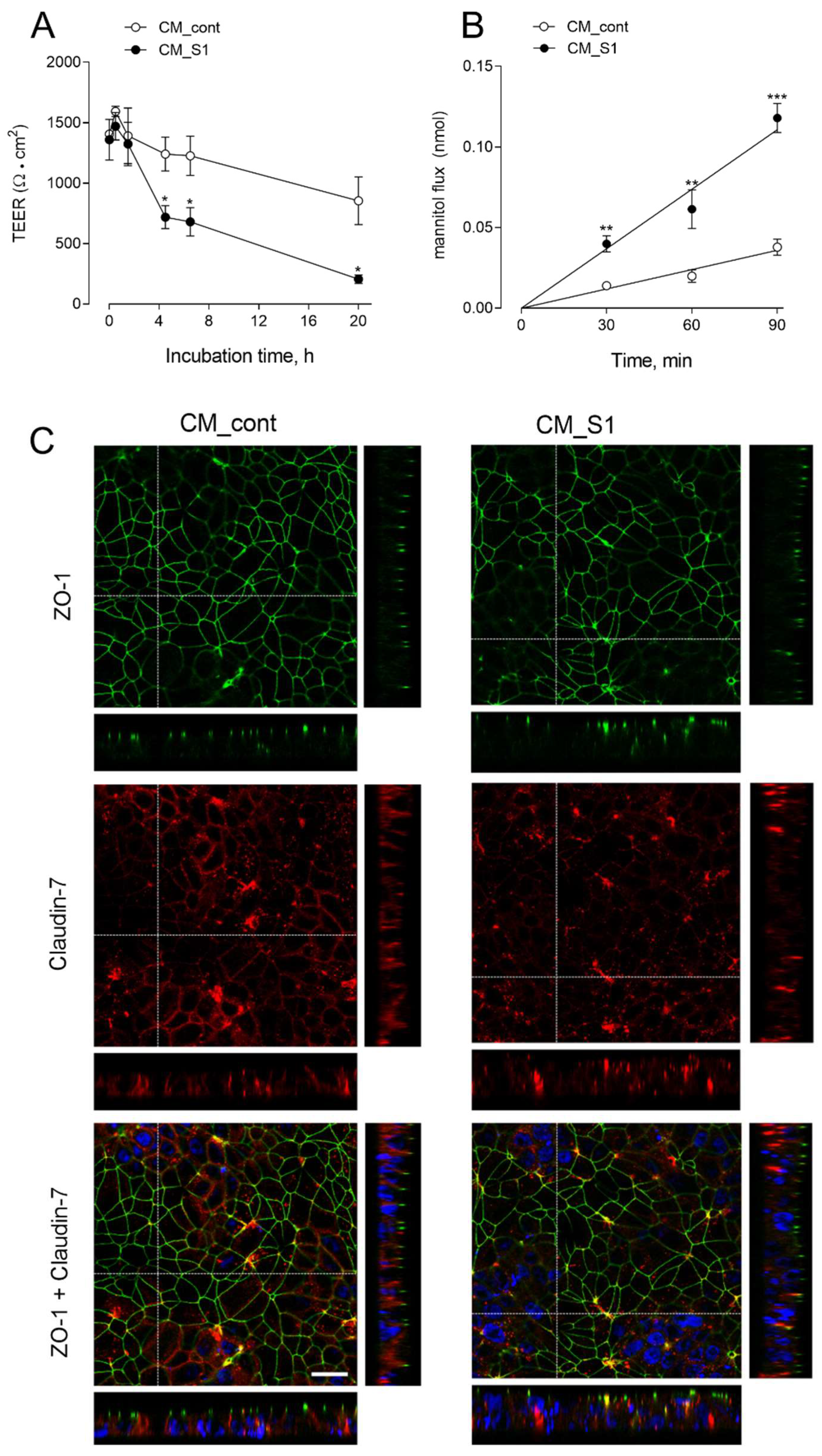
2.3. Measurements of TEER and Paracellular Permeability
2.4. RT-qPCR Analysis
2.5. Cytokine Analysis
2.6. Western Blot Analysis
2.7. Immunocytochemistry
2.8. Statistical Analysis
2.9. Materials
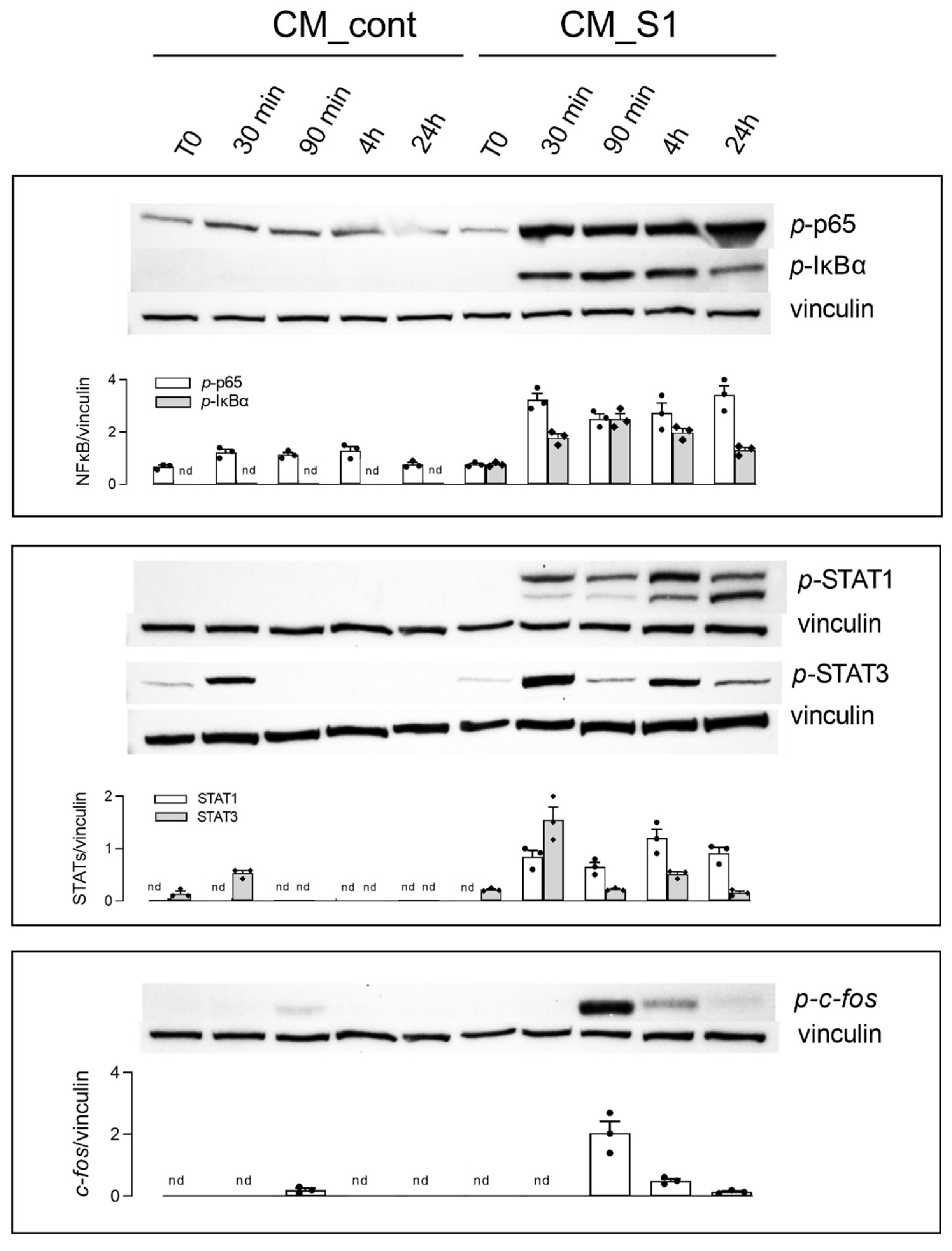
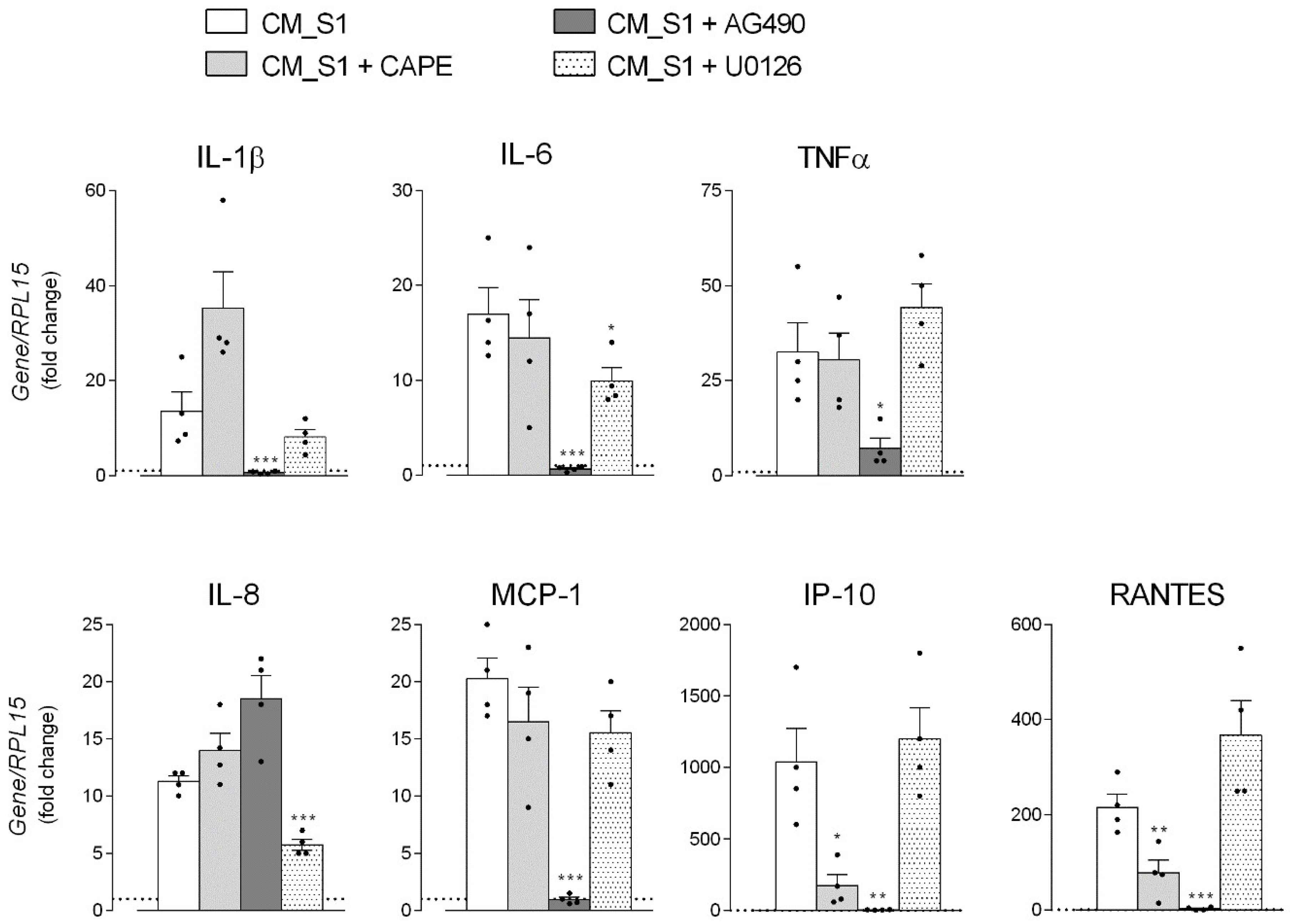
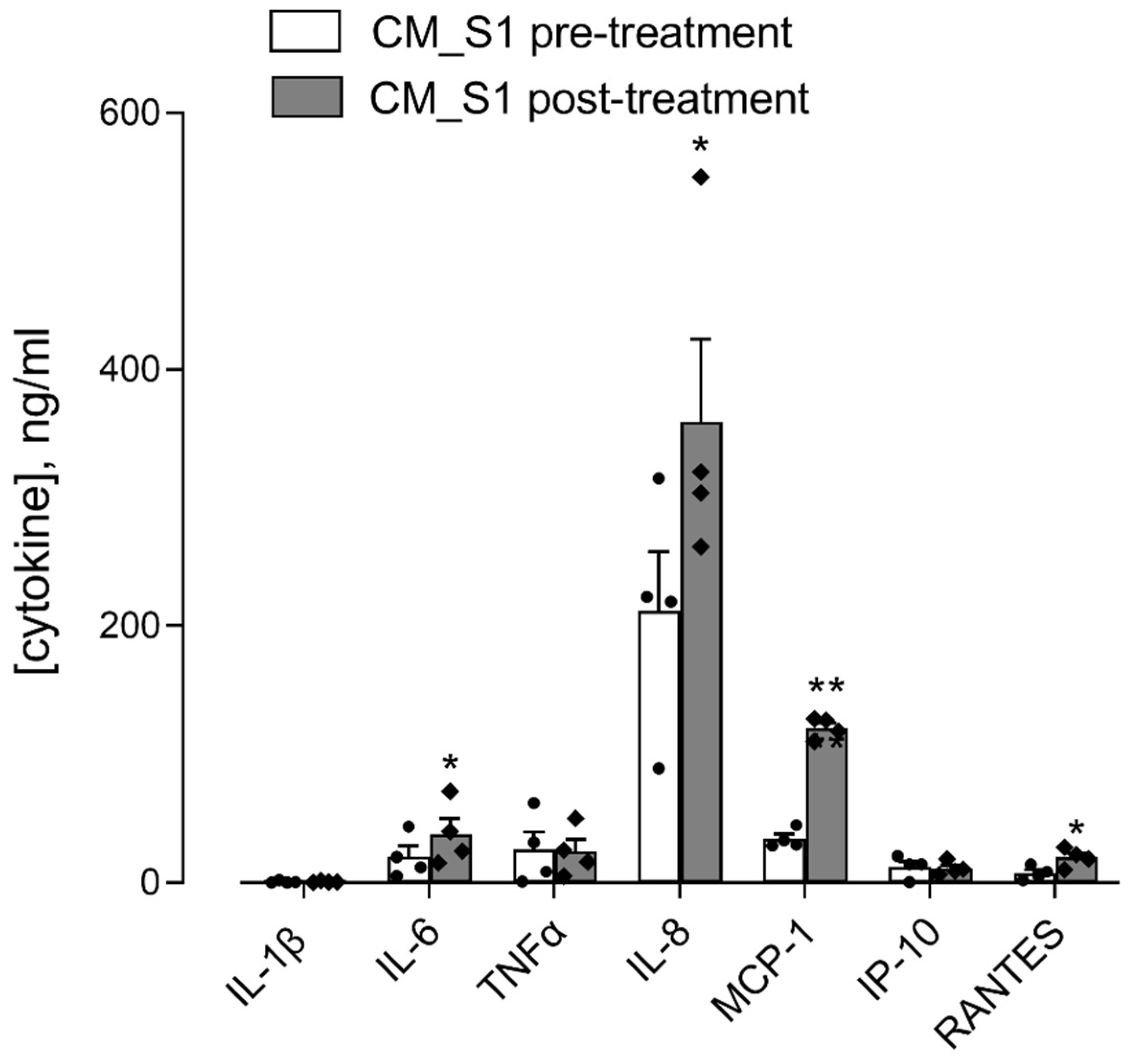
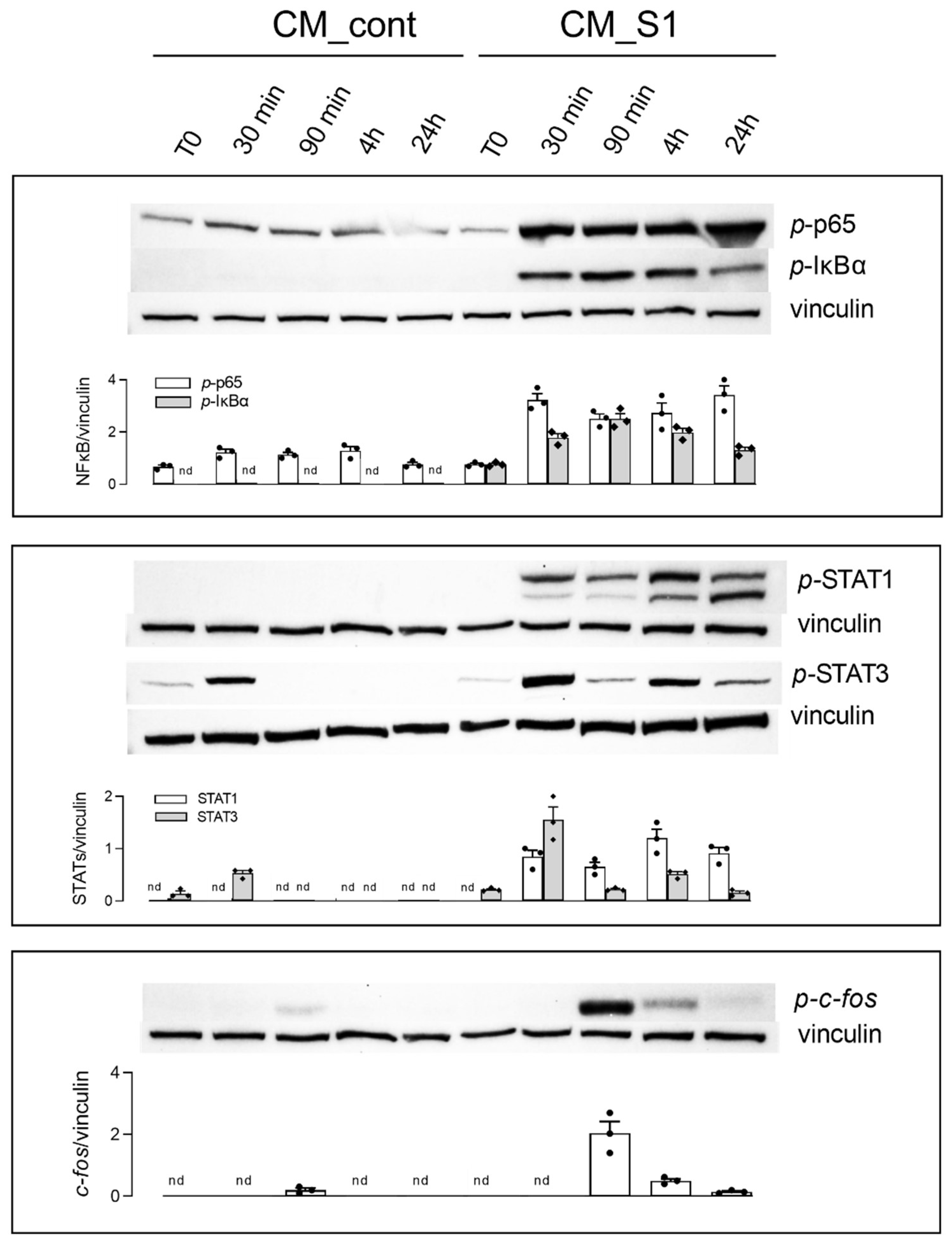
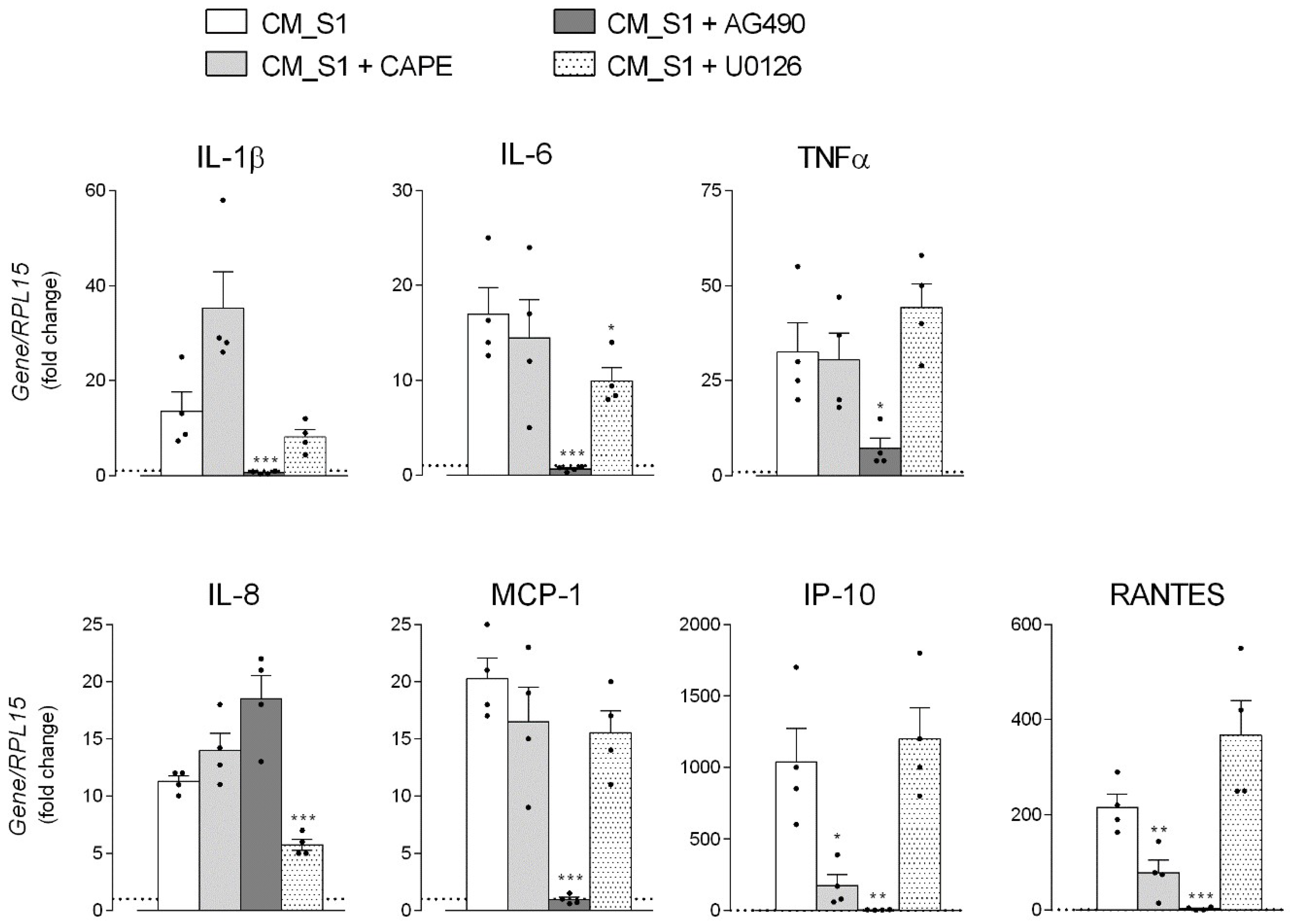
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in wuhan, china. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of covid-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Scialo, F.; Daniele, A.; Amato, F.; Pastore, L.; Matera, M.G.; Cazzola, M.; Castaldo, G.; Bianco, A. Ace2: The major cell entry receptor for sars-cov-2. Lung 2020, 198, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, A.; Geng, K.; Honnen, W.; Wang, X.; Bruiners, N.; Singh, S.; Ferrara, F.; D’Angelo, S.; Bradbury, A.R.; et al. Human immunodeficiency viruses pseudotyped with sars-cov-2 spike proteins infect a broad spectrum of human cell lines through multiple entry mechanisms. Viruses 2021, 13, 953. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the sars-cov-2 spike glycoprotein. Cell 2020, 183, 1735. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Chaudhuri, R.; Joshi, M.C.; Almatroudi, A.; Rahmani, A.H.; Ali, S.M. Covid-19: The emerging immunopathological determinants for recovery or death. Front. Microbiol. 2020, 11, 588409. [Google Scholar] [CrossRef]
- Bastard, P.; Zhang, Q.; Zhang, S.Y.; Jouanguy, E.; Casanova, J.L. Type i interferons and sars-cov-2: From cells to organisms. Curr. Opin. Immunol. 2022, 74, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Effort, C.H.G.; Cobat, A.; Casanova, J.L. Human genetic and immunological determinants of critical covid-19 pneumonia. Nature 2022. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with covid-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Ricci, D.; Etna, M.P.; Rizzo, F.; Sandini, S.; Severa, M.; Coccia, E.M. Innate immune response to sars-cov-2 infection: From cells to soluble mediators. Int. J. Mol. Sci. 2021, 22, 7017. [Google Scholar] [CrossRef] [PubMed]
- Anka, A.U.; Tahir, M.I.; Abubakar, S.D.; Alsabbagh, M.; Zian, Z.; Hamedifar, H.; Sabzevari, A.; Azizi, G. Coronavirus disease 2019 (covid-19): An overview of the immunopathology, serological diagnosis and management. Scand. J. Immunol. 2021, 93, e12998. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; HLH Across Speciality Collaboration. Covid-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Kosyreva, A.; Dzhalilova, D.; Lokhonina, A.; Vishnyakova, P.; Fatkhudinov, T. The role of macrophages in the pathogenesis of sars-cov-2-associated acute respiratory distress syndrome. Front. Immunol. 2021, 12, 682871. [Google Scholar] [CrossRef]
- Wang, J.; Yang, X.; Li, Y.; Huang, J.A.; Jiang, J.; Su, N. Specific cytokines in the inflammatory cytokine storm of patients with covid-19-associated acute respiratory distress syndrome and extrapulmonary multiple-organ dysfunction. Virol. J. 2021, 18, 117. [Google Scholar] [CrossRef]
- Rotoli, B.M.; Barilli, A.; Visigalli, R.; Ferrari, F.; Dall’Asta, V. Endothelial cell activation by sars-cov-2 spike s1 protein: A crosstalk between endothelium and innate immune cells. Biomedicines 2021, 9, 1220. [Google Scholar] [CrossRef]
- Karwaciak, I.; Salkowska, A.; Karas, K.; Dastych, J.; Ratajewski, M. Nucleocapsid and spike proteins of the coronavirus sars-cov-2 induce il6 in monocytes and macrophages-potential implications for cytokine storm syndrome. Vaccines 2021, 9, 54. [Google Scholar] [CrossRef]
- Olajide, O.A.; Iwuanyanwu, V.U.; Lepiarz-Raba, I.; Al-Hindawi, A.A. Induction of exaggerated cytokine production in human peripheral blood mononuclear cells by a recombinant sars-cov-2 spike glycoprotein s1 and its inhibition by dexamethasone. Inflammation 2021, 44, 1865–1877. [Google Scholar] [CrossRef]
- Shirato, K.; Kizaki, T. Sars-cov-2 spike protein s1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 2021, 7, e06187. [Google Scholar] [CrossRef]
- Ingoglia, F.; Visigalli, R.; Rotoli, B.M.; Barilli, A.; Riccardi, B.; Puccini, P.; Milioli, M.; Di Lascia, M.; Bernuzzi, G.; Dall’Asta, V. Human macrophage differentiation induces octn2-mediated l-carnitine transport through stimulation of mtor-stat3 axis. J. Leukoc. Biol. 2017, 101, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Barilli, A.; Visigalli, R.; Ferrari, F.; Borsani, G.; Dall’Asta, V.; Rotoli, B.M. Flagellin from pseudomonas aeruginosa stimulates atb(0,+) transporter for arginine and neutral amino acids in human airway epithelial cells. Front. Immunol. 2021, 12, 641563. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.; Singh, S.; Burke, T.R.; Grunberger, D., Jr.; Aggarwal, B.B. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor nf-kappa b. Proc. Natl. Acad. Sci. USA 1996, 93, 9090–9095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meydan, N.; Grunberger, T.; Dadi, H.; Shahar, M.; Arpaia, E.; Lapidot, Z.; Leeder, J.S.; Freedman, M.; Cohen, A.; Gazit, A.; et al. Inhibition of acute lymphoblastic leukaemia by a jak-2 inhibitor. Nature 1996, 379, 645–648. [Google Scholar] [CrossRef]
- Favata, M.F.; Horiuchi, K.Y.; Manos, E.J.; Daulerio, A.J.; Stradley, D.A.; Feeser, W.S.; Van Dyk, D.E.; Pitts, W.J.; Earl, R.A.; Hobbs, F.; et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Birch, N.P.; Suresh, V. An optimised human cell culture model for alveolar epithelial transport. PLoS ONE 2016, 11, e0165225. [Google Scholar] [CrossRef]
- Kuehn, A.; Kletting, S.; de Souza Carvalho-Wodarz, C.; Repnik, U.; Griffiths, G.; Fischer, U.; Meese, E.; Huwer, H.; Wirth, D.; May, T.; et al. Human alveolar epithelial cells expressing tight junctions to model the air-blood barrier. ALTEX 2016, 33, 251–260. [Google Scholar] [CrossRef]
- Capaldo, C.T.; Nusrat, A. Cytokine regulation of tight junctions. Biochim. Biophys. Acta 2009, 1788, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Metz, J.K.; Wiegand, B.; Schnur, S.; Knoth, K.; Schneider-Daum, N.; Gross, H.; Croston, G.; Reinheimer, T.M.; Lehr, C.-M.; Hittinger, M. Modulating the barrier function of human alveolar epithelial (haelvi) cell monolayers as a model of inflammation. Altern. Lab. Anim. 2020, 48, 252–267. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the dependency of cellular protein levels on mrna abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Grifoni, E.; Valoriani, A.; Cei, F.; Lamanna, R.; Gelli, A.M.G.; Ciambotti, B.; Vannucchi, V.; Moroni, F.; Pelagatti, L.; Tarquini, R.; et al. Interleukin-6 as prognosticator in patients with covid-19. J. Infect. 2020, 81, 452–482. [Google Scholar] [CrossRef]
- Herold, T.; Jurinovic, V.; Arnreich, C.; Lipworth, B.J.; Hellmuth, J.C.; von Bergwelt-Baildon, M.; Klein, M.; Weinberger, T. Elevated levels of il-6 and crp predict the need for mechanical ventilation in covid-19. J. Allergy Clin. Immunol. 2020, 146, 128–136 e4. [Google Scholar] [CrossRef]
- Ulhaq, Z.S.; Soraya, G.V. Interleukin-6 as a potential biomarker of covid-19 progression. Med. Mal. Infect. 2020, 50, 382–383. [Google Scholar] [CrossRef] [PubMed]
- Kesmez Can, F.; Ozkurt, Z.; Ozturk, N.; Sezen, S. Effect of il-6, il-8/cxcl8, ip-10/cxcl 10 levels on the severity in covid 19 infection. Int. J. Clin. Pract. 2021, 75, e14970. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts covid-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Gasparello, J.; d’Aversa, E.; Breveglieri, G.; Borgatti, M.; Finotti, A.; Gambari, R. In vitro induction of interleukin-8 by sars-cov-2 spike protein is inhibited in bronchial epithelial ib3-1 cells by a mir-93-5p agomir. Int. Immunopharmacol. 2021, 101, 108201. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Baker, C.S.; Evans, T.W.; Haslam, P.L. G-csf and il-8 but not gm-csf correlate with severity of pulmonary neutrophilia in acute respiratory distress syndrome. Eur. Respir. J. 2000, 15, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Sekido, N.; Akahoshi, T.; Wada, T.; Mukaida, N.; Matsushima, K. Essential involvement of interleukin-8 (il-8) in acute inflammation. J. Leukoc. Biol. 1994, 56, 559–564. [Google Scholar] [CrossRef]
- Kinnare, N.; Hook, J.S.; Patel, P.A.; Monson, N.L.; Moreland, J.G. Neutrophil extracellular trap formation potential correlates with lung disease severity in covid-19 patients. Inflammation 2021. [Google Scholar] [CrossRef]
- Iturricastillo, G.; Avalos Perez-Urria, E.; Counago, F.; Landete, P. Scientific evidence in the covid-19 treatment: A comprehensive review. World J. Virol. 2021, 10, 217–228. [Google Scholar] [CrossRef]
- Farahani, M.; Niknam, Z.; Mohammadi Amirabad, L.; Amiri-Dashatan, N.; Koushki, M.; Nemati, M.; Pouya, F.D.; Rezaei-Tavirani, M.; Rasmi, Y.; Tayebi, L. Molecular pathways involved in covid-19 and potential pathway-based therapeutic targets. Biomed. Pharmacother. 2022, 145, 112420. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, M. Nf-kappab signalling as a pharmacological target in covid-19: Potential roles for ikkbeta inhibitors. Naunyn. Schmiedebergs Arch. Pharmacol. 2021, 394, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Attiq, A.; Yao, L.J.; Afzal, S.; Khan, M.A. The triumvirate of nf-kappab, inflammation and cytokine storm in covid-19. Int. Immunopharmacol. 2021, 101, 108255. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. Sars-cov-2 spike protein induces inflammation via tlr2-dependent activation of the nf-kappab pathway. Elife 2021, 10, e68563. [Google Scholar] [CrossRef]
- Grant, A.H.; Estrada, A., 3rd; Ayala-Marin, Y.M.; Alvidrez-Camacho, A.Y.; Rodriguez, G.; Robles-Escajeda, E.; Cadena-Medina, D.A.; Rodriguez, A.C.; Kirken, R.A. The many faces of jaks and stats within the covid-19 storm. Front. Immunol. 2021, 12, 690477. [Google Scholar] [CrossRef]
- Salem, F.; Li, X.Z.; Hindi, J.; Casablanca, N.M.; Zhong, F.; El Jamal, S.M.; Al Rasheed, M.R.H.; Li, L.; Lee, K.; Chan, L.; et al. Activation of stat3 signaling pathway in the kidney of covid-19 patients. J. Nephrol. 2021, 1–9. [Google Scholar] [CrossRef]
- Hariharan, A.; Hakeem, A.R.; Radhakrishnan, S.; Reddy, M.S.; Rela, M. The role and therapeutic potential of nf-kappa-b pathway in severe covid-19 patients. Inflammopharmacology 2021, 29, 91–100. [Google Scholar] [CrossRef]
- Kircheis, R.; Haasbach, E.; Lueftenegger, D.; Heyken, W.T.; Ocker, M.; Planz, O. Nf-kappab pathway as a potential target for treatment of critical stage covid-19 patients. Front. Immunol. 2020, 11, 598444. [Google Scholar] [CrossRef]
- Seif, F.; Aazami, H.; Khoshmirsafa, M.; Kamali, M.; Mohsenzadegan, M.; Pornour, M.; Mansouri, D. Jak inhibition as a new treatment strategy for patients with covid-19. Int. Arch. Allergy Immunol. 2020, 181, 467–475. [Google Scholar] [CrossRef]
- Gatti, M.; Turrini, E.; Raschi, E.; Sestili, P.; Fimognari, C. Janus kinase inhibitors and coronavirus disease (covid)-19: Rationale, clinical evidence and safety issues. Pharmaceuticals 2021, 14, 738. [Google Scholar] [CrossRef]
- Limen, R.Y.; Sedono, R.; Sugiarto, A.; Hariyanto, T.I. Janus kinase (jak)-inhibitors and coronavirus disease 2019 (covid-19) outcomes: A systematic review and meta-analysis. Expert. Rev. Anti Infect. Ther. 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.C.; Li, S.; Yuan, L.X.; Chen, R.A.; Liu, D.X.; Fung, T.S. Induction of the proinflammatory chemokine interleukin-8 is regulated by integrated stress response and ap-1 family proteins activated during coronavirus infection. Int. J. Mol. Sci. 2021, 22, 5646. [Google Scholar] [CrossRef] [PubMed]
- Cesta, M.C.; Zippoli, M.; Marsiglia, C.; Gavioli, E.M.; Mantelli, F.; Allegretti, M.; Balk, R.A. The role of interleukin-8 in lung inflammation and injury: Implications for the management of covid-19 and hyperinflammatory acute respiratory distress syndrome. Front. Pharmacol. 2021, 12, 808797. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/Protein | Forward Primer | Reverse Primer |
|---|---|---|
| RPL15/RPL15 | Hs03855120_g1 (TaqMan® Assay, Thermo Fisher Scientific) | |
| IL1B/IL-1β | Hs99999029_m1 (TaqMan® Assay, Thermo Fisher Scientific) | |
| IL6/IL-6 | AACCTGAACCTTCCAAAGATGG | TCTGGCTTGTTCCTCACTACT |
| TNFA/TNFα | ATGAGCACTGAAAGCATGATCC | GAGGGCTGATTAGAGAGAGGTC |
| CXCL8/IL-8 | ACTGAGAGTGATTGAGAGTGGAC | AACCCTCTGCACCCAGTTTTC |
| CXCL10/IP-10 | GTGGCATTCAAGGAGTACCTC | TGATGGCCTTCGATTCTGGATT |
| CCL2/MCP-1 | CAGCCAGATGCAATCAATGCC | TGGAATCCTGAACCCACTTCT |
| CCL5/RANTES | CTCCCCATATTCCTCGGACA | GTTGATGTACTCCCGAACCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barilli, A.; Visigalli, R.; Ferrari, F.; Bianchi, M.G.; Dall’Asta, V.; Rotoli, B.M. Immune-Mediated Inflammatory Responses of Alveolar Epithelial Cells: Implications for COVID-19 Lung Pathology. Biomedicines 2022, 10, 618. https://doi.org/10.3390/biomedicines10030618
Barilli A, Visigalli R, Ferrari F, Bianchi MG, Dall’Asta V, Rotoli BM. Immune-Mediated Inflammatory Responses of Alveolar Epithelial Cells: Implications for COVID-19 Lung Pathology. Biomedicines. 2022; 10(3):618. https://doi.org/10.3390/biomedicines10030618
Chicago/Turabian StyleBarilli, Amelia, Rossana Visigalli, Francesca Ferrari, Massimiliano G. Bianchi, Valeria Dall’Asta, and Bianca Maria Rotoli. 2022. "Immune-Mediated Inflammatory Responses of Alveolar Epithelial Cells: Implications for COVID-19 Lung Pathology" Biomedicines 10, no. 3: 618. https://doi.org/10.3390/biomedicines10030618
APA StyleBarilli, A., Visigalli, R., Ferrari, F., Bianchi, M. G., Dall’Asta, V., & Rotoli, B. M. (2022). Immune-Mediated Inflammatory Responses of Alveolar Epithelial Cells: Implications for COVID-19 Lung Pathology. Biomedicines, 10(3), 618. https://doi.org/10.3390/biomedicines10030618