
Gene Variants Involved in Nonsense-Mediated mRNA Decay Suggest a Role in Autism Spectrum Disorder

 , ,
, ,  , and
, and
Abstract
:1. Introduction
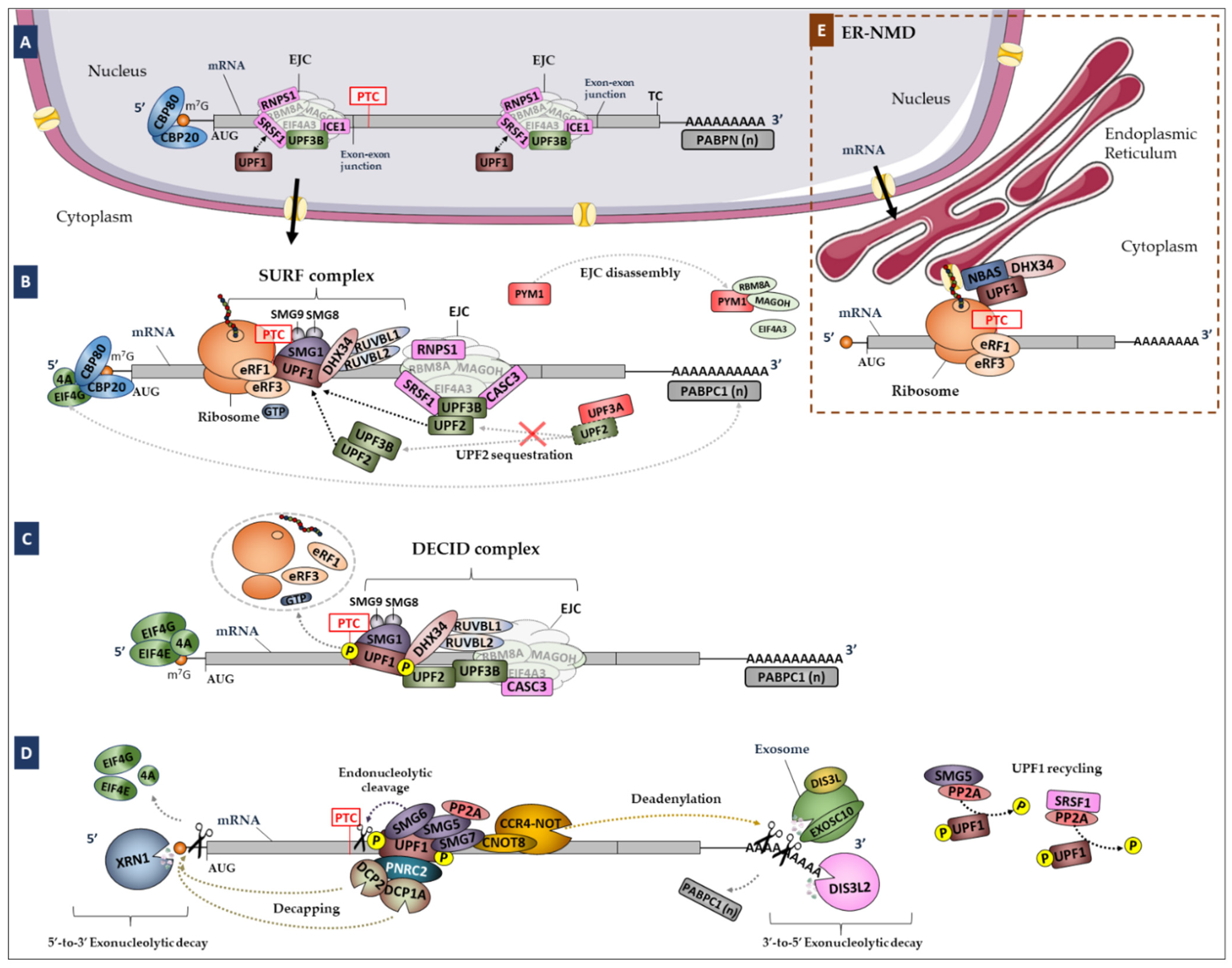
2. Materials and Methods
2.1. Identification of Genes Encoding NMD Factors and Regulators
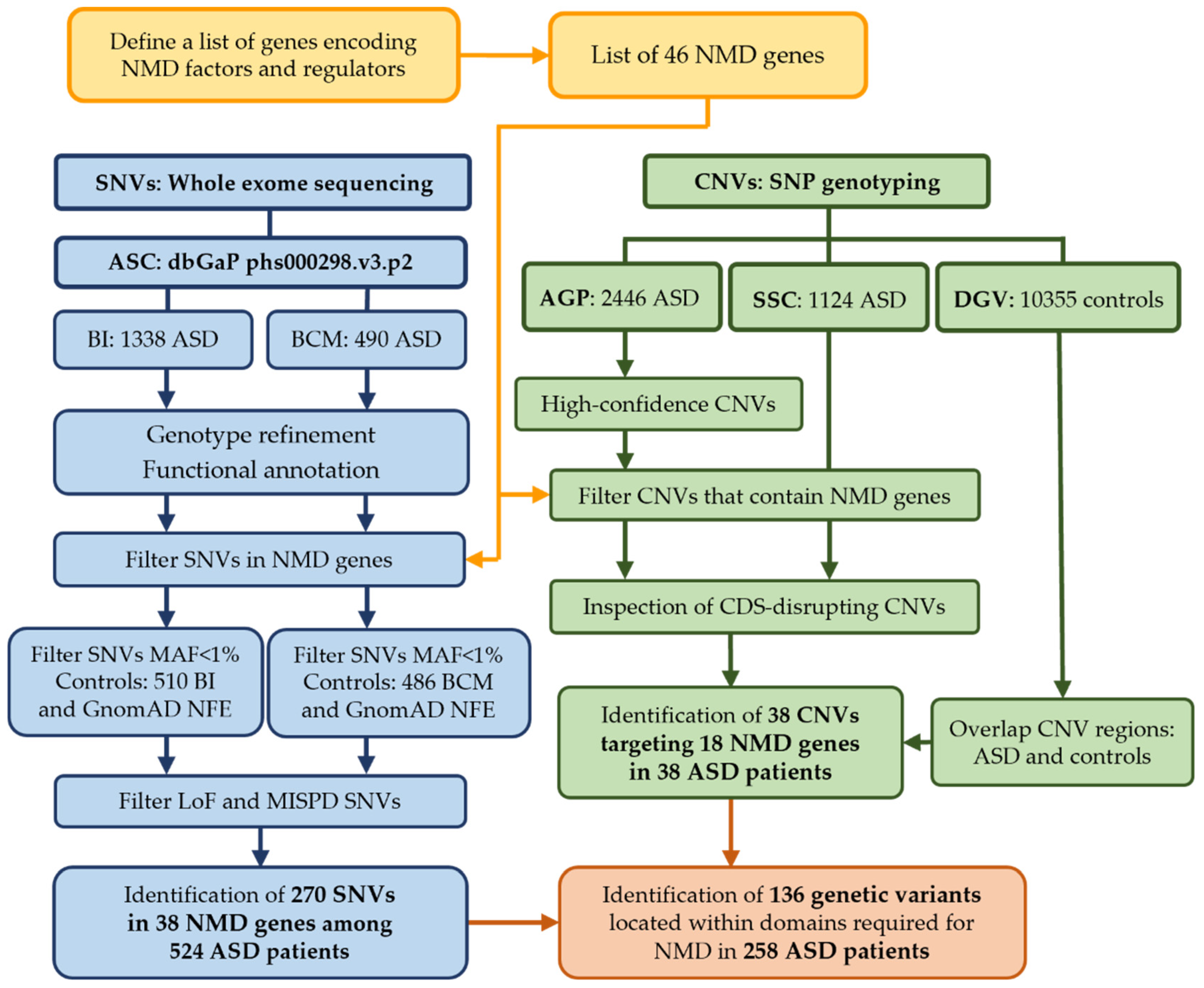
2.2. ASD Genomic Datasets
2.3. Sequencing Data Processing, Annotation and SNV Discovery
2.4. CNV Discovery
2.5. Protein Domains
2.6. ASD Candidate Genes
2.7. Brain Expression of NMD Genes
3. Results
3.1. Genes Encoding Proteins Involved in the NMD Pathway
Evidence for NMD Involvement in ASD Pathophysiology
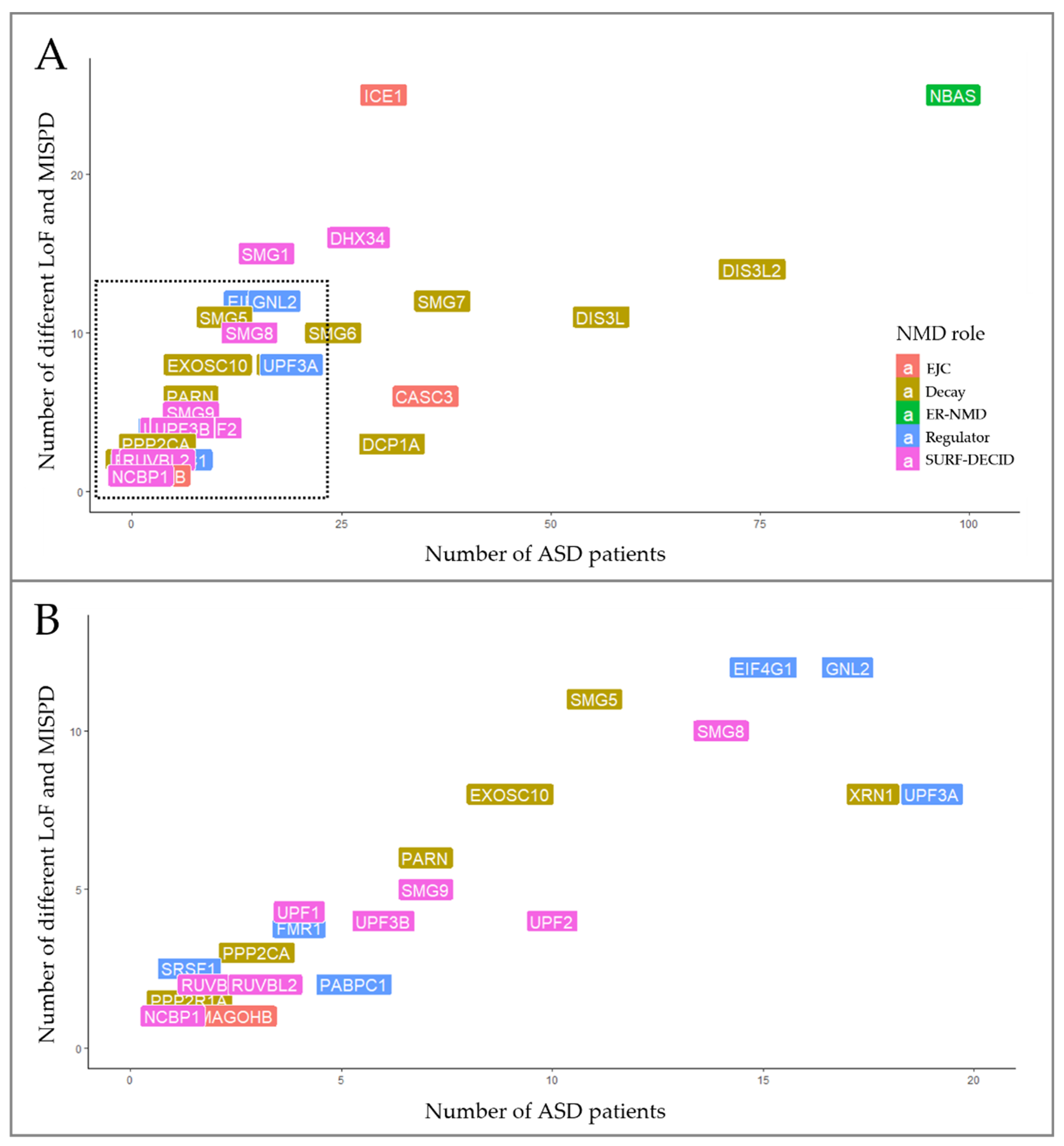
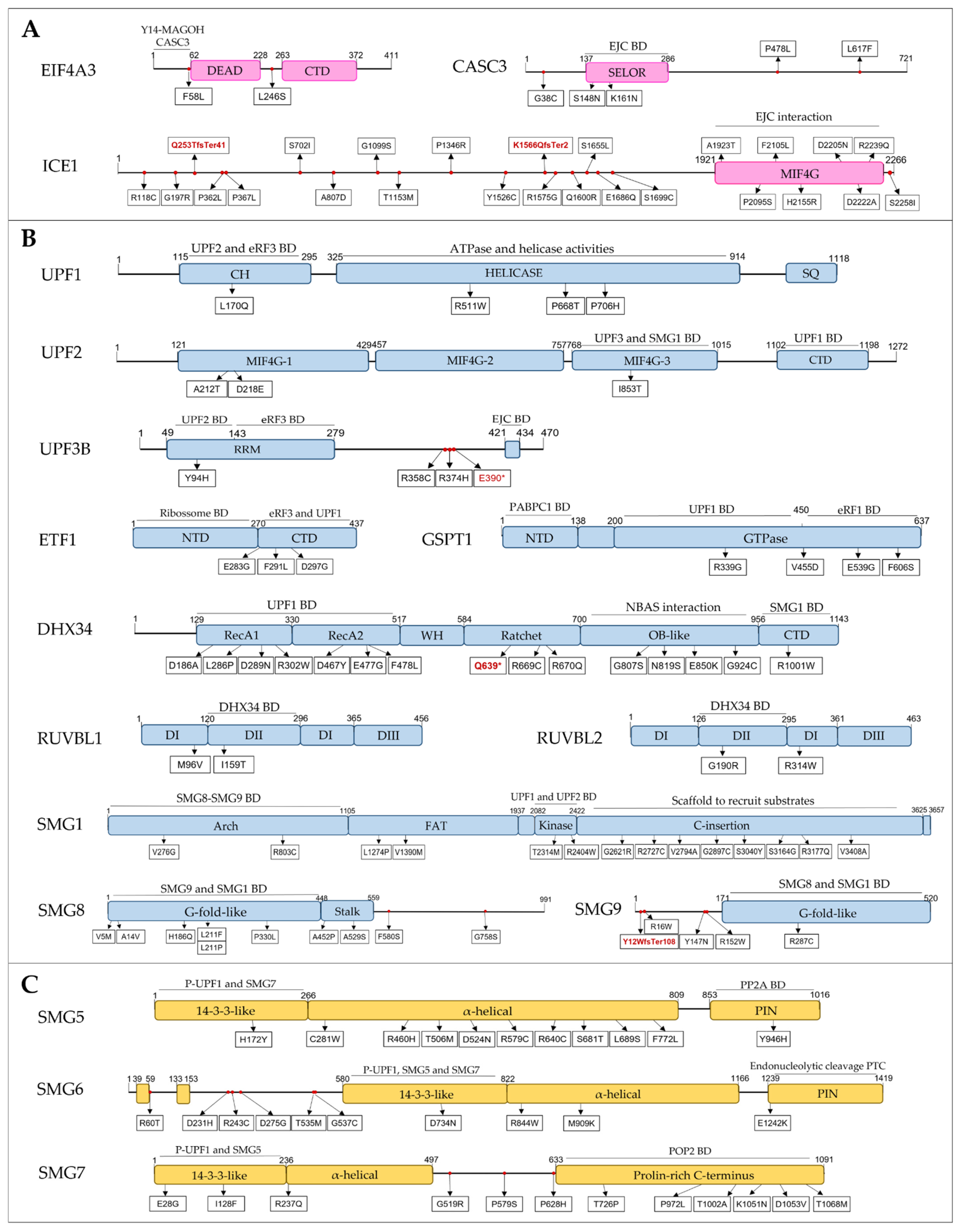
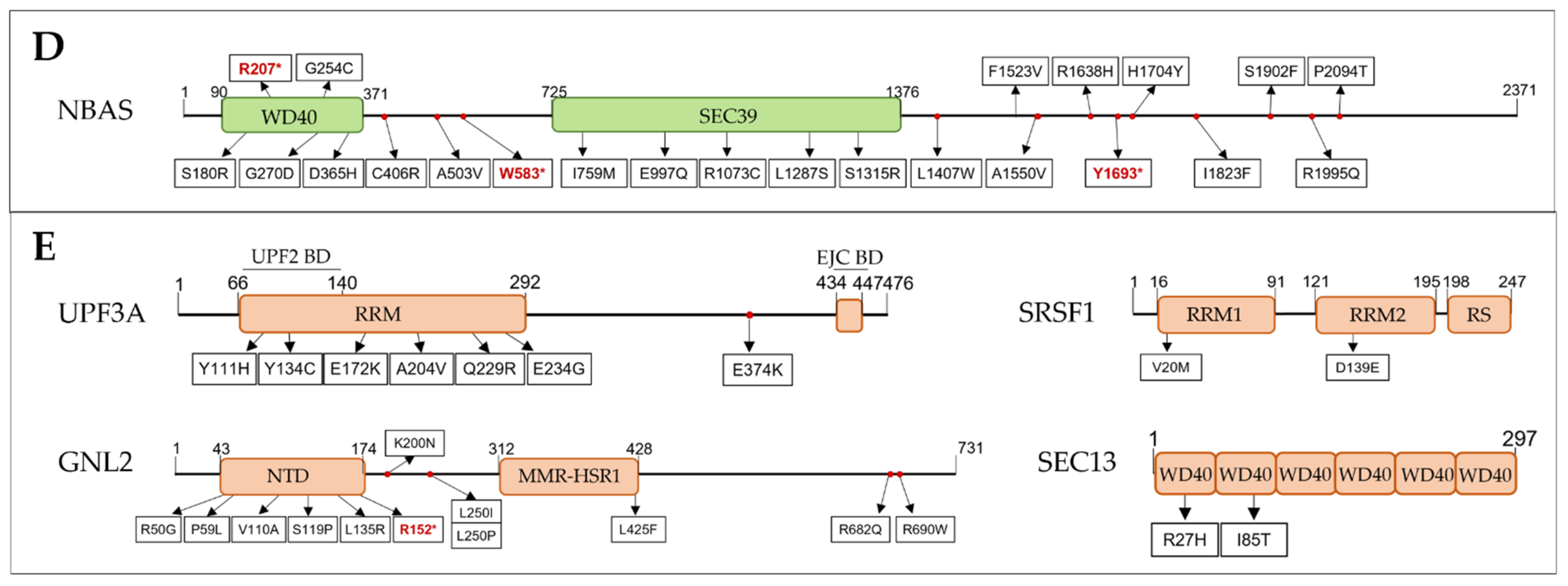
3.2. Discovery of SNVs in NMD Genes

3.2.1. EJC Components and Regulators

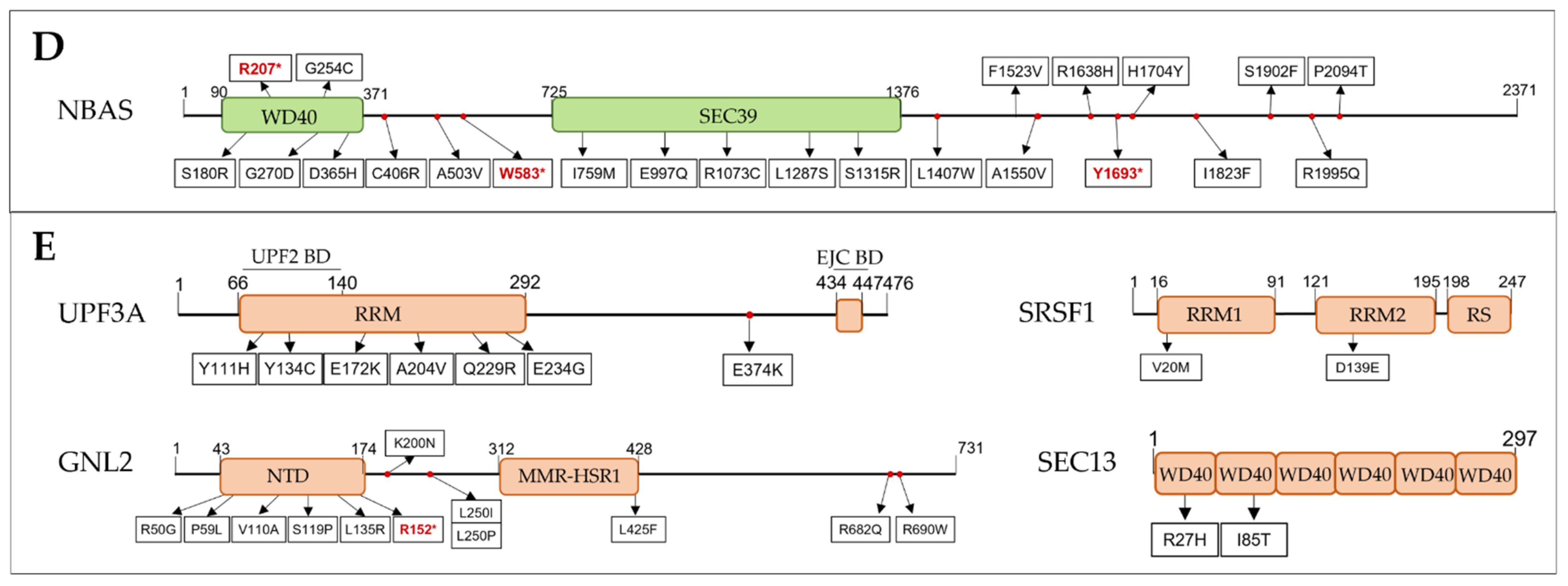
3.2.2. SURF and DECID Complexes
3.2.3. mRNA Decay
3.2.4. NMD Regulators
3.3. CNVs Encompassing NMD Genes in ASD Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; Am. Psychiatr. Publ.: Washington, DC, USA, 2013. [Google Scholar]
- Chiarotti, F.; Venerosi, A. Epidemiology of Autism Spectrum Disorders: A Review of Worldwide Prevalence Estimates Since 2014. Brain Sci. 2020, 10, 274. [Google Scholar] [CrossRef] [PubMed]
- Colvert, E.; Tick, B.; McEwen, F.; Stewart, C.; Curran, S.R.; Woodhouse, E.L.; Gillan, N.; Hallett, V.; Lietz, S.; Garnett, T.; et al. Heritability of Autism Spectrum Disorder in a UK Population-Based Twin Sample. JAMA Psychiatry 2015, 72, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Hultman, C.; Larsson, H.; Reichenberg, A. The Heritability of Autism Spectrum Disorder. JAMA 2017, 318, 1182–1184. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.; Ercan-sencicek, G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; State, M. Multiple recurrent de novo copy number variations (CNVs), including duplications of the 7q11.23 Williams-Beuren syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of Genes and Cellular Pathways Dysregulated in Autism Spectrum Disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [Green Version]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [Green Version]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Asif, M.; Martiniano, H.F.D.M.C.; Marques, A.R.; Santos, J.X.; Vilela, J.; Rasga, C.M.; Oliveira, G.; Couto, F.; Vicente, A.M. Identification of biological mechanisms underlying a multidimensional ASD phenotype using machine learning. Transl. Psychiatry 2020, 10, 43. [Google Scholar] [CrossRef] [Green Version]
- Parikshak, N.; Swarup, V.; Belgard, T.; Irimia, M.; Ramaswami, G.; Gandal, M.; Hartl, C.; Leppä, V.; Ubieta, L.D.L.T.; Huang, J.; et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Xavier Santos, J.; Rasga, C.; Moura Vicente, A. Exposure to Xenobiotics and Gene-Environment Interactions in Autism Spectrum Disorder: A Systematic Review. In Autism Spectrum Disorder—Profile, Heterogeneity, Neurobiology and Intervention; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Tarpey, P.S.; Raymond, F.L.; Nguyen, L.S.; Rodriguez, J.; Hackett, A.; Vandeleur, L.; Smith, R.; Shoubridge, C.; Edkins, S.; Stevens, C.; et al. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat. Genet. 2007, 39, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laumonnier, F.; Shoubridge, C.; Antar, C.; Nguyen, L.S.; Van Esch, H.; Kleefstra, T.; Briault, S.; Fryns, J.P.; Hamel, B.; Chelly, J.; et al. Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol. Psychiatry 2010, 15, 767–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Hoekzema, K.; Vecchio, D.; Wu, H.; Sulovari, A.; Coe, B.P.; Gillentine, M.A.; Wilfert, A.B.; Perez-Jurado, L.A.; Kvarnung, M.; et al. Large-scale targeted sequencing identifies risk genes for neurodevelopmental disorders. Nat. Commun. 2020, 11, 4932. [Google Scholar] [CrossRef]
- Forsyth, J.K.; Nachun, D.; Gandal, M.; Geschwind, D.H.; Anderson, A.E.; Coppola, G.; Bearden, C.E. Synaptic and Gene Regulatory Mechanisms in Schizophrenia, Autism, and 22q11.2 Copy Number Variant–Mediated Risk for Neuropsychiatric Disorders. Biol. Psychiatry 2020, 87, 150–163. [Google Scholar] [CrossRef] [Green Version]
- The Brainstorm Consortium; Anttila, V.; Bulik-Sullivan, B.; Finucane, H.K.; Walters, R.K.; Bras, J.; Duncan, L.; Escott-Price, V.; Falcone, G.J.; Gormley, P.; et al. Analysis of shared heritability in common disorders of the brain. Science 2018, 360, eaap8757. [Google Scholar] [CrossRef] [Green Version]
- Wittmann, J.; Hol, E.M.; Jäck, H.-M. hUPF2 Silencing Identifies Physiologic Substrates of Mammalian Nonsense-Mediated mRNA Decay. Mol. Cell. Biol. 2006, 26, 1272–1287. [Google Scholar] [CrossRef] [Green Version]
- Jolly, L.; Homan, C.; Jacob, R.; Barry, S.; Gecz, J. The UPF3B gene, implicated in intellectual disability, autism, ADHD and childhood onset schizophrenia regulates neural progenitor cell behaviour and neuronal outgrowth. Hum. Mol. Genet. 2013, 22, 4673–4687. [Google Scholar] [CrossRef] [Green Version]
- Alrahbeni, T.; Sartor, F.; Anderson, J.; Miedzybrodzka, Z.; McCaig, C.; Müller, B. Full UPF3B function is critical for neuronal differentiation of neural stem cells. Mol. Brain 2015, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Lou, C.-H.; Dumdie, J.; Goetz, A.; Shum, E.Y.; Brafman, D.; Liao, X.; Mora-Castilla, S.; Ramaiah, M.; Cook-Andersen, H.; Laurent, L.; et al. Nonsense-Mediated RNA Decay Influences Human Embryonic Stem Cell Fate. Stem Cell Rep. 2016, 6, 844–857. [Google Scholar] [CrossRef] [Green Version]
- Notaras, M.; Allen, M.; Longo, F.; Volk, N.; Toth, M.; Jeon, N.L.; Klann, E.; Colak, D. UPF2 leads to degradation of dendritically targeted mRNAs to regulate synaptic plasticity and cognitive function. Mol. Psychiatry 2020, 25, 3360–3379. [Google Scholar] [CrossRef] [PubMed]
- McSweeney, C.; Dong, F.; Chen, M.; Vitale, J.; Xu, L.; Crowley, N.; Luscher, B.; Zou, D.; Mao, Y. Full function of exon junction complex factor, Rbm8a, is critical for interneuron development. Transl. Psychiatry 2020, 10, 379. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, R.; Nogueira, G.; da Costa, P.J.; Pinto, F.; Romão, L. Nonsense-Mediated mRNA Decay in Development, Stress and Cancer. In The mRNA Metabolism in Human Disease; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2019; Volume 1157, ISBN 9783030199661. [Google Scholar] [CrossRef]
- Tejada, M.I.; Villate, O.; Ibarluzea, N.; De La Hoz, A.B.; Martínez-Bouzas, C.; Beristain, E.; Martinez, F.; Friez, M.J.; Sobrino, B.; Barros, F. Molecular and Clinical Characterization of a Novel Nonsense Variant in Exon 1 of the UPF3B Gene Found in a Large Spanish Basque Family (MRX82). Front. Genet. 2019, 10, 1074. [Google Scholar] [CrossRef] [PubMed]
- Domingo, D.; Nawaz, U.; Corbett, M.; Espinoza, J.L.; Tatton-Brown, K.; Coman, D.; Wilkinson, M.F.; Gecz, J.; Jolly, L.A. A synonymous UPF3B variant causing a speech disorder implicates NMD as a regulator of neurodevelopmental disorder gene networks. Hum. Mol. Genet. 2020, 29, 2568–2578. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Imamachi, N.; Pröschel, C.; Mitsutomi, S.; Nagao, R.; Akimitsu, N.; Maquat, L.E. Loss of the fragile X syndrome protein FMRP results in misregulation of nonsense-mediated mRNA decay. Nat. Cell Biol. 2021, 23, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, G.; Fernandes, R.; García-Moreno, J.F.; Romão, L. Nonsense-mediated RNA decay and its bipolar function in cancer. Mol. Cancer 2021, 20, 72. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S.; The AmiGO Hub; The Web Presence Working Group. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef]
- Tweedie, S.; Braschi, B.; Gray, K.; Jones, T.E.M.; Seal, R.L.; Yates, B.; Bruford, E.A. Genenames.org: The HGNC and VGNC resources in 2021. Nucleic Acids Res. 2021, 49, D939–D946. [Google Scholar] [CrossRef]
- Buxbaum, J.D.; Daly, M.J.; Devlin, B.; Lehner, T.; Roeder, K.; State, M.W. The Autism Sequencing Consortium: Large-Scale, High-Throughput Sequencing in Autism Spectrum Disorders. Neuron 2012, 76, 1052–1056. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Sabo, A.; Neale, B.M.; Nagaswamy, U.; Stevens, C.; Lim, E.; Bodea, C.A.; Muzny, D.; Reid, J.G.; Banks, E.; et al. Analysis of Rare, Exonic Variation amongst Subjects with Autism Spectrum Disorders and Population Controls. PLoS Genet. 2013, 9, e1003443. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischbach, G.D.; Lord, C. The Simons Simplex Collection: A Resource for Identification of Autism Genetic Risk Factors. Neuron 2010, 68, 192–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Samocha, K.; Robinson, E.; Sanders, S.; Stevens, C.; Sabo, A.; McGrath, L.; Kosmicki, J.A.; Rehnström, K.; Mallick, S.; Kirby, A.; et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014, 46, 944–950. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Sjöstedt, E.; Zhong, W.; Fagerberg, L.; Karlsson, M.; Mitsios, N.; Adori, C.; Oksvold, P.; Edfors, F.; Limiszewska, A.; Hikmet, F.; et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 2020, 367, eaay5947. [Google Scholar] [CrossRef]
- Lindsay, S.J.; Xu, Y.; Lisgo, S.N.; Harkin, L.F.; Copp, A.J.; Gerrelli, D.; Clowry, G.J.; Talbot, A.; Keogh, M.J.; Coxhead, J.; et al. HDBR Expression: A Unique Resource for Global and Individual Gene Expression Studies during Early Human Brain Development. Front. Neuroanat. 2016, 10, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehring, N.H.; Kunz, J.B.; Neu-Yilik, G.; Breit, S.; Viegas, M.H.; Hentze, M.W.; Kulozik, A.E. Exon-Junction Complex Components Specify Distinct Routes of Nonsense-Mediated mRNA Decay with Differential Cofactor Requirements. Mol. Cell 2005, 20, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Chamieh, H.; Ballut, L.; Bonneau, F.; Le Hir, H. NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat. Struct. Mol. Biol. 2008, 15, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Wachsmuth, L.; Kulozik, A.E.; Gehring, N.H. Two mammalian MAGOH genes contribute to exon junction complex composition and nonsense-mediated decay. RNA Biol. 2013, 10, 1291–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabin, J.W.; Woodward, L.A.; Patton, R.D.; Yi, Z.; Jia, M.; Wysocki, V.H.; Bundschuh, R.; Singh, G. The Exon Junction Complex Undergoes a Compositional Switch that Alters mRNP Structure and Nonsense-Mediated mRNA Decay Activity. Cell Rep. 2018, 25, 2431–2446.e7. [Google Scholar] [CrossRef] [Green Version]
- Bono, F.; Ebert, J.; Lorentzen, E.; Conti, E. The Crystal Structure of the Exon Junction Complex Reveals How It Maintains a Stable Grip on mRNA. Cell 2006, 126, 713–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerbracht, J.V.; Boehm, V.; Britto-Borges, T.; Kallabis, S.; Wiederstein, J.L.; Ciriello, S.; Aschemeier, D.U.; Krüger, M.; Frese, C.K.; Altmüller, J.; et al. CASC3 promotes transcriptome-wide activation of nonsense-mediated decay by the exon junction complex. Nucleic Acids Res. 2020, 48, 8626–8644. [Google Scholar] [CrossRef]
- Baird, T.D.; Cheng, K.C.-C.; Chen, Y.-C.; Buehler, E.; Martin, S.E.; Inglese, J.; Hogg, J.R. ICE1 promotes the link between splicing and nonsense-mediated mRNA decay. eLife 2018, 7, e33178. [Google Scholar] [CrossRef]
- Gehring, N.H.; Lamprinaki, S.; Kulozik, A.E.; Hentze, M.W. Disassembly of Exon Junction Complexes by PYM. Cell 2009, 137, 536–548. [Google Scholar] [CrossRef] [Green Version]
- Kashima, I.; Yamashita, A.; Izumi, N.; Kataoka, N.; Morishita, R.; Hoshino, S.; Ohno, M.; Dreyfuss, G.; Ohno, S. Binding of a novel SMG-1–Upf1–eRF1–eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006, 20, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Okada-Katsuhata, Y.; Yamashita, A.; Kutsuzawa, K.; Izumi, N.; Hirahara, F.; Ohno, S. N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res. 2012, 40, 1251–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neu-Yilik, G.; Raimondeau, E.; Eliseev, B.; Yeramala, L.; Amthor, B.; Deniaud, A.; Huard, K.; Kerschgens, K.; Hentze, M.W.; Schaffitzel, C.; et al. Dual function of UPF3B in early and late translation termination. EMBO J. 2017, 36, 2968–2986. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, Y.; Li, X.; Serin, G.; Maquat, L.E. Evidence for a Pioneer Round of mRNA Translation: mRNAs Subject to Nonsense-Mediated Decay in Mammalian Cells Are Bound by CBP80 and CBP20. Cell 2001, 106, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Hosoda, N.; Kim, Y.K.; Lejeune, F.; Maquat, L.E. CBP80 promotes interaction of Upf1 with Upf2 during nonsense-mediated mRNA decay in mammalian cells. Nat. Struct. Mol. Biol. 2005, 12, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Sato, H.; Tang, Y.; Matsuda, D.; Maquat, L.E. UPF1 Association with the Cap-Binding Protein, CBP80, Promotes Nonsense-Mediated mRNA Decay at Two Distinct Steps. Mol. Cell 2010, 39, 396–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rufener, S.C.; Mühlemann, O. eIF4E-bound mRNPs are substrates for nonsense-mediated mRNA decay in mammalian cells. Nat. Struct. Mol. Biol. 2013, 20, 710–717. [Google Scholar] [CrossRef]
- Yamashita, A.; Izumi, N.; Kashima, I.; Ohnishi, T.; Saari, B.; Katsuhata, Y.; Muramatsu, R.; Morita, T.; Iwamatsu, A.; Hachiya, T.; et al. SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev. 2009, 23, 1091–1105. [Google Scholar] [CrossRef] [Green Version]
- Deniaud, A.; Karuppasamy, M.; Bock, T.; Masiulis, S.; Huard, K.; Garzoni, F.; Kerschgens, K.; Hentze, M.W.; Kulozik, A.E.; Beck, M.; et al. A network of SMG-8, SMG-9 and SMG-1 C-terminal insertion domain regulates UPF1 substrate recruitment and phosphorylation. Nucleic Acids Res. 2015, 43, 7600–7611. [Google Scholar] [CrossRef] [Green Version]
- Longman, D.; Hug, N.; Keith, M.; Anastasaki, C.; Patton, E.E.; Grimes, G.; Cáceres, J.F. DHX34 and NBAS form part of an autoregulatory NMD circuit that regulates endogenous RNA targets in human cells, zebrafish and Caenorhabditis elegans. Nucleic Acids Res. 2013, 41, 8319–8331. [Google Scholar] [CrossRef] [Green Version]
- Hug, N.; Cáceres, J.F. The RNA Helicase DHX34 Activates NMD by Promoting a Transition from the Surveillance to the Decay-Inducing Complex. Cell Rep. 2014, 8, 1845–1856. [Google Scholar] [CrossRef] [Green Version]
- Melero, R.; Hug, N.; López-Perrote, A.; Yamashita, A.; Caceres, J.F.; Llorca, O. The RNA helicase DHX34 functions as a scaffold for SMG1-mediated UPF1 phosphorylation. Nat. Commun. 2016, 7, 10585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, N.; Yamashita, A.; Iwamatsu, A.; Kurata, R.; Nakamura, H.; Saari, B.; Hirano, H.; Anderson, P.; Ohno, S. AAA+ Proteins RUVBL1 and RUVBL2 Coordinate PIKK Activity and Function in Nonsense-Mediated mRNA Decay. Sci. Signal. 2010, 3, ra27. [Google Scholar] [CrossRef] [PubMed]
- López-Perrote, A.; Hug, N.; González-Corpas, A.; Rodríguez, C.F.; Serna, M.; García-Martín, C.; Boskovic, J.; Fernandez-Leiro, R.; Caceres, J.F.; Llorca, O. Regulation of RUVBL1-RUVBL2 AAA-ATPases by the nonsense-mediated mRNA decay factor DHX34, as evidenced by Cryo-EM. bioRxiv 2020, 9, e63042. [Google Scholar] [CrossRef]
- Anders, K.R.; Grimson, A.; Anderson, P. SMG-5, required for C.elegans nonsense-mediated mRNA decay, associates with SMG-2 and protein phosphatase 2A. EMBO J. 2003, 22, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, T.; Yamashita, A.; Kashima, I.; Schell, T.; Anders, K.; Grimson, A.; Hachiya, T.; Hentze, M.; Anderson, P.; Ohno, S. Phosphorylation of hUPF1 Induces Formation of mRNA Surveillance Complexes Containing hSMG-5 and hSMG-7. Mol. Cell 2003, 12, 1187–1200. [Google Scholar] [CrossRef]
- Huntzinger, E.; Kashima, I.; Fauser, M.; Saulière, J.; Izaurralde, E. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 2008, 14, 2609–2617. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, P.; Josi, C.; Kurosawa, H.; Yamashita, A.; Mühlemann, O. A novel phosphorylation-independent interaction between SMG6 and UPF1 is essential for human NMD. Nucleic Acids Res. 2014, 42, 9217–9235. [Google Scholar] [CrossRef] [Green Version]
- Eberle, A.B.; Lykke-Andersen, S.; Mühlemann, O.; Jensen, T.H. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat. Struct. Mol. Biol. 2009, 16, 49–55. [Google Scholar] [CrossRef]
- Loh, B.; Jonas, S.; Izaurralde, E. The SMG5–SMG7 heterodimer directly recruits the CCR4-NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev. 2013, 27, 2125–2138. [Google Scholar] [CrossRef] [Green Version]
- Lai, T.; Cho, H.; Liu, Z.; Bowler, M.W.; Piao, S.; Parker, R.; Kim, Y.K.; Song, H. Structural Basis of the PNRC2-Mediated Link between mRNA Surveillance and Decapping. Structure 2012, 20, 2025–2037. [Google Scholar] [CrossRef] [Green Version]
- Lejeune, F.; Li, X.; Maquat, L.E. Nonsense-Mediated mRNA Decay in Mammalian Cells Involves Decapping, Deadenylating, and Exonucleolytic Activities. Mol. Cell 2003, 12, 675–687. [Google Scholar] [CrossRef]
- Gregersen, L.H.; Schueler, M.; Munschauer, M.; Mastrobuoni, G.; Chen, W.; Kempa, S.; Dieterich, C.; Landthaler, M. MOV10 Is a 5′ to 3′ RNA Helicase Contributing to UPF1 mRNA Target Degradation by Translocation along 3′ UTRs. Mol. Cell 2014, 54, 573–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staals, R.; Bronkhorst, A.W.; Schilders, G.; Slomovic, S.; Schuster, G.; Heck, A.; Raijmakers, R.; Pruijn, G.J.M. Dis3-like 1: A novel exoribonuclease associated with the human exosome. EMBO J. 2010, 29, 2358–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Costa, P.J.; Menezes, J.; Saramago, M.; García-Moreno, J.; Santos, H.A.; Carvalho, M.D.G.; Arraiano, C.M.; Viegas, S.C.; Romão, L. A role for DIS3L2 over natural nonsense-mediated mRNA decay targets in human cells. Biochem. Biophys. Res. Commun. 2019, 518, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Longman, D.; Jackson-Jones, K.A.; Maslon, M.; Murphy, L.C.; Young, R.S.; Stoddart, J.J.; Hug, N.; Taylor, M.S.; Papadopoulos, D.K.; Cáceres, J.F. Identification of a localized nonsense-mediated decay pathway at the endoplasmic reticulum. Genes Dev. 2020, 34, 1075–1088. [Google Scholar] [CrossRef]
- Ivanov, P.; Gehring, N.H.; Kunz, J.B.; Hentze, M.; Kulozik, A.E. Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J. 2008, 27, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Peixeiro, I.; Inácio, A.; Barbosa, C.; Silva, A.L.; Liebhaber, S.A.; Romão, L. Interaction of PABPC1 with the translation initiation complex is critical to the NMD resistance of AUG-proximal nonsense mutations. Nucleic Acids Res. 2011, 40, 1160–1173. [Google Scholar] [CrossRef] [Green Version]
- Fatscher, T.; Boehm, V.; Weiche, B.; Gehring, N.H. The interaction of cytoplasmic poly(A)-binding protein with eukaryotic initiation factor 4G suppresses nonsense-mediated mRNA decay. RNA 2014, 20, 1579–1592. [Google Scholar] [CrossRef] [Green Version]
- Joncourt, R.; Eberle, A.B.; Rufener, S.C.; Mühlemann, O. Eukaryotic Initiation Factor 4G Suppresses Nonsense-Mediated mRNA Decay by Two Genetically Separable Mechanisms. PLoS ONE 2014, 9, e104391. [Google Scholar] [CrossRef] [Green Version]
- Shum, E.Y.; Jones, S.H.; Shao, A.; Dumdie, J.; Krause, M.D.; Chan, W.-K.; Lou, C.-H.; Espinoza, J.L.; Song, H.-W.; Phan, M.H.; et al. The Antagonistic Gene Paralogs Upf3a and Upf3b Govern Nonsense-Mediated RNA Decay. Cell 2016, 165, 382–395. [Google Scholar] [CrossRef] [Green Version]
- Morris, C.; Wittmann, J.; Jäck, H.-M.; Jalinot, P. Human INT6/eIF3e is required for nonsense-mediated mRNA decay. EMBO Rep. 2007, 8, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Aznarez, I.; Nomakuchi, T.T.; Tetenbaum-Novatt, J.; Rahman, M.A.; Fregoso, O.; Rees, H.; Krainer, A.R. Mechanism of Nonsense-Mediated mRNA Decay Stimulation by Splicing Factor SRSF1. Cell Rep. 2018, 23, 2186–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casadio, A.; Longman, D.; Hug, N.; Delavaine, L.; Baier, R.V.; Alonso, C.R.; Cáceres, J.F. Identification and characterization of novel factors that act in the nonsense-mediated mRNA decay pathway in nematodes, flies and mammals. EMBO Rep. 2015, 16, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Pratt, G.A.; Martinez, F.J.; Yeo, G.W.; Lykke-Andersen, J. Target Discrimination in Nonsense-Mediated mRNA Decay Requires Upf1 ATPase Activity. Mol. Cell 2015, 59, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Clerici, M.; Deniaud, A.; Boehm, V.; Gehring, N.H.; Berger-Schaffitzel, C.H.; Cusack, S. Structural and functional analysis of the three MIF4G domains of nonsense-mediated decay factor UPF2. Nucleic Acids Res. 2014, 42, 2673–2686. [Google Scholar] [CrossRef]
- Jonas, S.; Weichenrieder, O.; Izaurralde, E. An unusual arrangement of two 14-3-3-like domains in the SMG5–SMG7 heterodimer is required for efficient nonsense-mediated mRNA decay. Genes Dev. 2013, 27, 211–225. [Google Scholar] [CrossRef] [Green Version]
- O’Rahilly, R.; Müller, F. Significant features in the early prenatal development of the human brain. Ann. Anat. 2008, 190, 105–118. [Google Scholar] [CrossRef]
- Gamazon, E.; Stranger, B.E. The impact of human copy number variation on gene expression. Brief. Funct. Genom. 2015, 14, 352–357. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.S.; Kim, H.-G.; Rosenfeld, J.A.; Shen, Y.; Gusella, J.F.; Lacassie, Y.; Layman, L.C.; Shaffer, L.G.; Gécz, J. Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum. Mol. Genet. 2013, 22, 1816–1825. [Google Scholar] [CrossRef] [Green Version]
- Correia, C.T.; Conceição, I.C.; Oliveira, B.; Coelho, J.; Sousa, I.; Sequeira, A.F.; Almeida, J.; Café, C.; Duque, F.; Mouga, S.; et al. Recurrent duplications of the annexin A1 gene (ANXA1) in autism spectrum disorders. Mol. Autism 2014, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Albers, C.A.; Paul, D.S.; Schulze, H.; Freson, K.; Stephens, J.C.; Smethurst, P.A.; Jolley, J.D.; Cvejic, A.; Kostadima, M.; Bertone, P.; et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat. Genet. 2012, 44, 435–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzahrani, F.; Kuwahara, H.; Long, Y.; Al-Owain, M.; Tohary, M.; AlSayed, M.; Mahnashi, M.; Fathi, L.; Alnemer, M.; Al-Hamed, M.H.; et al. Recessive, Deleterious Variants in SMG8 Expand the Role of Nonsense-Mediated Decay in Developmental Disorders in Humans. Am. J. Hum. Genet. 2020, 107, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Stoica, L.; Liu, Y.; Zhu, P.J.; Bhattacharya, A.; Buffington, S.A.; Huq, R.; Eissa, N.T.; Larsson, O.; Porse, B.T.; et al. Inhibition of Upf2-Dependent Nonsense-Mediated Decay Leads to Behavioral and Neurophysiological Abnormalities by Activating the Immune Response. Neuron 2019, 104, 665–679.e8. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NMD Group | Gene SYMBOL (1) | Alternative Name (2) | Role in NMD | References |
|---|---|---|---|---|
| EJC | EIF4A3 | eIF4AIII DDX48 | RNA helicase eukaryotic initiation factor 4A3 is a core EJC factor that interacts with the Y14-MAGOH heterodimer to provide a stable and direct binding site for the UPF3B protein and activate NMD | [45,46] |
| RBM8A | Y14 | RNA-binding motif protein 8A is a core EJC factor that interacts with eIF4A3 and MAGOH to provide a stable and direct binding site for the UPF3B protein and activate NMD | [45,46] | |
| MAGOH | MAGOH1 | Mago nashi homolog protein is a core EJC factor that interacts with eIF4A3 and Y14 to provide a stable and direct binding site for the UPF3B protein and activate NMD | [45,46,47] | |
| MAGOHB | MGN2 | Mago nashi protein homolog B is a paralog of MAGOH that interacts with eIF4A3 and Y14 forming the trimeric EJC core to activate NMD | [47] | |
| RNPS1 | RNA-binding protein S1 is a component of the SR-rich EJCs that enhances NMD in early phase of the pathway | [45,48] | ||
| CASC3 | MLN51 BTZ | The peripheral EJC component CASC3 activates NMD and promotes SMG6-dependent endonucleolytic cleavage | [48,49,50] | |
| ICE1 | KIAA0947 | Component of the little elongation complex promotes the association of EJC with UPF3B and activates NMD | [51] | |
| PYM1 | WIGB | Ribosome-associated protein PYM interacts with Y14-MAGOH triggering EJC disassembly which leads to NMD inhibition | [52] | |
| SURF-DECID | UPF1 | RENT1 smg-2 | Up-frameshift protein 1 is the central component of the NMD pathway; its helicase and ATPase activities are essential to trigger NMD | [46,53,54] |
| UPF2 | RENT2 smg-3 | Up-frameshift protein 2 and UPF3B interact with UPF1 favoring its phosphorylation by SMG1 and formation of DECID complex | [46,53] | |
| UPF3B | UPF3X | Up-frameshift protein 3B and UPF2 interact with UPF1 favoring its phosphorylation by SMG1 and formation of DECID complex; UPF3B also forms a stable trimeric complex with eRF1-eRF3a to promote dissociation of the termination complexes and triggers NMD | [46,53,54,55] | |
| ETF1 | eRF1 | Eukaryotic release factor 1 is part of the eRF1–eRF3 translation termination complex that associates with UPF1 and SMG1-SMG8-SMG9 to form SURF and activate NMD | [53] | |
| GSPT1 | eRF3a | Eukaryotic release factor 3 is part of the eRF1–eRF3 translation termination complex that associates with UPF1 and SMG1-SMG8-SMG9 to form SURF and activate NMD | [53] | |
| NCBP1 | CBP80 | Component of the cap-binding complex (CBC) directly binds to UPF1, promoting the interaction with UPF2 to form SURF and activate NMD | [56,57,58] | |
| NCBP2 | CBP20 | Component of the CBC is essential for the stability of complex | [56,57,58] | |
| EIF4E | eIF4E | Eukaryotic translation initiation factor 4E binds to UPF1 and activates NMD | [59] | |
| SMG1 | ATX | Suppressor of morphogenesis in genitalia-1 associate with SMG8-SMG9 to form the SMG1c kinase complex that catalyzes UPF1 phosphorylation | [53,60,61] | |
| SMG8 | Suppressor of morphogenesis in genitalia-8 and suppressor of morphogenesis in genitalia-9 are co-factors that regulate SMG1 kinase activity | [53,60,61] | ||
| SMG9 | ||||
| DHX34 | KIAA0134 | RNA helicase DHX34 binds SMG1 and promotes UPF1 phosphorylation, triggering the conversion from the SURF to the DECID complex | [62,63,64] | |
| RUVBL1 | RVB1 | AAA-ATPases RUVBL1 and RUVBL2 form a hetero-hexameric ring promoting the transition from SURF to the DECID complex | [65,66] | |
| RUVBL2 | RVB2 | |||
| mRNA decay | SMG5 | EST1B | Suppressor of morphogenesis in genitalia-5 and -7 form a heterodimer that binds p-UPF1 and recruit decapping enzymes, the CCR4-NOT complex (through CNOT8) and PP2A | [54,67,68] |
| SMG7 | EST1C | |||
| SMG6 | EST1A | Endonuclease that interacts both with UPF1 and p-UPF1 and cleaves NMD targets close to the PTC | [54,69,70,71] | |
| CNOT8 | POP2 CAF1 | CCR4-NOT transcription complex subunit 8 is recruited by SMG7 to degrade NMD targets | [72] | |
| DCP1A | mRNA-decapping enzyme 1A is a decapping activator and together with PNRC2 stimulate the decapping activity of DCP2 | [73] | ||
| PNRC2 | Proline rich nuclear receptor coactivator 2 binds p-UPF1 and stimulate the decapping activity of DCP2 | [73] | ||
| DCP2 | Decapping protein engaged in the 5′→3′ mRNA degradation | [74] | ||
| MOV10 | gb110 | RNA helicase contributes to degradation of UPF1-regulated transcripts | [75] | |
| PPP2CA | PP2AC | Protein phosphatase 2 (PP2A) promotes dephosphorylation of UPF1; both structural (PPP2R1A) and catalytic (PPP2CA) subunits of PP2A interact with SMG5 | [54,67,68] | |
| PPP2R1A | PP2AA | |||
| XRN1 | Exonuclease involved in 5′→3′ mRNA degradation | [74] | ||
| DIS3L | DIS3L1 | Core exosome-associated factor involved in the 3′→5′ mRNA degradation | [76] | |
| DIS3L2 | Exoribonuclease that degrades mRNA from 3′→5′ | [77] | ||
| EXOSC10 | PM/Scl100 Rrp6p | Exosome catalytic subunit involved in the 3′→5′ mRNA degradation | [74] | |
| PARN | Ribonuclease engaged in the 3′→5′ mRNA degradation | [74] | ||
| ER–NMD | NBAS | NAG | Protein involved in Golgi-to-endoplasmic reticulum (ER) retrograde transport recruits UPF1 to the membrane of the ER and regulates a subset of NMD targets translated at the ER | [62,78] |
| Regulator | PABPC1 | PABP1 | Polyadenylate-binding protein 1 inhibits the interaction of UPF1 with eRF3, repressing NMD | [79,80] |
| EIF4G1 | EIF4G | Eukaryotic initiation factor 4G inhibits NMD | [81,82] | |
| UPF3A | UPF3 | Up-frameshift protein 3A compete with UPF3B for UPF2-binding and inhibits NMD | [83] | |
| FMR1 | FMRP | Fragile X mental retardation protein binds directly to UPF1 and acts as an NMD repressor | [28] | |
| EIF3E | INT6 EIF3S6 | Eukaryotic translation initiation factor 3 subunit E is a non-core eIF3 subunit that interacts with UPF2 and triggers NMD | [84] | |
| SRSF1 | SFRS1 | Serine/arginine-rich splicing factor 1 promotes NMD by enhancing UPF1-binding to the mRNA in the nucleus and it is also involved in UPF1 dephosphorylation | [85] | |
| SEC13 | GNL2 and SEC13 are conserved NMD factors that regulate endogenous NMD targets but their exact role is unknown | [86] | ||
| GNL2 | Ngp-1 |
| NMD Group | Gene | Location | CNV Type | Gene Region | Protein Domains Affected | ASD N (1) |
|---|---|---|---|---|---|---|
| EJC | RBM8A | 1q21.1 | Deletion | Whole gene | All domains | 1 |
| RBM8A | 1q21.1 | Duplication | Whole gene | All domains | 3 | |
| SURF-DECID | UPF2 | 10p14 | Duplication | Partial | MIF4G domains | 1 |
| UPF3B | Xq24 | Duplication | Whole gene | All domains | 1 | |
| GSPT1 | 16p13.13 | Duplication | Whole gene | All domains | 1 | |
| NCBP2 | 3q29 | Deletion | Whole gene | All domains | 1 | |
| DHX34 | 19q13.32 | Deletion | Whole gene | All domains | 1 | |
| RUVBL2 | 19q13.33 | Duplication | Partial | DI domain | 1 | |
| mRNA decay | DIS3L | 15q22.31 | Duplication | Whole gene | All domains | 1 |
| DIS3L2 | 2q37.1 | Deletion | Partial | Part of CSD2 and RNB domains | 1 | |
| DIS3L2 | 2q37.1 | Duplication | Partial | RNB and C-terminal S1 domain | 1 | |
| EXOSC10 | 1p36.22 | Deletion | Partial | PMC2NT, EXO1 and HRCD domains | 1 | |
| EXOSC10 | 1p36.22 | Duplication | Partial | EXO and HRCD domain | 1 | |
| MOV10 | 1p13.2 | Duplication | Whole gene | All domains | 2 | |
| PARN | 16p13.12 | Deletion | Partial | All domains | 1 | |
| PPP2R1A | 19q13.41 | Duplication | Partial | PP2A subunit B binding | 1 | |
| SMG6 | 17p13.3 | Duplication | Partial | PIN domain | 1 | |
| XRN1 | 3q23 | Duplication | Partial | XRN1 SH3-like domain | 13 | |
| ER-NMD | NBAS | 2p24.3 | Duplication | Partial | Sec39-like domain | 1 |
| Regulator | FMR1 | Xq27.3 | Deletion | Partial | KH2, NES and RGG domains | 1 |
| UPF3A | 13q34 | Duplication | Whole gene | All domains | 2 | |
| UPF3A | 13q34 | Duplication | Partial | EJC-binding domain | 1 |
| NMD Group | Gene | Location | SNVs (1) | CNVs (1) | N ASD (2) | pLI | mis_Z | SFARI (3) | |
|---|---|---|---|---|---|---|---|---|---|
| LoF | MISPD | ||||||||
| EJC | EIF4A3 | 17q25.3 | 1 | 1 | 1.00 | 4.02 | |||
| RBM8A | 1q21.1 | 2 | 4 | 0.57 | 2.16 | 1q21.1 region | |||
| CASC3 | 17q21.1 | 3 | 3 | 0.61 | 1.32 | ||||
| ICE1 | 5p15.32 | 2 | 7 | 10 | 1.00 | 0.73 | |||
| SURF-DECID | UPF1 | 19p13.11 | 4 | 4 | 1.00 | 5.63 | |||
| UPF2 | 10p14 | 1 | 3 | 1 | 11 | 1.00 | 3.19 | category 3 | |
| UPF3B | Xq24 | 1 | 3 | 1 | 7 | 0.98 | 1.84 | category 1 | |
| ETF1 | 5q31.2 | 1 | 3 | 6 | 1.00 | 4.39 | |||
| GSPT1 | 16p13.13 | 4 | 1 | 7 | 1.00 | 3.32 | |||
| DHX34 | 19q13.32 | 2 | 12 | 1 | 25 | 0.00 | −0.08 | ||
| RUVBL1 | 3q21.3 | 1 | 1 | 1.00 | 3.39 | ||||
| RUVBL2 | 19q13.33 | 1 | 1 | 2 | 1.00 | 3.11 | |||
| SMG1 | 16p12.3 | 1 | 12 | 13 | 1.00 | 3.30 | |||
| SMG8 | 17q22 | 6 | 8 | 0.01 | 1.73 | ||||
| SMG9 | 19q13.31 | 1 | 1 | 4 | 0.00 | 1.60 | |||
| mRNA decay | SMG5 | 1q22 | 2 | 2 | 0.01 | 1.13 | |||
| SMG6 | 17p13.3 | 2 | 1 | 6 | 0.98 | 0.18 | category 3 | ||
| SMG7 | 1q25.3 | 8 | 14 | 1.00 | 2.19 | ||||
| ER-NMD | NBAS(4) | 2p24.3 | 5 | 20 | 1 | 99 | 0.00 | −0.87 | |
| Regulator | UPF3A | 13q34 | 1 | 2 | 2 | 11 | 0.00 | −0.60 | |
| SRSF1 | 17q22 | 1 | 1 | 0.98 | 3.96 | ||||
| SEC13(4) | 3p25.3 | 2 | 2 | 0.02 | 0.62 | ||||
| GNL2(4) | 1p34.3 | 1 | 11 | 17 | 0.00 | 0.28 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, A.R.; Santos, J.X.; Martiniano, H.; Vilela, J.; Rasga, C.; Romão, L.; Vicente, A.M. Gene Variants Involved in Nonsense-Mediated mRNA Decay Suggest a Role in Autism Spectrum Disorder. Biomedicines 2022, 10, 665. https://doi.org/10.3390/biomedicines10030665
Marques AR, Santos JX, Martiniano H, Vilela J, Rasga C, Romão L, Vicente AM. Gene Variants Involved in Nonsense-Mediated mRNA Decay Suggest a Role in Autism Spectrum Disorder. Biomedicines. 2022; 10(3):665. https://doi.org/10.3390/biomedicines10030665
Chicago/Turabian StyleMarques, Ana Rita, João Xavier Santos, Hugo Martiniano, Joana Vilela, Célia Rasga, Luísa Romão, and Astrid Moura Vicente. 2022. "Gene Variants Involved in Nonsense-Mediated mRNA Decay Suggest a Role in Autism Spectrum Disorder" Biomedicines 10, no. 3: 665. https://doi.org/10.3390/biomedicines10030665
APA StyleMarques, A. R., Santos, J. X., Martiniano, H., Vilela, J., Rasga, C., Romão, L., & Vicente, A. M. (2022). Gene Variants Involved in Nonsense-Mediated mRNA Decay Suggest a Role in Autism Spectrum Disorder. Biomedicines, 10(3), 665. https://doi.org/10.3390/biomedicines10030665